

Abstract
The power of an SNP‐based genome‐wide association study (GWAS) was first demonstrated in Japan using the JSNP database and is currently a major strategy adopted around the world for a number of common diseases including cancers. The hypothesis‐free strategy can lead us to a novel hypothesis for carcinogenesis and may contribute to identifying a high risk group for research and, in the future, practice of personalized prevention. We performed a GWAS on diffuse‐type gastric cancer and identified a significant association with SNPs in the PSCA (prostate stem cell antigen) gene. The association was validated by a Korean gastric case‐control analysis. The PSCA protein is expressed predominantly in the stem cell/precursor‐rich region of the gastric epithelium, which is considered as the origin of diffuse‐type gastric cancer, and showed tumor suppressor‐like characteristics. Individuals with a low PSCA promoter activity are susceptible to diffuse‐type gastric cancer. By contrast, the polymorphism does not significantly predispose to intestinal‐type gastric cancer, congruous to the hypothesis of the two distinct carcinogenesis pathways for the two major types of gastric cancer. In addition to publication on a specific gene, the sharing of GWAS data through a database on the web is expected to accelerate validation and discovery by other investigators. GeMDBJ (Genome Medicine Database of Japan), started in 2005 in Japan, is one of such attempts. Moreover, the advent of “next generation” sequencers may herald a new era in which the poorly explored domains of the genetic architecture of disease susceptibility may be unveiled. (Cancer Sci 2010)
In general, individual cancer susceptibility is determined by a combination of lifestyle/environmental factors, genetic factors, and age. Among the three, the effect of lifestyle/environmental factors and age may be inflicted upon somatic cells as genomic and epigenomic damage, which can be altered in the life course, whereas the germline genetic factors confer a fixed, stable probability for disease development, obeying a basic principle of genetics expressed in well‐established mathematical formulas. First, this stability is an important, albeit not well‐appreciated, feature which is required as a reliable and practical biomarker for the assessment of an individual risk of disease development. For many, if not all, diseases, the genetic factors are able to serve as a fundamental upon which the effects of the non‐genetic risk factors are combined to capture a high risk group, which in turn is a target of research and practice of individualized prevention. Second, the identification of genes related to disease susceptibility may lead to elucidation of the molecular pathway of pathogenesis, which in turn may contribute to the development of molecular target prevention.
In cancer, extreme cases of the three‐factor combination are obviously hereditary cancer syndromes, which roughly account for 5% or less of all cancer cases. Starting from the cloning of the RB1 (retinoblastoma 1) gene for hereditary retinoblastoma in 1986, most, but not all, causative genes for the major monogenic, hereditary cancer syndromes have been identified to date. Typically, large pedigree‐based linkage analyses have played a major role in the gene hunting for Mendelian phenotypes. By contrast, >95% of cancer cases are considered multifactorial, polygenic diseases, in which the relative disease‐predisposing effect of each individual genetic factor is small (relative risk of 1.5 or less), and thus an association study is generally preferred to linkage analysis.( 1 ) A search for such genetic susceptibility factors has long been dependent on a candidate‐gene approach based on existing knowledge and hypotheses on human genes. Although several well‐known successes have been noted, such as ALDH2 (aldehyde dehydrogenase 2 family (mitochondrial), see ref. 2 for review) and hOGG1 ( 3 ) (8‐oxoguanine DNA glycosylase) polymorphisms for alcohol‐related esophageal cancers and lung cancers, respectively, human knowledge and insights on genes, their variations, and functions are far from perfect. Moreover, accumulating evidence suggests that non‐coding domains of the genome may be involved in a number of important biological processes and phenotypes. Therefore, a genome‐wide systematic, hypothesis‐free screen is an essential strategy to complement candidate gene approaches for identifying genetic susceptibility factors for polygenic diseases. However, such a robust genome scan had to wait for the advent of powerful genome‐wide markers and their high‐throughput analytical methods.
Single Nucleotide Polymorphism (SNP)‐Based Genome‐Wide Association Study: First Success From Japan
Currently, an SNP has been widely used as a de facto standard for an excellent and robust genetic marker for a hypothesis‐free, genome‐wide association study (GWAS) based on linkage disequilibrium mapping.( 4 , 5 ) Single nucleotide polymorphisms (SNPs), not only being genetic markers, could themselves be functionally responsible for disease susceptibility. One of the essential elements of a GWAS is the database providing comprehensive information on common variations of human DNA sequences. A group led by Yusuke Nakamura at RIKEN, and The Institute of Medical Science, The University of Tokyo, started a systematic resequencing of genomic DNA of Japanese individuals and constructed the JSNP database in 2002 (http://snp.ims.u‐tokyo.ac.jp/index.html).( 6 , 7 ) Using that database, his group first demonstrated a proof of principle of the SNP‐based GWAS by identifying the association between the genetic polymorphism of the lymphotoxin‐α gene and myocardial infarction.( 8 )
Following their pioneering efforts, The International HapMap Consortium was launched in 2002 to expand the polymorphism catalog to other ethnic groups. The HapMap phase 1 and 2 data were released in 2005 (http://snp.cshl.org/).( 9 ) In January 2008, the 1000 Genomes Project was started to sequence the genomes of at least 1000 people around the world, supported by Wellcome Trust Sanger Institute, UK, the Beijing Genomics Institute, China, and the National Human Genome Research Institute (NHGRI), USA (http://www.1000genomes.org/).
Genome‐Wide Association Study (GWAS) On Cancer
Genome‐wide association studies (GWAS) on various types of cancers started to appear in leading journals around 2007. To aid in an overview of the field, Nature Genetics was chosen as a representative journal, and the GWAS papers published there have been summarized (Table S1). The Table shows that the current standard for a GWAS requires a thousand or more subjects for both cases and controls, with cohort‐based nested case‐control studies preferred, and multiple replications, often over different ethnic groups. The increased power makes it possible to detect genetic factors with small effect size. Although the strength of a GWAS lies in its hypothesis‐free nature, which may lead us to an unexpected new discovery, it may also pose a significant difficulty for investigators; many a GWAS has led investigators to genomic regions where either no protein‐coding genes are known, or, alternatively, multiple genes are present in the contiguous linkage disequilibrium block, making the pinpointing of a single responsible gene impossible by statistical genetics alone (Table S1). Even if the genomic region identified by a GWAS contains a single known gene, proving its role in the carcinogenesis process by an animal model is generally expected to be difficult because of the small effect size, and polygenic and multifactorial nature of the pathogenesis (i.e. human‐specific gene–gene/gene–environment interactions).
Nevertheless, we believe that a GWAS should be carried out for major human diseases, because so little is known about their genetic architecture. Following RIKEN’s successes, a JSNP‐based GWAS was continued as a part of the Millennium Genome Project( 10 ) organized by the Japanese government. A GWAS on diffuse‐type gastric cancer has been reported from our group,( 11 ) and is reviewed briefly in the following section.
A GWAS on Diffuse‐Type Gastric Cancer
Classification of gastric cancer. Gastric cancer is still the most common cancer in Japanese males (about 20% of all cancers) and the second most frequent cancer among Japanese females after breast cancer (including carcinoma in situ).( 12 ) Japan has a long history of advanced gastric cancer research, and the Japanese Classification of Gastric Carcinoma by the Japanese Gastric Cancer Association( 13 ) recognizes seven histological types for gastric adenocarcinoma, which corresponds to about 90% of all gastric cancer: papillary adenocarcinoma (pap); tubular adenocarcinoma, well‐differentiated type (tub1); tubular adenocarcinoma, moderately differentiated type (tub2); poorly differentiated adenocarcinoma, solid type (por1); poorly differentiated adenocarcinoma, non‐solid type (por2); signet‐ring cell carcinoma (sig); and mucinous adenocarcinoma (muc). Although this is a “classification,” it should always be kept in mind that different types may coexist in an actual clinical specimen at different proportions.
However, in many clinico‐epidemiological studies, the old but simple two‐category classification has often been used.( 14 ) The Lauren classification focuses on the growth pattern and divides gastric adenocarcinomas into the intestinal type, which forms a glandular structure, and the diffuse type, which shows diffuse infiltrative growth. Although the degree of differentiation is not the primary consideration in the Lauren classification, the intestinal type generally corresponds to pap, tub1, tub2, and por1, while the diffuse type is por2 or sig. muc may be either intestinal‐ or diffuse‐type. It is plausible that the success and usefulness of the Lauren classification is related to the distinctive difference in the underlying carcinogenesis mechanisms between the two types (Fig. 1). The major pathway leading to the intestinal type may start with Helicobacter pylori (H. pylori) infection, followed by atrophic gastritis and intestinal metaplasia, which is also a senescent change of gastric mucosa. By contrast, genuine, or de novo, diffuse‐type gastric cancer is believed to originate from stem cells or precursors for gastric epithelial cells in the background of normal gastric mucosa,( 15 , 16 ) although many undifferentiated gastric adenocarcinomas may represent a de‐differentiated stage of the intestinal type.
Figure 1.
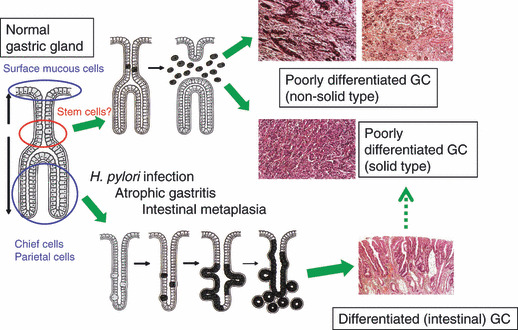
Gastric carcinogenesis: a hypothesis. In clinicopathological or epidemiological studies, two‐major category classification has often been employed for gastric adenocarcinomas (GCs): intestinal or differentiated type, and diffuse or undifferentiated type. It has been suggested that the two types have distinct carcinogenesis pathways. H. pylori, Helicobacter pylori. Modified from K. Nakamura. Igan‐no‐Kozo (in Japanese), 3rd edn. Tokyo: Igaku‐shoin, 2005.
The intestinal type is more common in males and older age groups and predominates in a high‐risk geographic area such as a high H. pylori infection rate. The diffuse type, though, occurs in a more equal male‐to‐female ratio and more frequently in the younger population than does the intestinal type, and its incidence has been increasing in contrast to the decreasing trend of the intestinal type.( 17 , 18 , 19 , 20 ) We have been especially interested in diffuse‐type gastric cancer, because this type includes a distinct form called linitis plastica.( 18 ) Linitis plastica, or type 4 advanced gastric cancer by the Japanese Classification,( 13 ) accounts for approximately 10% of all gastric cancer in Japan, but its 5‐year survival rate there is 10–20%( 21 ) with no significant improvement in the past decade.
JSNP‐based GWAS by the Millennium Genome Project. Var‐ious environmental factors, including diet and smoking, have been suggested in relation to a predisposition to gastric cancer.( 17 ) In Japan, H. pylori infection appears almost as a prerequisite for the development of this cancer, because 99% of Japanese with gastric cancer and 90% of the Japanese adult (>40 years old) population were seropositive for H. pylori, and the infection was significantly associated with both the differentiated (≈ intestinal) and undifferentiated (≈ diffuse) types of gastric cancers.( 22 ) Overall, however, development of the intestinal type is greatly influenced by environmental factors, while it is presumed that for the diffuse type a non‐environmental factor, such as genetic predisposition, is also important. Several genetic and epigenetic alterations in gastric cancer were reported, including the CDH1 (E‐cadherin) gene mutation, which is causal to hereditary diffuse‐type gastric cancer.( 23 ) However, the pathogenic germline CDH1 mutation is rare among Japanese pedigrees of familial gastric cancer,( 24 ) and little is known about genetic factors involved in the polygenic, common type of diffuse‐type gastric cancer. For instance, the role of the CDH1 polymorphism is not clear in the Asian meta‐analysis.( 25 ) Therefore, we performed the first GWAS for this type of gastric cancer as a part of the five‐disease joint GWAS (other diseases: Alzheimer’s disease, type 2 diabetes, hypertension, and asthma) in the Millennium Genome Project.
The design and results of the GWAS were published previously,( 11 , 26 ) and only their essential points are summarized here (Fig. 2 and Table 1). Briefly, the first stage of this two‐stage GWAS analyzed 85576 JSNPs in 188 cases with linitis plastica and 752 references (a 752 case mix of four other common diseases and another 752 population control data from the JSNP database). We selected 2753 SNPs from the first‐stage screening purely by statistical criteria, without relying on gene annotation, and genotyped another 749 cases with diffuse‐type gastric cancer and 750 controls. The screening and a subsequent high‐density typing identified a significant association with an SNP (rs2294008) in the first exon of the PSCA (prostate stem cell antigen) gene: gender‐ and age‐adjusted odds ratio (OR) by dominant model = 4.18, 95% confidence interval (CI) = 2.88–6.21, P = 1.5 × 10−17 for a total of 925 cases with diffuse‐type gastric cancer and 1396 controls. Because case‐control association studies are so prone to error and bias, validating in independent populations is critical. The association of the PSCA SNP was replicated on diffuse‐type gastric cancer in 454 cases and 390 controls in Korea (adjusted dominant model OR = 3.61, 95% CI = 2.41–5.51, P = 3.2 × 10−11). The SNP was far less significant in 599 and 417 cases with intestinal‐type gastric cancer in Japan (P = 0.0041) and Korea (P = 0.0011), respectively. Moreover, Matsuo et al. ( 27 ) reported an independent replication of the association with the diffuse‐type gastric cancer in Japan. They also detected a significant heterogeneity between the diffuse and intestinal types (P‐heterogeneity = 0.007).
Figure 2.
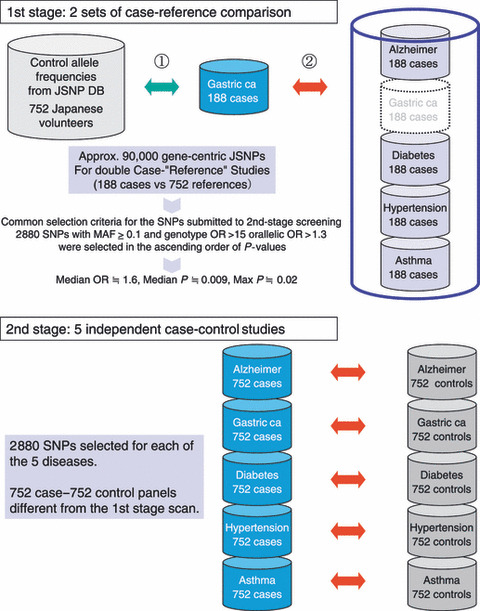
Design of the two‐stage genome‐wide association study (GWAS) by Millennium Genome Project. As shown in Supplementary Table S1 online, GWAS is often performed in multiple stages. In the Millennium Genome Project, five diseases, including gastric cancer, were analyzed simultaneously in the same GWAS. Therefore, one of the reference (control) groups in the first screening was a case mix of the other four diseases, while the second stage was a more conventional case‐control study. DB, database; MAF, minor allele frequency; OR, odds ratio.
Table 1.
Association of the PSCA SNP (rs2294008) and gastric cancers in Japan and Korea( 11 )
| Allele OR | 95% CI | P‐value (Fisher) | Dominant model OR | 95% CI | P‐value (logistic) | |
|---|---|---|---|---|---|---|
| Risk allele frequency among 1396 Japanese control individuals = 0.617 | ||||||
| Diffuse type 925 cases | 1.67 | 1.47–1.90 | 2.2 × 10−15 | 4.18 | 2.88–6.21 | 1.5 × 10−17 |
| Intestinal type 599 cases | 1.29 | 1.11–1.49 | 5.1 × 10−4 | 1.59 | 1.15–2.21 | 0.0041 |
| Risk allele frequency among 390 Korean control individuals = 0.462 | ||||||
| Diffuse type 454 cases | 1.91 | 1.57–2.33 | 6.3 × 10−11 | 3.61 | 2.41–5.51 | 3.2 × 10−11 |
| Intestinal type 417 cases | 1.37 | 1.12–1.68 | 0.0017 | 1.85 | 1.27–2.71 | 0.0011 |
CI, confidence interval; OR, odds ratio; PSCA, prostate stem cell antigen.
Functional studies on PSCA and its SNPs. The discovery of the PSCA SNP as a novel genetic susceptibility factor for gastric cancer was totally unexpected. The gene was originally identified as a prostate‐specific antigen overexpressed in prostate cancers,( 28 , 29 ) but it was also expressed strongly in the stomach and less intensely in the bladder, gallbladder, and tonsils.( 30 ) It is noteworthy that the PSCA SNP identified for gastric cancer was recently found to be associated also with bladder cancer.( 31 ) To explore a biological basis for this association, we performed a series of functional analyses of the gene and its SNPs as detailed previously.( 11 )
First, our anti‐PSCA monoclonal antibody localized PSCA protein expression in differentiating epithelial cells in the isthmus and neck regions of normal gastric epithelium (Fig. 3). This was a very interesting observation, because these regions of the gastric gland are considered to harbor stem cells and precursors for the two‐directional differentiation of gastric epithelial cells (Fig. 3) and because diffuse‐type gastric cancer is suggested to arise from the stem cells and/or progenitors of the isthmus region.( 16 ) Second, immunohistochemistry and quantitative RT‐PCR revealed a frequent silencing of the gene in gastric cancer tissues. We analyzed 19 diffuse‐type and 21 intestinal‐type gastric cancers, and no PSCA staining was detectable in any of the diffuse‐type cancers, while 20 of the 21 intestinal‐type cancers appeared to retain some, but definitely reduced, PSCA expression, as compared to the surrounding normal gastric epithelium on the same section. Third, the transfection and overexpression of the PSCA cDNA induced inhibition of in vitro colony‐formation and growth of the HSC57 gastric cancer cells, which do not detectably express the endogenous PSCA gene (Fig. 4). Fourth, a reporter assay on the 3.2‐kb upstream fragment of the gene revealed that the fragment containing C allele at SNP (rs2294008) residing in the first exon has higher transcriptional activity than that containing T, the risk allele. In particular, a single substitution of the C allele with the risk allele T of rs2294008 reduces transcriptional activity of an upstream fragment of the PSCA gene, suggesting that the SNP is functionally responsible for the observed association with diffuse‐type gastric cancer.
Figure 3.
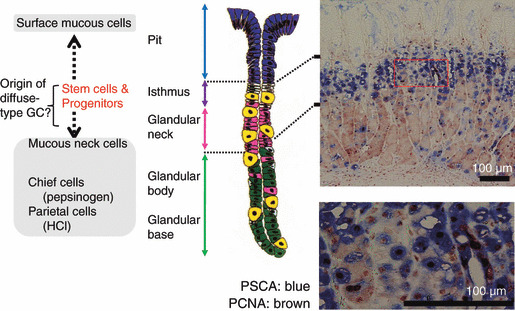
PSCA (prostate stem cell antigen) expression at isthmus and neck region of gastric epithelium.( 11 ) Center and left, a schematic architecture of the gastric gland. The isthmus is considered to contain stem cells and precursors of three major cell lineages of the gastric gland: pit, parietal, and zymogenic cells. Right, an immunohistochemistry by anti‐PSCA monoclonal antibody (stained blue) and by anti‐PCNA antibody (brown). Bar, 100 μm. GC, gastric adenocarcinomas; HCI, hydrochloric acid; PCNA, proliferating cell nuclear antigen. Modified from G.R. Van Den Brink et al. Gastroenterol 2001; 121:317.
Figure 4.
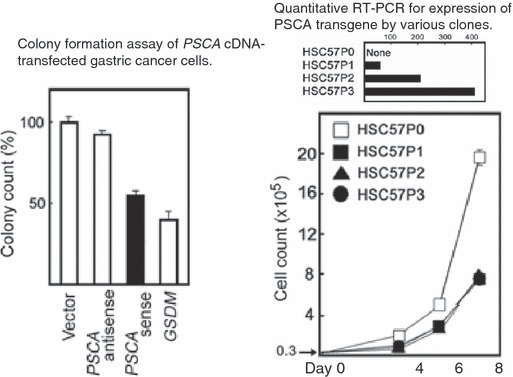
In vitro tumor suppressor‐like activities of PSCA (prostate stem cell antigen) cDNA.( 11 ) Left, the PSCA cDNA was cloned into an expression vector driven by CMV promoter in the sense and anti‐sense orientations and transfected to HSC57 gastric cancer cells. Colonies of stable transfectants selected by G418 selection were counted. The GSDM (gasdermin) cDNA was used as a positive control for the tumor suppressor‐like activity.( 47 ) Right, cell growth inhibition of the stable clones of the PSCA‐transfected HSC57. The parental HSC57P0 cells do not detectably express endogenous PSCA.
PSCA (prostate stem cell antigen) hypothesis generated by a hypothesis‐free GWAS on diffuse‐type gastric carcinogenesis. These functional studies suggest a simple but attractive hypothesis regarding the PSCA gene and diffuse‐type gastric carcinogenesis as follows: “The PSCA protein is expressed predominantly at the stem cell/precursor‐rich region of the gastric epithelium (which is also considered as the origin of diffuse type gastric cancer), has a tumor suppressor‐like activity, and is involved in the regulation of gastric epithelial‐cell proliferation. Individuals with a low promoter activity of the tumor‐suppressive PSCA gene are susceptible to diffuse‐type gastric cancer development. By contrast, the polymorphism does not significantly predispose to intestinal‐type gastric cancer, congruous to the hypothesis of the two distinctive carcinogenesis pathways for the two major types of gastric cancer as depicted in Figure 1. PSCA (prostate stem cell antigen) appears to play a different role in a different tissue context, because the protein is overexpressed in prostate cancer cells.” This study may become another example in which an agnostic GWAS has unveiled an unexpected molecular mechanism of a disease development.
Genome‐Wide Association Study (GWAS) Databases and GeMDBJ
Genome‐wide association study (GWAS) databases in the world. Collaboration, as we experienced at the validation stage on the Korean gastric cancer cases and controls in the PSCA research, has been not only crucial, as in much other human disease research, but also potentially robust for genetic association studies, because the genotype part of the relevant data is less prone to ascertainment error, unlike many other molecular phenotypes or clinico‐epidemiological and lifestyle‐related information. One step forward to such an ad hoc, gene‐ or marker‐specific collaboration for a replication purpose would be the sharing of genome scan data through public accessible databases. In fact, there has been an increasing demand for an efficient use of research resources, such as invaluable human genome samples and phenotype information, especially those obtained by public funded research. The mission of such a database includes, at least (i) validation and evaluation of published research results and conclusions by allowing access to the authors’ original data; (ii) offering reference data to allow comparison with the users’ own experimental data; (iii) hypothesis generation through a biostatistical/bioinformatics exploration of the database data; and (iv) development and validation of tools and methods in biostatistics/bioinformatics. Therefore, a database should present data in two ways simultaneously: (i) through an easily navigatable, reference‐dictionary type interface to respond to each user’s specific data search; and (ii) as a downloadable flat file of as‐raw‐as‐possible data.
A number of omics databases, especially those offering genome and/or transcriptome datasets, have been established on the web. For germline genomic data for human diseases, examples include: dbGaP (database of Genotyping and Phenotypes), which is presented by NCBI (National Center for Biotechnology Information) and archives and distributes the results of genome‐wide association studies, medical sequencing, and molecular diagnostic assays, as well as the association between genotype and non‐clinical traits (http://www.ncbi.nlm.nih.gov/gap/); CGEMS (Cancer Genetic Markers of Susceptibility), which presents GWAS data on various cancers such as breast, prostate, pancreatic, and lung from the NCI (National Cancer Institute) Cohort Consortium as well as collaborative case‐control epidemiologic studies with biospecimens (http://cgems.cancer.gov/); WTCCC (Wellcome Trust Case Control Consortium), which provides GWAS data on case‐control and cohort studies on various diseases, mostly on UK samples (http://www.wtccc.org.uk/); and GWAS DB and Mutation Database (http://gwas.lifesciencedb.jp/), which is a part of the Integrated Database project by the Ministry of Education, Culture, Sports, Science and Technology of Japan (http://lifesciencedb.mext.go.jp/en/index.html) and offers GWAS data on several diseases. Availability of GWAS databases representing different ethnic groups is important, because a minor allele frequency (MAF) of disease susceptibility locus is one of the critical factors determining the statistical power of a GWAS, and a significant ethnic difference in MAF is often observed among Caucasians, Africans, and Asians; GWAS in each ethnic group has actually detected a different set of SNPs in the same disease.( 32 )
Genome Medicine Database of Japan (GeMDBJ). GeMDBJ (Genome Medicine Database of Japan, http://gemdbj.nibio.go.jp/) was originally developed to release GWAS data from the Millennium Genome Project in Japan. Since the Project was finished in 2005, the database has been maintained by the National Institute of Biomedical Innovation (NiBio), a funding agency supported by the Ministry of Health, Labour and Welfare of Japan, and the contents of GeMDBJ have been further developed by a collaboration of five national centers and a national research institute: National Center for Neurology and Psychiatry (NCNP), National Cancer Center (NCC), National Center for Global Health and Medicine (NCGM, formerly International Medical Center of Japan), National Cardiovascular Center (NCVC), National Center for Child Health and Development (NCCHD), and National Institute of Health Sciences (NIHS) (Fig. 5). The database offers genome‐wide SNP typing data (allele and genotype frequency data) for five major diseases (Alzheimer’s disease, gastric cancer, type 2 diabetes, hypertension, and asthma) targeted by the Millennium Genome Project. In addition to the SNP data, the database contains Affymetrix GeneChip transcriptome data on >1000 samples, most of which are various types of clinical cancer tissues. A recent addition to the database is GeMDBJ Proteomics,( 33 ) which presents cancer proteome analysis data based on the standardized protocol( 34 ) with protein spot quantification by 2D‐DIGE (two‐dimensional difference gel electrophoresis) and protein identification by mass spectrometry.
Figure 5.
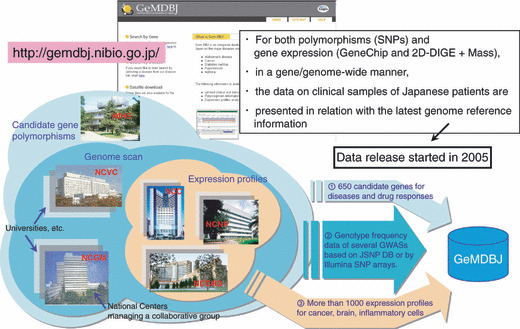
Status of GeMDBJ (Genome Medicine Database of Japan). Since the completion of the Millennium Genome Project, GeMDBJ (http://gemdbj.nibio.go.jp/) has been developed by a joint effort of five national centers (NCNP, NCC, NCGM, NCVC, and NCCHD) and a national research institute (NIHS) (see text for full names). The left figure shows that expression profile data have been provided by three centers, genome scan data from two additional (total five) centers, and candidate gene SNP data from all six institutions, each specializing in different diseases (five national centers) or on drug responses (NIHS). GeMDBJ Proteomics was constructed by NCC researchers.( 33 )
Privacy protection issue with GWAS databases. In August 2008, a shockwave spread among GWAS database constructors. A paper by Homer et al. ( 35 ) demonstrated that it is feasible to infer whether an individual DNA sample was included in an aggregate genotype dataset, such as genotype and allele frequency data of a group of patients, which had been considered safe for posting in the publicly accessible database. Their simulation suggested that 50 000 SNP data can detect an individual within aggregate data of 1000 people. To make such a group assignment (case or control) possible for an individual, it is necessary to have high‐density genomic data for that specific individual, as well as the allele frequencies of the control (or reference) population. While the latter population control genomic data are available for many major ethnic groups, the former individual genome‐wide data are currently unlikely to be easily obtained outside the research context. However, such a situation will rapidly change by the introduction of commercial or medical massive genotyping or resequencing services. In response to the Homer paper, the major GWAS databases removed aggregate datasets from the open access tier and moved them into the controlled access category.( 36 ) In Japan, the Integrated Database Project has recently established a policy to share the GWAS and resequencing data for disease‐associated genes (http://gwas.lifesciencedb.jp/gwasdb/db_policy_en.html), which we have decided to adopt for GeMDBJ, because the policy was very well contemplated and also because we believe that policy standardization is important.
Towards an integrated database. Currently, GeMDBJ should be considered still in its infancy in the sense that each dataset is presented relatively separately, and we are exploring a way for a powerful synthetic analysis by correlating the multiple types of the omics and phenotype data. However, the potential power of such omics data integration was already hinted at in our first experience with gastric cancer GWAS. Because the data from the Millennium Genome Project was posted in the database as soon as possible, the first screening data from the two‐stage JSNP genome scan had been already made available to the public when we were still in the midst of narrowing down our target to the PSCA gene through extensive second‐stage screening, validation by the Korean researchers, and functional analyses. At that time, hundreds of Affymetrix GeneChip data were also accessible at GeMDBJ on various types of cells and tissues, including those of the stomach. When we combined the database search for a low P‐value in the GWAS first‐screening data and the increased expression in the gastric tissue, we were surprised to see that the PSCA gene was actually selected, without the then unpublished second‐screening data. Of course, this could not be generalized for all of the susceptibility genes, because some of them may not necessarily be expressed in the tissues from which the cancer arises; for instance, some cancer susceptibility genes may not be expressed in the target tissues but in the liver or kidney involved in carcinogen metabolism and transport. Moreover, the PSCA case should be considered a rather fortunate case, because, as described in the Introduction, a hypothesis‐free GWAS often results in a genome region in which a protein‐coding gene was not known or cannot be singled out from many possible candidates. Integration of various data, knowledge, and information by an interactive database is expected to play a crucial role in such difficult GWAS cases and may accelerate a novel discovery in science and medicine.
Prospects for the Next Generation Germline Analyses
The genetic architecture of disease susceptibility has not yet been fully resolved for any common disease. Even for cases with identified causative gene mutations of classical Mendelian diseases, inter‐pedigree differences are often observed regarding disease penetrance and expressivity, which may be explained by an involvement of modifier genes. For common diseases, one popular hypothesis for genetic architecture is a CD‐CV, or common disease‐common variant, hypothesis which predicts a relatively frequent (common) disease susceptibility allele at each of the major underlying disease loci.( 37 ) Because disease susceptibility alleles have been able to achieve a high equilibrium frequency in a given population, they should at least have little or no effect on reproductive fitness,( 38 ) and the overall effect size (such as risk ratio) may also be small. The classical example of the CD‐CV model is the APOE e4 allele for Alzheimer’s disease.( 39 ) Although common diseases are believed to be polygenic, the number of common alleles involved in a disease is still a matter of speculation; for instance, as suggested by one simulation for breast cancer,( 40 ) there may be 30–40 alleles with a relative risk of 1.5 and a minor allele frequency of 0.1. However, there are several cases against the CD‐CV hypothesis, with the alternative model being called CD‐MRV (multiple rare variants) or by other related names.( 41 ) The simple but strong argument for CD‐MRV is that a common disease is common because of highly prevalent non‐genetic factors, not because of common “susceptibility alleles” in a given population.( 39 ) It has been predicted that “neutral” susceptibility alleles without evolutionary selection pressure become non‐polymorphic, while susceptibility alleles under weak selection tend to remain polymorphic, especially at loci with high mutation rates. In this model, there will be extensive allelic heterogeneity in the genetic architecture of a common disease. Although some of the susceptibility alleles may be rare in the population, the collective frequency of these alleles may be quite high.( 39 , 41 , 42 )
Figure 6 illustrates an image of the genetic architecture of cancer (modified from ref. 39). The current GWAS covers a relatively limited range of allele frequency (i.e. common variants) and effect size (OR of approximately 1.2 or higher) for most diseases. To capture the entire landscape of genetic factors, the current GWAS can be extended along the upper left direction (higher frequency and lower effect size), if a sufficient research resource is available, but the clinical/public health significance is doubtful.( 41 ) On the other hand, it is conceivable that a number of significant genetic factors are yet to be disclosed on the right lower area; in addition to some cases with clinically definitive, but mutation‐undetectable, hereditary cancer syndromes, many cases with familial clustering, young age at onset, and/or multiple/double cancers may be due to underlying genetic causes.
Figure 6.
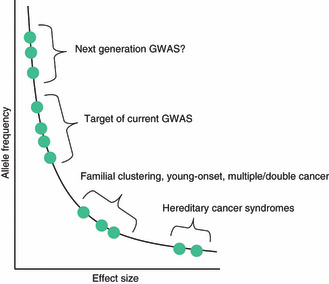
Genetic architecture of disease susceptibility (an image). It is assumed that multiple genetic polymorphisms with a range of allele frequencies and effect sizes constitute an individual and population genetic architecture of disease risk. However, current and past linkage and association studies could access only limited segments of the curve. Personal genome sequencing by next or future generation sequencers is expected to fill, at least some, gaps. GWAS, genome‐wide association study. Adapted from ref. 39.
For these cases, the recent advent of whole genome or even exome sequencing is beginning to show several early successes on recessive non‐cancer hereditary diseases.( 43 , 44 , 45 ) Unlike classical linkage analyses on large pedigrees, it is remarkable that these studies reached the causative genes on a small number of subjects, who were not necessarily related,( 43 ) although a family‐based sequencing would be more powerful.( 44 ) The hurdles will be higher for dominant Mendelian traits,( 43 ) which may account for most of the hereditary cancer syndromes. However, many of them are “dominant” because of the second‐hit somatic inactivation of a causative tumor suppressor gene. The combination of the exomic sequencing of the tumor and blood (germline) DNAs identified a novel susceptibility gene for a familial pancreatic cancer.( 46 ) In the coming era of personal genome sequencing, we will witness amazing progress in unveiling the missing portion of the genetic architecture of cancer (Fig. 6), which will be followed by intervention research for the development of personalized prevention based on genomic factors.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- CI
confidence interval
- OR
odds ratio
- PSCA
prostate stem cell antigen
Supporting information
Table S1. Genome‐wide association study (GWAS) and related studies on cancers reported in Nature Genetics (as of December 2009).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank all the co‐authors of reference 11, which is the major original work for this review article. As regards GeMDBJ, we are deeply grateful to Drs Yuichi Goto, Kazuki Yasuda, Naoharu Iwai, Kenji Matsumoto, Yoshiro Saito, Tadashi Kondo, and all the other investigators who kindly contributed their data and ideas to the database. We also thank the engineers and scientists of Mitsui Knowledge Industry Co., Ltd., for their efforts in the database construction. This work was supported by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NiBio) and by a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan.
References
- 1. Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science 1996; 273: 1516–17. [DOI] [PubMed] [Google Scholar]
- 2. Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Jpn J Clin Oncol 2003; 33: 111–21. [DOI] [PubMed] [Google Scholar]
- 3. Li H, Hao X, Zhang W, Wei Q, Chen K. The hOGG1 Ser326Cys polymorphism and lung cancer risk: a meta‐analysis. Cancer Epidemiol Biomarkers Prev 2008; 17: 1739–45. [DOI] [PubMed] [Google Scholar]
- 4. Horvath S, Baur MP. Future direction of research in statistical genetics. Stat Med 2000; 19: 3337–43. [DOI] [PubMed] [Google Scholar]
- 5. Pritchard JK, Przeworski M. Linkage disequilibrium in humans: models and data. Am J Hum Genet 2001; 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirakawa M, Tanaka T, Hashimoto Y, Kuroda M, Takagi T, Nakamura Y. JSNP: a database of common gene variations in the Japanese population. Nucleic Acids Res 2002; 30: 158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haga H, Yamada R, Nakamura Y, Tanaka T. Gene‐based SNP discovery as part of the Japanese Millennium Genome Project: identification of 190562 genetic variations in the human genome. J Hum Genet 2002; 47: 605–10. [DOI] [PubMed] [Google Scholar]
- 8. Ozaki K, Ohnishi Y, Iida A et al. Functional SNPs in the lymphotoxin‐α gene that are associated with susceptibility to myocardial infarction. Nat Genet 2002; 32: 650–4. [DOI] [PubMed] [Google Scholar]
- 9. The International HapMap Consortium . The International HapMap project. Nature 2003; 426: 789–96. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida T, Yoshimura K. Outline of disease gene hunting approaches in the Millennium Genome Project of Japan. Proc Jpn Acad 2003; 79: 34–50. [Google Scholar]
- 11. Sakamoto H, Yoshimura K, Saeki N et al. Genetic variation in PSCA is associated with susceptibility to diffuse‐type gastric cancer. Nat Genet 2008; 40: 730–40. [DOI] [PubMed] [Google Scholar]
- 12. http://www.fpcr.or.jp/publication/pdf/statistics2009/date04.pdf
- 13. Japanese Gastric Cancer Association . Japanese classification of gastric carcinoma. 2nd English edition. Gastric Cancer 1998; 1: 10–24. [DOI] [PubMed] [Google Scholar]
- 14. Lauren P. The two histological main types of gastric carcinoma: diffuse and so‐called intestinal‐type carcinoma. An attempt at a histo‐clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31–49. [DOI] [PubMed] [Google Scholar]
- 15. Hohenberger P, Gretschel S. Gastric cancer. Lancet 2003; 362: 305–15. [DOI] [PubMed] [Google Scholar]
- 16. Schier S, Wright NA. Stem cell relationships and the origin of gastrointestinal cancer. Oncology 2005; 1 (Suppl 1): 9–13. [DOI] [PubMed] [Google Scholar]
- 17. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosai J. Gastrointesitinal tract–stomach. In: Rosai and Ackerman’s Surgical Pathology, Rosai J, ed. Edinburgh: Mosby, 2004; 648–711. [Google Scholar]
- 19. Yokota T, Teshima S, Saito T, Kikuchi S, Kunii Y, Yamauchi H. Borrmann’s type IV gastric cancer: clinicopathologic analysis. Can J Surg 1999; 42: 371–6. [PMC free article] [PubMed] [Google Scholar]
- 20. Henson DE, Dittus C, Younes M, Nguyen H, Albores‐Saavedra J. Differential trend in the intestinal and diffuse types of gastric carcinoma in United States, 1973–2000–increase in the signet ring cell type. Arch Pathol Lab Med 2004; 128: 765–70. [DOI] [PubMed] [Google Scholar]
- 21. Japanese Gastric Cancer Association Registration Committee , Maruyama K, Kaminishi M et al. Gastric cancer treated in 1991 in Japan: data analysis of nationwide registry. Gastric Cancer 2006; 9: 51–66. [DOI] [PubMed] [Google Scholar]
- 22. Sasazuki S, Inoue M, Iwasaki M et al. Effect of Helicobacter pylori infection combined with CagA and pepsinogen status on gastric cancer development among Japanese men and women: a nested case‐control study. Cancer Epidemiol Biomarkers Prev 2006; 15: 1341–7. [DOI] [PubMed] [Google Scholar]
- 23. Guilford P, Hopkins J, Harraway J et al. E‐cadherin germline mutations in familial gastric cancer. Nature 1998; 392: 402–5. [DOI] [PubMed] [Google Scholar]
- 24. Shinmura K, Kohno T, Takahashi M et al. Familial gastric cancer: clinicopathological characteristics, RER phenotype and germline p53 and E‐cadherin mutations. Carcinogenesis 1999; 20: 1127–31. [DOI] [PubMed] [Google Scholar]
- 25. Wang G‐Y, Lu C‐Q, Zhang R‐M, Hu X‐H, Luo ZW. The E‐cadherin gene polymorphism ‐160C/A and Cancer Risk: a HuGE review and meta‐analysis of 26 case‐control studies. Am J Epidemiol 2008; 167: 7–14. [DOI] [PubMed] [Google Scholar]
- 26. Sato Y, Suganami H, Hamada C, Yoshimura I, Yoshida T, Yoshimura K. Designing a multistage, SNP‐based, genome screen for common diseases. J Hum Genet 2004; 49: 669–76. [DOI] [PubMed] [Google Scholar]
- 27. Matsuo K, Tajima K, Suzuki T et al. Association of prostate stem cell antigen gene polymorphisms with the risk of stomach cancer in Japanese. Int J Cancer 2009; 125: 1961–4. [DOI] [PubMed] [Google Scholar]
- 28. Reiter RE, Gu Z, Watabe T et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci USA 1998; 95: 1735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gu Z, Thomas G, Yamashiro J et al. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000; 19: 1288–96. [DOI] [PubMed] [Google Scholar]
- 30. Bahrenberg G, Brauers A, Joost H‐G, Jakse G. Reduced expression of PSCA, a member of the LY‐6 family of cell surface antigen, in bladder, esophagus, and stomach tumors. Biochem Biophys Res Commun 2000; 275: 783–8. [DOI] [PubMed] [Google Scholar]
- 31. Wu X, Ye Y, Kiemeney LA et al. Genetic variation in the prostate stem cell antigen gene PSCA confers susceptibility to urinary bladder cancer. Nat Genet 2009; 41: 991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yasuda K, Miyake K, Horikawa Y et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008; 40: 1092–7. [DOI] [PubMed] [Google Scholar]
- 33. Kondo T. Proteome expression database; Genome Medicine Database of Japan Proteomics. Expert Rev Proteomics 2010; 7: 21–7. [DOI] [PubMed] [Google Scholar]
- 34. Kondo T, Hirohashi S. Application of highly sensitive fluorescent dyes (CyDye DIGE Fluor saturation dyes) to laser microdissection and two‐dimensional difference gel electrophoresis (2D‐DIGE) for cancer proteomics. Nat Protoc 2007; 1: 2940–56. [DOI] [PubMed] [Google Scholar]
- 35. Homer N, Szelinger S, Redman M et al. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high‐density SNP genotyping microarrays. PLoS Genet 2008; 4: e1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Couzin J. Genetic privacy. Whole‐genome data not anonymous, challenging assumptions. Science 2008; 321: 1278. [DOI] [PubMed] [Google Scholar]
- 37. Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet 2001; 17: 502–10. [DOI] [PubMed] [Google Scholar]
- 38. Kimura M, Ohta T. The age of a neutral mutant persisting in a finite population. Genetics 1973; 75: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wright AF, Hastie ND. Complex genetic diseases: controversy over the Croesus code. Genome Biol 2001;2: commnet2007.1‐2007.8. Epub 2001 Aug 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ponder BA. Cancer genetics. Nature 2001; 411: 336–41. [DOI] [PubMed] [Google Scholar]
- 41. Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 2008; 40: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 2001; 69: 124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ng SB, Buckingham KJ, Lee C et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010; 42: 30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roach JC, Glusman G, Smit AF et al. Analysis of genetic inheritance in a family quartet by whole‐genome sequencing. Science 2010; 328: 636–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lupski JR, Reid JG, Gonzaga‐Jauregui C et al. Whole‐genome sequencing in a patient with Charcot‐Marie‐Tooth neuropathy. N Engl J Med 2010; 362: 1181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones S, Hruban RH, Kamiyama M et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009; 324: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saeki N, Kim DH, Usui T et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF‐β‐dependent apoptotic signalling. Oncogene 2007; 26: 6488–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome‐wide association study (GWAS) and related studies on cancers reported in Nature Genetics (as of December 2009).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item