

Abstract
Aberrant transactivation of a certain set of target genes by the β‐catenin and T‐cell factor‐4 nuclear complex has been considered crucial for the initiation of colorectal carcinogenesis. We previously identified splicing factor‐1 (SF1) as a novel component of the β‐catenin and T‐cell factor‐4 complex, and showed that the overexpression of SF1 inhibited the gene transactivational activity of the complex and markedly suppressed β‐catenin‐evoked colony formation by human embryonic kidney 293 cells. However, the involvement of SF1 in the process of carcinogenesis in vivo remains unclear. In the present study, we established SF1‐knockout mice using the gene trapping method. Homozygous mice (Sf1 −/–) died during embryonic development before embryonic day (E)8.5, whereas heterozygous (Sf1 +/–) mice were born alive and developed normally. Azoxymethane (AOM) was given at a dose of 10 mg/kg body weight once a week for 6 weeks to 7‐week‐old Sf1+/– and Sf1+/+ mice. At 23 weeks after the start of AOM the average number (5.5 ± 0.6 versus 2.2 ± 0.2 in females [P = 0.003, Mann–Whitney U‐test], 3.7 ± 0.2 versus 1.7 ± 0.7 in males [P = 0.014]) and volume of colon tumors per mouse (8.7 ± 1.6 versus 2.2 ± 0.5 mm3 per female [P = 0.0008], 11.3 ± 3.4 versus 0.6 ± 0.2 mm3 per male [P = 0.001]) were significantly higher in Sf1+/– than in Sf1+/+ mice. The increased susceptibility of Sf1 +/– mice to AOM‐induced colon tumorigenesis indicates the crucial involvement of SF1 in the β‐catenin‐mediated regulation of proliferation and differentiation of intestinal epithelial cells. (Cancer Sci 2007; 98: 1862–1867)
Abbreviations:
- AOM
azoxymethane
- APC
adenomatous polyposis coli
- Ct
threshold cycle
- E
embryonic day
- ES
embryonic stem
- HEK
human embryonic kidney
- LEF
lymphoid enhancer factor
- PCR
polymerase chain reaction
- premRNA
premessenger RNA
- RT
reverse transcription
- SA
splice acceptor
- SF1
splicing factor‐1
- TCF
T‐cell factor
Mutational inactivation of the tumor suppressor gene APC is the earliest and most frequent genetic event in colorectal cancer.( 1 ) The APC gene product forms a complex with axin/axin‐2, β‐catenin, casein kinase I, and glycogen synthase kinase 3β,( 2 , 3 ) and this multiprotein complex is essential for the phosphorylation of β‐catenin and subsequent degradation of phosphorylated β‐catenin through the ubiquitin–proteasome pathway.( 4 , 5 )β‐Catenin is the downstream effector of the Wnt signaling pathway.( 6 ) Mutation of the APC or β‐catenin (CTNNB1) gene leads to activation of Wnt signaling and accumulation of β‐catenin in the cytoplasm.( 5 , 7 ) The accumulated β‐catenin forms complexes with TCF/LEF family transcription factors.( 8 ) TCF‐4 is a TCF/LEF family transcription factor that regulates the gene expression necessary for the maintenance of the undifferentiated status of intestinal epithelial cells.( 9 , 10 , 11 , 12 ) Constitutive transactivation of a certain set of target genes of TCF‐4 by accumulation of β‐catenin protein imposes a crypt progenitor phenotype on intestinal epithelial cells that is considered crucial for colorectal carcinogenesis.( 11 )
In our previous study we carried out large‐scale protein profiling to identify proteins whose expression is regulated by the TCF‐4 and β‐catenin nuclear complex.( 13 ) We examined more than 4000 peptides derived from colorectal cancer cells, and identified 87 proteins whose expression was significantly upregulated or downregulated by induction of dominant negative TCF‐4. A zinc finger protein SF1/ZNF162/ZFM1 was one of the proteins negatively regulated by β‐catenin. The expression of SF1 was correlated with the differentiation status of intestinal epithelial cells and inversely correlated with tumorigenesis. Furthermore, we found that SF1 was a novel component of the TCF‐4 and β‐catenin complex. SF1 cDNA transfection markedly inhibited the transcriptional activity of the TCF‐4 and β‐catenin complex and suppressed β‐catenin‐evoked colony formation by HEK293 cells. All these results indicated that SF1 is a negative regulator of the oncogenic activity of the TCF‐4 and β‐catenin complex, but it has remained undetermined whether SF1 actually exerts a protective function during the process of colorectal carcinogenesis in vivo.
In the present study we adopted a genetic approach to clarify the functional involvement of SF1 in colon carcinogenesis. We found that the haploinsufficiency of the Sf1 gene that encodes SF1 dramatically enhances the tumorigenic effects of a well‐characterized organotropic colon carcinogen, AOM.
Materials and Methods
Generation of Sf1‐knockout mice. Animal experiments were carried out according to the guidelines of the National Cancer Center Research Institute (Tokyo, Japan), which meets all the ethical requirements stipulated by Japanese law. The experimental protocol was reviewed and approved by the institutional ethics committee.
Sf1‐knockout mice were generated by the gene trapping method.( 14 , 15 ) The gene trap vector pU17( 15 ) contains 1.8 kb of an intron and an SA sequence from the mouse En2 gene, the β‐galactosidase/neomycin phosphotransferase fusion (βgeo) gene and a polyadenylation signal. A lox71 site is located within the intron sequence, and loxP, lox2272 and lox511 sites are located downstream from the βgeo, polyadenylation signal and pSP73 vector sequences, respectively (Fig. 1a). An ES cell line TT2( 16 ) was used for gene trapping. For electroporation with the pU17 gene trap vector, 80 µg of SpeI‐digested DNA and 2 × 107 cells were used. The cells were suspended in 0.8 mL of phosphate‐buffered saline, electroporated using a Bio‐Rad Gene Pulser (Bio‐Rad Laboratories, Hercules, CA) set at 800 V and 3 µF, then fed with medium supplemented with 200 µg/mL G418 after 48 h. Selection was maintained for 7 days, and the colonies were then picked out and placed in 24‐well plates. Genomic sequences flanking the gene trap vector were determined by the modified rapid amplification of cDNA 5′‐ends method.( 14 ) Chimeras were produced by aggregation of isolated trapped ES cells, which had only a single copy of the vector integrated, with eight‐cell embryos of ICR mice (Kyudo, Kumamoto, Japan). Chimeric mice were mated with C57BL/6 females (Clea Japan, Tokyo, Japan) to obtain F1 heterozygotes. A sperm suspension was prepared from the vasa deferentia and cauda epididymis of 8‐week‐old F1 heterozygous males, and eggs were collected from superovulated F1 heterozygous females. Twenty‐four hours after fertilization, two‐cell embryos were surgically transferred to pseudopregnant ICR recipients to obtain F2 mice.( 17 )
Figure 1.
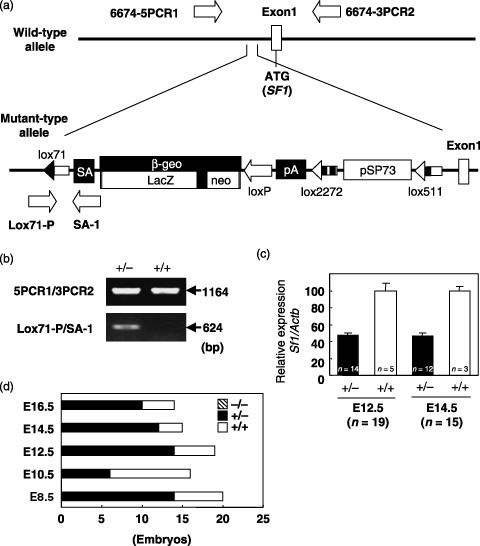
Generation of Sf1‐knockout mice. (a) Integration site of the trap vector. Clear boxes on the maps represent the first exon of the Sf1 gene. The trap vector was inserted 215 bp upstream from the first exon. The arrows indicate the primers used for genotyping. (b) Genotyping by polymerase chain reaction. The primer pairs Lox71‐P/SA‐1 and 5PCR1/3PCR2 were used to detect the Sf1geo and wild‐type alleles, respectively. (c) Expression of the Sf1 gene in Sf1+/+ and Sf1+/– embryos (E12.5 and E14.5). The levels of expression of Sf1 mRNA relative to β‐actin mRNA (Actb) (ΔΔCt) are expressed as percentages (ΔΔCt) of the values in the controls (Sf1 +/+). Data are expressed as mean ± SD. n, number of embryos examined. (d) Genotype distribution of E8.5, E10.5, E12.5, E14.5, and E16.5 F2 embryos. E, embryonic day.
Genotyping. Genomic DNA was isolated by proteinase K digestion, phenol–chloroform extraction, and ethanol precipitation from biopsy samples of newborn pups, and E16.5, E14.5, E12.5, E10.5 and E8.5 embryos. To identify the mutant allele, PCR was carried out using the primer pair Lox71‐P (5′‐AGGTCGAGGGACCTATACCG‐3′) and SA‐1 (5′‐GAGGCCGCTTGTCCTCTTTG‐3′). The PCR conditions were 35 cycles of 96°C for 30 s, 62°C for 42 s, and 72°C for 90 s, using AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA). To identify the wild‐type allele, the primers 6674‐5PCR1 (5′‐CTCTCACGTCACAGACTT‐3′) and 6674‐3PCR2 (5′‐ACTCAAGCATCCCTAGTAGC‐3′) were used. The PCR conditions were 30 cycles of 96°C for 15 s, 62°C for 30 s, and 68°C for 4 min, using KOD‐Plus DNA Polymerase (Toyobo, Osaka, Japan).
Real‐time RT‐PCR analysis. Total RNA was prepared from formalin‐fixed embryos or tumors with the RecoverAll Total Nucleic Acid Isolation Kit (Ambion, Austin, TX). DNase‐I‐treated total RNA was random primed and reverse transcribed with SuperScript reverse transcriptase (Invitrogen, Carlsbad, CA). The TaqMan universal PCR master mix and the predesigned TaqMan Gene Expression probe and primer sets were purchased from Applied Biosystems. Amplification data measured as an increase in reporter fluorescence were collected in real time with the PRISM 7000 Sequence Detection system (Applied Biosystems). Relative mRNA expression level was calculated by the comparative Ct method.
AOM treatment. Mice were housed in plastic cages in an air‐conditioned room with a 12‐h light–dark cycle. Water and a fat‐rich diet (AIN‐93G; Clea Japan) were available ad libitum. Because a sufficient number of male Sf1+/+ littermates was not obtained by in vitro fertilization, age‐matched additional control C57BL/6J (Sf1+/+ ) mice were purchased from Charles River Japan (Tokyo, Japan). Ten Sf1+/– F2 mice (4 females and 6 males), 10 Sf1+/+ littermates (8 females and 2 males), and 10 C57BL/6J controls (5 females and 5 males) received intraperitoneal injections of 10 mg AOM (Nard, Osaka, Japan)/kg body weight once a week for 6 weeks.( 18 ) The mean body weights of Sf1+/– , Sf1+/+ , and C57BL/6J (Sf1+/+ ) mice at the start of AOM treatment (7 weeks old) were not significantly different (data not shown).
Evaluation of tumorigenesis. Twenty‐three weeks after the first AOM treatment, the gut was filled with 10% buffered formalin through the anus immediately after the animals were killed, then opened longitudinally. After overnight fixation, specimens were stained briefly with 0.5% methylene blue (Sigma‐Aldrich, St Louis, MO).( 19 ) The numbers and two diameters (largest and smallest dimension) of polyps in the colon were measured in the ×20 and ×4 power field of a dissecting microscope (Nikon, Tokyo, Japan). Tumor volumes were determined according to V = 1/2A × B2, where A denotes the largest dimension of the tumor and B represents the smallest dimension.( 20 ) Formalin‐fixed, paraffin‐embedded sections were stained by the standard hematoxylin–eosin technique.( 19 )
Immunohistochemistry. Immunohistochemistry was carried out as previously described.( 13 , 21 ) Anti‐β‐catenin (clone 14) mouse monoclonal antibody was purchased from BD Transduction Laboratories (Palo Alto, CA). Anti‐SF1 goat (sc‐21157) polyclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Statistical analysis. The statistical significance of differences was evaluated by Mann–Whitney U‐test using a tool available in the R statistical package (http://www.r‐project.org/).
Results
Mice lacking SF1 are embryonic lethal. In order to identify the involvement of SF1 in colon tumorigenesis, we established an Sf1‐knockout mouse line using the gene trapping method.( 14 , 15 ) The trap vector was found to be inserted in the promoter region, 215 bp upstream from the first exon of the Sf1 gene (Fig. 1a). Genotyping by PCR using the primer pairs 5PCR1/3PCR2 and Lox71‐P/SA‐1 clearly identified wild‐type and mutant alleles, respectively (Fig. 1b). Real‐time RT‐PCR revealed an approximately 50% reduction of Sf1 mRNA expression in embryos with the Sf1 +/– genotype (Fig. 1c).
Although we examined 40 newborns, no animal was homozygous for the mutant Sf1 allele. To determine the lethal point, 20 E8.5, 16 E10.5, 19 E12.5, 15 E14.5, and 14 E16.5 embryos were genotyped, but no Sf1 −/– was identified (Fig. 1d), indicating that homozygous mice died before E8.5. These results indicate the indispensable role of SF1 in early embryonic development, although sagittal sections of whole E8.5 and E10.5 Sf1+/– embryos showed no apparent morphological abnormality (data not shown).
Susceptibility of SF1‐knockout mice to colon tumorigenesis. To evaluate the effects of SF1 haploinsufficiency on colon tumorigenesis, mice with the Sf1 +/– and Sf1 +/+ genotypes were treated with AOM, a known carcinogen organotropic to the colon,( 22 ) once a week for 6 weeks, and killed 23 weeks after the first injection of AOM (Fig. 2). As shown in Fig. 3a, Sf1 heterozygotes showed an increase in colon tumor multiplicity (5.5 ± 0.6 tumors/female, 3.7 ± 0.2 tumors/male) compared with wild‐type littermates (2.4 ± 0.3 tumors/female, 3.0 ± 0.7 tumors/male) as well as control C57BL/6J mice (1.8 ± 0.2 tumors/female, 1.2 ± 0.4 tumors/male). There was also an increase in tumor volume in Sf1 heterozygotes (8.7 ± 1.6 mm3/female, 11.3 ± 3.4 mm3/male) compared with wild‐type littermates (1.7 ± 0.5 mm3/female, 0.3 ± 0.1 mm3/male) as well as C57BL/6J controls (2.9 ± 1.0 mm3/female, 0.7 ± 0.2 mm3/male) (Fig. 3b).
Figure 2.
Induction of colon tumorigenesis by azoxymethane (AOM). Sf1 +/–, Sf1 +/+, and C57BL/6J (Sf1 +/+) mice received intraperitoneal injections of 10 mg AOM/kg body weight once a week for 6 weeks. Twenty‐three weeks after the first injection, the mice were killed.
Figure 3.
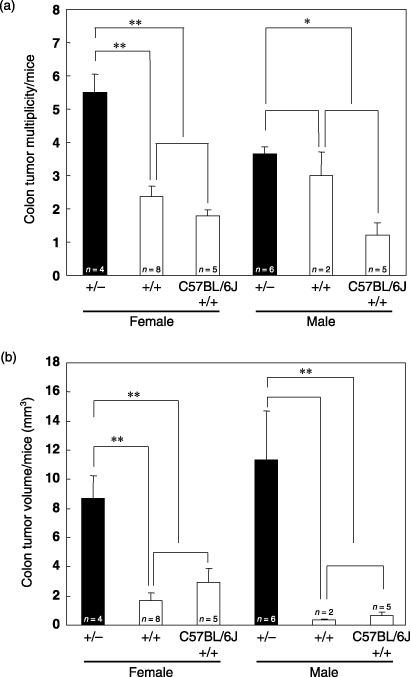
Increased tumorigeneis of Sf1 +/– mice treated with azoxymethane. The numbers (a) and volumes (b) of tumors that developed in Sf1+/– , Sf1+/+ , and C57BL/6J (Sf1 +/+) mice were determined. Data were expressed as mean ± SE. Statistical analysis was conducted using the Mann–Whitney U‐test. n, number of mice examined. *Significantly different with a P‐value of < 0.05; **significantly different with a P‐value of <0.01.
In addition to the increase in tumor number and volume, we noticed morphological alterations in the tumors of Sf1 heterozygous mice (Fig. 4a–d). The tumors that developed in AOM‐treated Sf1+/– mice showed more marked histological atypia with irregular glandular structures and inverted papillary protrusions (Fig. 4c,d) than those of Sf1+/+ littermates (Fig. 4a,b).
Figure 4.
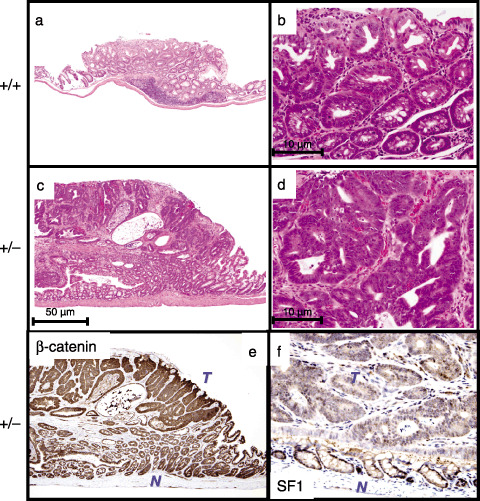
Histological differences of tumors that developed in Sf1 +/+ and Sf1 +/– mice. (a–d) Histology of colon tumors that developed in azoxymethane (AOM)‐treated Sf1+/+ (a,b) and Sf1+/– (c,d) mice (hematoxylin–eosin staining). Original magnification: ×40 (a,c) and ×200 (b,d). (e,f) Expression of the β‐catenin (e) and SF1 (f) proteins in colon tumors that developed in AOM‐treated Sf1+/– mice. T, tumor; N, colon mucosa.
Tumor cells of Sf1+/– mice showed increased expression of the β‐catenin protein in comparison with colonic epithelial cells (Fig. 4e), indicating that active Wnt/β‐catenin signaling is involved in the tumor development in AOM‐treated Sf1+/– mice. Consistent with our previous observation,( 13 ) the nuclear expression of SF1 in tumor cells that developed in AOM‐treated Sf1+/– mice was reduced in comparison with nuclear expression in colonic epithelial cells (Fig. 4f).
SF1 is a component of the β‐catenin and TCF‐4 complex, and it functions as a negative regulator of its transcriptional activity.( 13 ) Higher levels of expression of known target genes of TCF/LEF, including the genes encoding c‐Jun (Jun), c‐myc (Myc), cyclin D1 (Ccnd1), and axis inhibitor‐2 (Axin2),( 23 ) were found in the tumors that developed in Sf1+/– mice than in the tumors that developed in Sf1+/+ mice (Fig. 5).
Figure 5.
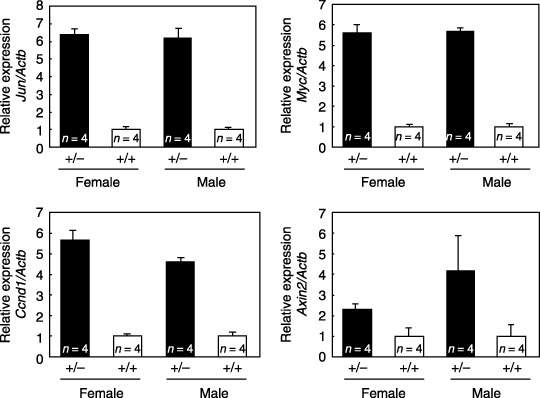
Expression of target genes of T‐cell factor/lymphoid enhancer factor in tumors that developed in Sf1 +/+ and Sf1 +/– mice treated with azoxymethane. The mRNA expression levels of c‐Jun (Jun), c‐myc (Myc), cyclin D1 (Ccnd1), and axis inhibitor‐2 (Axin2) relative to β‐actin (Actb) (ΔCt) are expressed as ratios (ΔΔCt) to the values in the controls (Sf1 +/+). Data are expressed as mean ± SD. n, number of tumors examined.
Discussion
The organization of the intestinal epithelium is maintained under a strict balance of cell proliferation and differentiation. Because intestinal epithelial stem cells continuously proliferate, the same proportion of cells must differentiate, die, and exfoliate.( 11 ) The β‐catenin and TCF‐4 complex has been implicated in the maintenance of the undifferentiated status of intestinal epithelial cells.( 11 ) Aberrant activation of Wnt signaling results in formation of the complex and blocks the differentiation program of intestinal epithelial cells. TCF‐4‐knockout (Tcf7l2−/– ) mice have been found to lack proliferating undifferentiated compartments in their intestinal crypts, and die shortly after birth.( 24 )
We previously identified SF1 as a protein whose expression was negatively regulated by the TCF‐4 and β‐catenin complex.( 13 ) Conversely, SF1 negatively regulated β‐catenin‐evoked gene transactivation and cell proliferation. SF1 protein is expressed in differentiated epithelial cells of the intestinal villi, but not in undifferentiated cells in the crypts or in adenoma cells of multiple intestinal polyposis (Min) (Apc Min/+) mice. The SF1 protein was strongly induced in colon carcinoma cells that had been exposed to a differentiation inducer, sodium butyrate. Based on these observations we concluded that SF1 is a differentiation‐associated tumor suppressor. Although the metabolites of AOM cause general DNA damage by the formation of adducts such as O 6‐methylguanine and 3‐methyladenine,( 18 ) injection of AOM into rodents seems to induce colon tumorigenesis invariably through the activation of Wnt signaling.( 18 , 25 ) The β‐catenin protein frequently accumulates in the cytoplasm and nucleus of tumor cells that develop in rats and mice treated with AOM.( 25 ) Mutation of the Apc gene is infrequent in the tumors induced by AOM,( 26 ) but missense mutations in the glycogen synthase kinase 3β phosphorylation sites of the β‐catenin gene have been frequently detected.( 25 ) The increased tumorigenesis of AOM‐treated Sf1+/– mice thus seems to be a direct consequence of the increased transcriptional activity of the β‐catenin and TCF‐4 complex. Actually, the levels of expression of known target genes of TCF/LEF were increased in the tumors of Sf1+/– mice in comparison with the tumors of Sf1+/+ mice (Fig. 5).
Although expression of SF1 was reduced in the colon tumors that developed in Sf1+/– mice (Fig. 4f), the fact that it was expressed at all suggested that the normal allele of Sf1 had been retained. SF1 is a suppressor of the β‐catenin‐evoked transcriptional activity of TCF‐4,( 13 ) and the increased susceptibility of Sf1 +/– mice to AOM‐induced colon tumorigenesis seems be a reflection of the dose‐dependent reduction of the suppressive effect.
Because only two male Sf1+/+ littermates were obtained by in vitro fertilization, age‐matched control male C57BL/6J (Sf1+/+ ) mice were included. Although there was no significant difference in tumor number or volume between the female Sf1+/+ littermates and C57BL/6J female mice (Fig. 3), the difference in genetic background influences the susceptibility of mice to carcinogens.( 27 ) The results obtained in C57BL/6J male mice must be interpreted with caution.
SF1 is known to be involved in biological processes other than the regulation of gene transactivation.( 13 , 28 ) SF1 recognizes the intron branch point sequence UACUAAC in the premRNA during splicesome assembly and regulates the premRNA splicing reaction.( 29 ) SF1 cDNA transfection induces splice variants,( 13 ) such as ERβΔ5‐6 (estrogen receptor‐β lacking exons 5 and 6),( 30 ) WISP (Wnt‐induced secreted protein)‐1v,( 31 ) and FGF3R (fibroblast growth factor receptor‐3)‐ATII.( 32 ) Haploinsufficiency of SF1 might result in the generation and/or suppression of splice variants that play crucial roles during the processes of intestinal epithelial differentiation and colon tumorigenesis.
The phenotypic manifestation of SF1‐knockout mice seems to be generally more severe than that of TCF‐4‐knockout mice.( 24 ) The expression of SF1 was not limited to cells of the intestinal epithelial lineage, and was ubiquitously expressed in various embryonic organs/tissues (data not shown). TCF‐4 homozygous (Tcf7l2−/– ) mutant mice were born in a Mendelian ratio,( 24 ) but SF1 homozygous mutant (Sf1−/– ) embryos died before E8.5 (Fig. 1c). Alternative mRNA splicing no doubt plays important roles in embryonic development. Knockout mice for other splicing‐related genes have also been reported to be embryonic lethal.( 33 , 34 ) To examine the direct effects of SF1 deficiency on tumorigenesis, conditional knockout of Sf1 in colon epithelial cells would be necessary.
These findings of the present study provide solid evidence for the role of SF1 in colon tumorigenesis and might open up new diagnostic and/or therapeutic avenues for colon cancer. However, we could not exclude the possibility that the increased susceptibility of Sf1 +/– mice to AOM‐induced colon tumorigenesis might be attributable to mechanisms unrelated to β‐catenin‐mediated gene transactivation or premRNA splicing. Further studies will be necessary to clarify the fundamental mechanisms whereby SF1 impacts on the regulation of differentiation and proliferation of intestinal epithelial cells.
Acknowledgments
We thank Dr Masako Ochiai and Dr Hitoshi Nakagama (National Cancer Center Research Institute, Tokyo, Japan) for technical advice on the protocol of AOM treatment. This work was supported by the Program for Promotion of Fundamental Studies in Health Sciences conducted by the National Institute of Biomedical Innovation of Japan, the Third‐Term Comprehensive Control Research for Cancer conducted by the Ministry of Health, Labor and Welfare of Japan and the Ministry of Education, Culture, Sports, Science and Technology of Japan, and generous grants from the Naito Foundation and the Princess Takamatsu Cancer Research Fund.
References
- 1. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996; 87: 159–70. [DOI] [PubMed] [Google Scholar]
- 2. Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the β‐catenin degradation complex. Proc Natl Acad Sci USA 2002; 99: 1182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci 2003; 94: 225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β‐Catenin is a target for the ubiquitin–proteasome pathway. Embo J 1997; 16: 3797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis – a look outside the nucleus. Science 2000; 287: 1606–9. [DOI] [PubMed] [Google Scholar]
- 6. Miller JR, Moon RT. Signal transduction through β‐catenin and specification of cell fate during embryogenesis. Genes Dev 1996; 10: 2527–39. [DOI] [PubMed] [Google Scholar]
- 7. Korinek V, Barker N, Morin PJ et al . Constitutive transcriptional activation by a β‐catenin‐Tcf complex in APC−/– colon carcinoma. Science 1997; 275: 1784–7. [DOI] [PubMed] [Google Scholar]
- 8. Morin PJ, Sparks AB, Korinek V et al . Activation of β‐catenin‐Tcf signaling in colon cancer by mutations in β‐catenin or APC. Science 1997; 275: 1787–90. [DOI] [PubMed] [Google Scholar]
- 9. He TC, Sparks AB, Rago C et al . Identification of c‐MYC as a target of the APC pathway. Science 1998; 281: 1509–12. [DOI] [PubMed] [Google Scholar]
- 10. Tetsu O, McCormick F. Beta‐catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422–6. [DOI] [PubMed] [Google Scholar]
- 11. Van De Wetering M, Sancho E, Verweij C et al . The β‐catenin/TCF‐4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111: 241–50. [DOI] [PubMed] [Google Scholar]
- 12. Yamada T, Takaoka AS, Naishiro Y et al . Transactivation of the multidrug resistance 1 gene by T‐cell factor 4/β‐catenin complex in early colorectal carcinogenesis. Cancer Res 2000; 60: 4761–6. [PubMed] [Google Scholar]
- 13. Shitashige M, Naishiro Y, Idogawa M et al . Involvement of splicing factor‐1 in β‐catenin/T‐cell factor‐4‐mediated gene transactivation and pre‐mRNA splicing. Gastroenterology 2007; 132: 1039–54. [DOI] [PubMed] [Google Scholar]
- 14. Araki K, Imaizumi T, Sekimoto T et al . Exchangeable gene trap using the Cre/mutated lox system. Cell Mol Biol 1999; 45: 737–50. [PubMed] [Google Scholar]
- 15. Taniwaki T, Haruna K, Nakamura H et al . Characterization of an exchangeable gene trap using pU‐17 carrying a stop codon‐β geo cassette. Dev Growth Differ 2005; 47: 163–72. [DOI] [PubMed] [Google Scholar]
- 16. Yagi T, Tokunaga T, Furuta Y et al . A novel ES cell line, TT2, with high germline‐differentiating potency. Anal Biochem 1993; 214: 70–6. [DOI] [PubMed] [Google Scholar]
- 17. Fraser LR, Drury LM. The relationship between sperm concentration and fertilization in vitro of mouse eggs. Biol Reprod 1975; 13: 513–8. [DOI] [PubMed] [Google Scholar]
- 18. Nozaki T, Fujihara H, Watanabe M et al . Parp‐1 deficiency implicated in colon and liver tumorigenesis induced by azoxymethane. Cancer Sci 2003; 94: 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamada T, Mori Y, Hayashi R et al . Suppression of intestinal polyposis in Mdr1‐deficient ApcMin/+ mice. Cancer Res 2003; 63: 895–901. [PubMed] [Google Scholar]
- 20. Yanase T, Tamura M, Fujita K, Kodama S, Tanaka K. Inhibitory effect of angiogenesis inhibitor TNP‐470 on tumor growth and metastasis of human cell lines in vitro and in vivo . Cancer Res 1993; 53: 2566–70. [PubMed] [Google Scholar]
- 21. Seike M, Kondo T, Mori Y et al . Proteomic analysis of intestinal epithelial cells expressing stabilized β‐catenin. Cancer Res 2003; 63: 4641–7. [PubMed] [Google Scholar]
- 22. Papanikolaou A, Wang QS, Delker DA, Rosenberg DW. Azoxymethane‐induced colon tumors and aberrant crypt foci in mice of different genetic susceptibility. Cancer Lett 1998; 130: 29–34. [DOI] [PubMed] [Google Scholar]
- 23. Idogawa M, Masutani M, Shitashige M et al . Ku70 and poly (ADP‐ribose) polymerase‐1 competitively regulate β‐catenin and T‐cell factor‐4‐mediated gene transactivation: possible linkage of DNA damage recognition and Wnt signaling. Cancer Res 2007; 67: 911–8. [DOI] [PubMed] [Google Scholar]
- 24. Korinek V, Barker N, Moerer P et al . Depletion of epithelial stem‐cell compartments in the small intestine of mice lacking Tcf‐4. Nat Genet 1998; 19: 379–83. [DOI] [PubMed] [Google Scholar]
- 25. Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the β‐catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis 2000; 21: 1117–20. [PubMed] [Google Scholar]
- 26. De Filippo C, Caderni G, Bazzicalupo M et al . Mutations of the Apc gene in experimental colorectal carcinogenesis induced by azoxymethane in F344 rats. Br J Cancer 1998; 77: 2148–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate‐induced colon carcinogenesis in mice. Carcinogenesis 2006; 27: 162–9. [DOI] [PubMed] [Google Scholar]
- 28. Goldstrohm AC, Albrecht TR, Sune C, Bedford MT, Garcia‐Blanco MA. The transcription elongation factor CA150 interacts with RNA polymerase II and the pre‐mRNA splicing factor SF1. Mol Cell Biol 2001; 21: 7617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Z, Luyten I, Bottomley MJ et al . Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science 2001; 294: 1098–102. [DOI] [PubMed] [Google Scholar]
- 30. Sato S, Idogawa M, Honda K et al . β‐Catenin interacts with the FUS proto‐oncogene product and regulates pre‐mRNA splicing. Gastroenterology 2005; 129: 1225–36. [DOI] [PubMed] [Google Scholar]
- 31. Tanaka S, Sugimachi K, Kameyama T et al . Human WISP1v, a member of the CCN family, is associated with invasive cholangiocarcinoma. Hepatology 2003; 37: 1122–9. [DOI] [PubMed] [Google Scholar]
- 32. Jang JH, Shin KH, Park YJ, Lee RJ, McKeehan WL, Park JG. Novel transcripts of fibroblast growth factor receptor 3 reveal aberrant splicing and activation of cryptic splice sequences in colorectal cancer. Cancer Res 2000; 60: 4049–52. [PubMed] [Google Scholar]
- 33. Xu X, Yang D, Ding JH et al . ASF/SF2‐regulated CaMKIIδ alternative splicing temporally reprograms excitation–contraction coupling in cardiac muscle. Cell 2005; 120: 59–72. [DOI] [PubMed] [Google Scholar]
- 34. Isono K, Mizutani‐Koseki Y, Komori T, Schmidt‐Zachmann MS, Koseki H. Mammalian polycomb‐mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev 2005; 19: 536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]