

Abstract
A family of transcription factors, the interferon regulatory factors (IRF), was identified originally in the context of the regulation of the type I interferon (IFN)‐α/β system. The IRF family has now expanded to nine members, and gene‐disruption studies have revealed the critical involvement of these members in multiple facets of host defense systems, such as innate and adaptive immune responses and tumor suppression. In the present review article, we aim at summarizing our current knowledge of the roles of IRF in host defense, with special emphasis on their involvement in the regulation of oncogenesis. (Cancer Sci 2008; 99: 467–478)
The original discovery of the first two members of the interferon (IFN) regulatory factor (IRF) family, IRF1 and IRF2, opened up new avenues of research in immunity and oncogenesis, which we may call ‘the IRF world’. The transcription factor IRF1 was identified originally as a regulator of the IFN system.( 1 ) Subsequent gene‐disruption studies revealed that IRF1 plays various roles in the host immune system against microbial infection: it is essential in IFN‐induced antiviral and antibacterial responses,( 2 , 3 ) the Th1‐type adaptive immune response, and the development of natural killer (NK) cells.( 4 , 5 , 6 ) Following the first identification of IRF1, at least nine structurally related members have been identified thus far, and currently constitute a family of IRF transcription factors. In particular, along with recent extensive studies of signaling pathways mediated by microbial pattern recognition receptors (PRR), much attention has been focused on the roles of the IRF family members in innate immunity. Certain microbial products, such as double‐stranded (ds) RNA, lipopolysaccharides, and oligodeoxyribonucleotides containing unmethylated CpG motifs, activate the IRF‐mediated induction of IFN‐α and IFN‐β genes through Toll‐like receptors (TLR). In addition, the recent identification of receptors for cytosolic nucleic acid recognition, such as retinoic acid inducible gene (RIG)‐I, melanoma differentiation‐associated gene (MDA) 5, and DNA‐dependent activator of IRF (DAI, previously known as DLM‐1 or ZBP‐1),( 7 , 8 ) has delineated the downstream activation pathways of IRF3 and IRF7 for type I IFN induction. In addition, it has been found that IRF members participate in the PRR‐mediated induction of proinflammatory cytokines. It has been shown that IRF5, which associates with MyD88, an adaptor of most TLR, is a critical regulator of the induction of proinflammatory cytokine genes.( 9 ) Interestingly, this IRF5 function was found to be negatively regulated by another member, IRF4.( 10 ) IRF1, which is induced by IFN‐γ, is another mediator that is activated downstream of the TLR‐MyD88 pathway for the induction of specific genes such as IFN‐β, inducible NO synthase, and IL‐12p35.( 11 ) It was also reported that IRF4 and IRF8 participate in TLR‐mediated signaling in dendritic cells (DC). Thus, many of the IRF family members are essential regulators in PRR‐mediated signaling. In relation to these findings on the roles of IRF in immune responses against infection, much attention has also been focused on their involvement in DNA damage‐induced responses and regulation of oncogenesis. Loss of expression or function of IRF is observed in human cancers, whereas a certain IRF member is overexpressed in hematological malignancy. Interestingly, human herpesvirus (HHV)‐8 encodes several proteins, termed vIRF, that are analogous to human IRF proteins and may be involved in the pathogenesis of Kaposi's sarcoma or other cancers.( 12 , 13 )
In the context of oncogenesis, we can therefore categorize several IRF family members into two types: antioncogenic IRF and oncogenic IRF. In the present review article, we summarize the contribution of IRF to the regulation of immune responses, cell growth, apoptosis, and oncogenesis. Understanding the molecular mechanisms by which the IRF members regulate cellular growth and tumor suppression will contribute to a better understanding of pathogenic processes leading to human immune and malignant diseases, and will provide novel therapeutic strategies.
Interferon regulatory factor family members: Fundamental characteristics and evolutionary implications
Interferon regulatory factor transcription factors have thus far been shown to be transcriptional mediators in many biological processes, including innate and adaptive immune responses, cell growth regulation and apoptosis, and hematopoietic development.( 14 , 15 , 16 ) To date, nine members have been identified, as well as virus‐encoded analogs of cellular IRF (Fig. 1): (1) cellular IRF (IRF1, IRF2, IRF3, IRF4 [Pip, PU.1‐interacting partner: ICSAT, IFN consensus sequence‐binding protein in adult T‐cell leukemia cell line or activated T cells; lymphoid‐specific member of the IRF family], IRF5, IRF6, IRF7, IRF8 ([ICSBP, IFN consensus sequence binding protein], and IRF9 [ISGF3γ, also refered to as p48]); and (2) viral IRF (vIRF1, vIRF2, and vIRF3/LANA2). A phylogenetic analysis( 17 ) indicates that the IRF proteins can be classified into four subfamilies: IRF1, IRF3, IRF4, and IRF5. IRF10, which was also identified in chicken (see below for details), belongs to the IRF4 subfamily.
Figure 1.
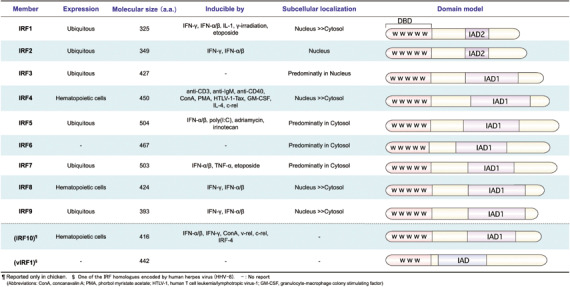
Interferon regulatory factor (IRF) family members. The fundamental characteristics of the nine human IRF family members (IRF1 to IRF9) and one avian IRF10 are shown in the table (left), and their schematic domain models are shown in the right panel. All IRF carry the N‐terminal DNA‐binding domain (DBD), which contains repeated tryptophan residues (represented by ‘W’) similar to c‐myb. All IRF, except IRF1 and IRF2, have an IRF association domain (IAD1) that is responsible for interaction with other family members or transcription factors such as PU.1, E47, and Stat. Another association domain (IAD2) that is present in IRF1 and IRF2 is important for their interaction with IRF8. Mammalian IRF10 has not been reported. Among the four viral IRF (vIRF) encoded by human herpesvirus‐8, vIRF1 is shown as a representative. vIRF1 also contains an IAD domain. (157)
Interferon regulatory factors share significant homology within the conserved N‐terminal DNA‐binding domain, which is characterized by having a winged‐type helix‐loop‐helix motif with a signature tryptophan pentad (Fig. 1). The crystal structure of IRF1 and IRF2 revealed that three of these repeats contact DNA with specific recognition of the GAAA and AANNGAAA sequences (recognized bases are underlined).( 18 , 19 ) The consensus DNA sequences that IRF recognizes were determined in several contexts. (1) The IRF‐binding element (IRF‐E, G[A]AAAG/CT/CGAAAG/CT/C),( 20 ) which was determined as the consensus sequence for IRF1 and IRF2 binding; (2) the IFN‐stimulated response element (ISRE; A/GNGAAANNGAAACT),( 21 ) which is recognized by IRF9; and (3) the IFN consensus sequence,( 22 , 23 ) for the site of recognition by IRF8. The secondary structures of the DNA‐binding domains of IRF are similar to each other, suggesting that IRF members recognize similar, if not identical, DNA sequences. The C‐terminal portion varies among these members and promotes versatile biological functions. In addition to their intrinsic transactivation potential, some IRF acquire a specific function by associating with another IRF member, other transcriptional factors, or cofactors. In addition, their transcriptional activities vary, resulting in activation, repression, or dual activity on their target genes. This is partly attributed to the partner proteins associated with IRF. These interactions are mediated by two types of association module of the C‐terminal region: (1) IRF‐associated domain 1,( 24 ) which was initially found in IRF8 and is conserved in all IRF (excluding IRF1 and IRF2); and (2) IAD2, which is shared only by IRF1 and IRF2. In most cases, these protein complexes enhance the ability of IRF to bind to target DNA sequences such as ISRE or IRF‐E. For example, IRF9 acts as a DNA‐binding subunit that associates with Stat1 and Stat2 to form the ISGF3 heterotrimeric complex in response to type I IFN signaling.( 25 ) IRF8 forms multiple protein complexes with both IRF1 and IRF2, resulting in increased binding activity to ISRE.( 24 , 26 ) IAD2 of IRF1 and IRF2 is an independent module for this interaction with IRF8. The IRF8 and IRF1 complex generally functions as a suppressor of transcription. IRF4 and IRF8 interact with PU.1, a member of the ETS family, and this interaction allows them to bind to the immunoglobulin light‐chain enhancer λB( 27 , 28 ) for the subsequent activation of gene transcription. IRF1, IRF3, and IRF7 form part of large protein complexes (IFN‐β enhanceosome, DRAF1) including CREB binding protein (CBP)/p300 proteins.( 29 , 30 , 31 , 32 ) Similarly, IRF1 and IRF2 were reported to form a complex with multiple histone acetylases, PCAF, CBP, and p300/CREB to bind to ISRE.( 33 ) On the other hand, IRF2, IRF4, IRF8, and IRF7 suppress transcription from several ISRE promoters.( 29 , 30 , 31 ) However, these IRF also function as activators in other promoters.( 28 , 34 )
Although some IRF are expressed predominantly in hematological tissues, others are expressed ubiquitously (Fig. 1). Unlike type I IFN genes, the gene locus of each IRF member is not clustered in the same region (Fig. 2a). From an evolutionary viewpoint, it was reported that type I IFN and IRF family members are found in vertebrates.( 35 , 36 ) Interestingly, IRF family members appear to show similar features to nuclear factor (NF)‐κB members (which are also found in non‐vertabrates) in various aspects: each binding element contains the identical core motif GAAA, and both factors are activated for the PRR‐mediated induction of cytokine genes. This process of activating each factor is mediated by a similar class of IκΒ kinases. Moreover, its has been shown that both factors interact with each other for the induction of certain genes.( 37 , 38 , 39 ) Together with recent findings about these factors in the context of PRR‐mediated pathways, it can be postulated that IRF members might have evolved during vertebrate evolution so as to modulate the functions of the NF‐κB family, such as the robust activation of type I IFN genes. In parallel with the possible evolution of IRF genes, type I IFN genes seem to have evolved from IFN‐β to multiple IFN‐α subtypes. In fact, there is evidence that the IFN‐α gene is controlled by IFN‐β( 40 ) (A. Takaoka, unpublished data, 2000). Although the IFN‐β gene is activated by both IRF3 and IRF7, IFN‐α genes are targeted mainly by IRF7,( 41 , 42 ) which may be considered as a newly evolved IRF. It is interesting to note that the promoter of the IFN‐β gene contains multiple factor binding sites, such as those for NF‐κB, IRF, and AP‐1, whereas only the IRF‐binding site is found in IFN‐α genes. Indeed, the IFN‐β gene is activated by the c‐Fos transcription factor, and this activation is critical to the negative regulation of osteoclast differentiation.( 43 ) Further approaches from the evolutional viewpoint may provide some clue to elucidating signaling pathways for the activation of IRF family members.
Figure 2.
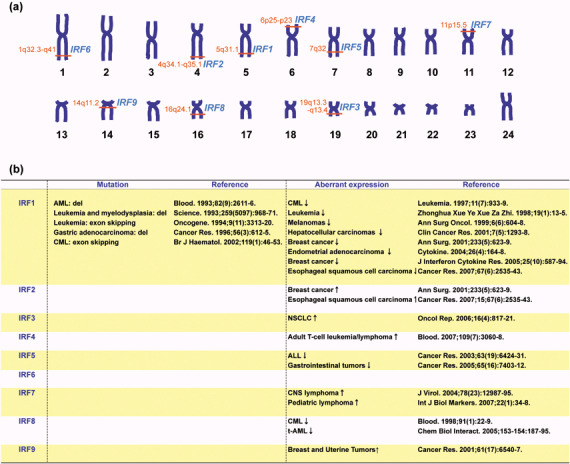
Mutations and aberrant expression of interferon regulatory factor (IRF) genes in human cancers. (a) Schematic representation of human chromosomes and gene loci of IRF genes. (b) Summary table of previous reports about mutations and aberrant expression of the IRF genes in primary specimens derived from human malignancies. Expression levels included are based on both mRNA and protein levels. AML, acute myelogenous leukemia; ALL, acute lymphocytic leukemia; CML, chronic myelogenous leukemia; NSCLC, non‐small cell lung cancer; CNS, central nervous system; t‐AML, therapy‐related acute myelogenous leukemia.
In light of the hitherto‐defined, most distinctive function of each IRF (Table 1), the IRF family members might be classified tentatively into four categories: (1) interferonic IRF (IRF3 and IRF7 as regulators of IFN production, and IRF2 and IRF9 as negative and positive mediators in IFN signaling, respectively); (2) stress‐responsive IRF (IRF1 and IRF5); (3) hematopoietic IRF (IRF4 and IRF8); and (4) morphogenic IRF (IRF6). However, as described below, it is clear that most if not all of these IRF are involved in oncogenesis and immunity. We will summarize each class of IRF; it is recommended that readers refer to other recent review articles for further details.( 44 , 45 , 46 , 47 , 48 )
Table 1.
Subfamily of the interferon regulatory factor (IRF) family members
| Subfamily | Member |
|---|---|
| 1. Interferonic IRF | |
| (i) Regulators of type I interferon induction | IRF3, IRF7 |
| (ii) Mediators in interferon signaling | IRF2, IRF9 |
| 2. Stress‐responsive IRF | IRF1, IRF5 |
| 3. Hematopoietic IRF | IRF4, IRF8 |
| 4. Morphogenic IRF | IRF6 |
Interferonic IRF
Regulators of IFN induction: IRF3 and IRF7. IRF3 and IRF7 are the key regulators of type I IFN gene expression upon viral infection. IRF3 is expressed constitutively and resides in the cytosol. When cells are infected with viruses, the activation of certain PRR including TLR3, TLR4, RIG‐I, MDA5 and DAI (DLM‐1/ZBP1) results in the phosphorylation of IRF3 on a cluster of serine and threonine residues in the C‐terminal region, leading to its nuclear translocation and subsequent induction of type I IFN genes. IRF7 is highly homologous to IRF3, but unlike IRF3, IRF7 is expressed at a low level in most cells and is IFN‐inducible in an IRF9‐dependent pathway. Similarly to IRF3, IRF7 is activated downstream of certain PRR including TLR3, TLR4, TLR7, TLR8, TLR9, RIG‐I, MDA5 and DAI (DLM‐1/ZBP1), and undergoes nuclear translocation for type IFN I induction. IRF3 and IRF7 form a homodimeric or a heterodimeric complex with each other and act differentially on the type I IFN gene family members. It has been shown that IRF3 potently activates the IFN‐β gene rather than the IFN‐α genes, except the IFN‐α4 gene, whereas IRF7 preferentially activates the IFN‐α and IFN‐α5 genes.( 49 ) During viral infection, PRR‐mediated signaling activates IRF kinases such as TBK1 and IKK∈/i, leading to the induction of type I IFN genes, but it was also reported that IRF3 and IRF7 are phosphorylated in response to DNA damage caused by ultraviolet irradiation.( 50 , 51 ) However, the signaling pathways leading to their activation following viral infection and DNA damage are clearly different. IRF3 is phosphorylated by DNA‐dependent protein kinase (DNA‐PK) at threonine residue‐135,( 50 ) which differs from TBK1‐mediated phosphorylation sites. This post‐translational modification likely occurs in the nucleus, and downregulates the export of IRF3 from the nucleus to the cytoplasm for degradation by the ubiquitin–proteasome pathway. There was also a study showing that DNA‐damaging agents and ultraviolet irradiation can activate IRF7 via the mitogen‐activated protein kinase kinase‐4–c‐Jun N‐terminal kinase pathway.( 51 ) IRF7 is localized on human chromosome 11p15.5 in a region that is CpG rich. Hypermethylation of the CpG island in the IRF7 promoter was shown to be responsible for silencing of the IRF7 gene in the 2fTGH fibrosarcoma cell line and human astrocytoma tissues.( 52 , 53 ) These results suggest that IRF7 plays a role in the maintenance of genomic stability. Further studies are needed to establish whether IRF7 is necessary for tumor suppression. Relevant to these observations, IRF7 was identified as a breast‐cancer susceptibility gene 1 (BRCA1) transcriptional target.( 54 , 55 ) BRCA1 is a tumor‐suppressor gene, the mutation of which is implicated in hereditary predisposition to breast and ovarian cancer, and it has been shown to be essential in a number of cellular processes including DNA repair and recombination, cell cycle checkpoint control, chromatin remodeling, and ubiquitination, as well as transcriptional regulation.( 56 , 57 ) In this context, IRF7 was shown to be upregulated synergistically by BRCA1, specifically in the presence of IFN‐γ, coincident with the synergistic induction of apoptosis.( 55 ) However, further analyses will be required to clarify whether IRF7 affects the tumor‐suppressive function of BRCA1, because the possibility that IRF7 acts as an apoptotic regulator or a tumor suppressor is currently controversial.
Recently, it has been demonstrated that these IRF also participate in the induction of another new class of IFN family members, type III IFN, including IFN‐λ1 (interleukin [IL]‐29), IFN‐λ2 (IL‐28A), and IFN‐λ3 (IL‐28B), which show similar biological antiviral functions to type I IFN. It seems that the IFN‐λ1 gene is regulated by virus‐activated IRF3 and IRF7, thus resembling the IFN‐βgene, whereas IFN‐λ2 and IFN‐λ3 gene expression is controlled mainly by IRF7, thus resembling the expression of IFN‐α genes.( 58 )
Mediators in IFN signaling: IRF2 and IRF9. IRF2 was found to play a role as a negative regulator, attenuating IFN‐α‐ and IFN‐β‐induced gene transcription in ISRE, where IRF9 acts as a component of the tertiary complex ISGF3.( 41 ) IRF2‐deficient mice develop an inflammatory skin disease with CD8+ T‐cell abnormality, which is due to hyperactivation of IFN‐α and IFN‐β signaling. IRF2 was initially assumed to function as a transcriptional repressor of the IFN‐βgene, whereas IRF1 functions as an activator. Further evidence has shown that IRF2 can actively induce certain genes such as vascular cell adhesion molecule‐1,( 59 ) the cell cycle‐regulated histone H4 genes,( 34 ) and the transporter of antigenic peptides to major histocompatibility complex (MHC) class I gene.( 60 ) The overexpression of IRF2 in NIH 3T3 cells results in oncogenic transformation, which can be reverted by the concomitant expression of IRF1.( 61 ) IRF2 was also shown to reverse N‐Ras‐induced growth inhibition in a myeloid cell line.( 62 ) IRF2 functions in a late phase by maintaining CD11blowDx5high cells expressing Ly49 receptors for efficient NK cell maturation,( 63 ) whereas IRF1 regulates the induction of IL‐15, which is involved in the expansion of immature NK cells at an earlier stage.( 5 ) IRF2 was also found to have a role in the development of myeloid DC.( 64 , 65 )
Interferon regulatory factor 9 plays a major role in multiple ISGF3‐dependent gene induction by type I and type II IFN.( 66 ) However, an emerging body of evidence suggests non‐ISGF3 roles for IRF9. IRF9 can also form a DNA‐binding complex with the STAT1 homodimer (Stat1–IRF9), which is required for IFN‐γ‐stimulated induction of the human IP‐10 gene.( 67 ) IRF9 is efficiently targeted to the nucleus via an intrinsic bipartite localization signal that functions in the absence of IFN signaling,( 68 ) and the ectopic expression of IRF9 confers resistance to antimicrotubule agents in breast cancer cell lines independent of IFN signals.( 69 ) Increased expression levels were observed in half of the breast and uterine cancer samples tested (Fig. 2b),( 69 ) which suggests its potential role in cancer biology.
Stress‐responsive IRF: IRF1 and IRF5. Intricate signaling networks have evolved to sense various stresses and to repair stress‐induced damage, and these networks are critical to the elimination of severely damaged cells, particularly for the prevention of malignant transformation. IRF1 and IRF2 were initially identified as factors that bind to positive regulatory domain 1 in the virus responsive element (VRE) of the IFN‐β gene. Although IFN‐α and IFN‐βgenes are normally induced in IRF1‐deficient mouse embryonic fibroblasts (MEF), the dsRNA‐mediated induction of type I IFN is downregulated in these mutant cells, and IRF1 has recently been shown to participate in type I IFN gene induction in some facets of TLR signaling.( 11 ) However, many studies have revealed that IRF1 is involved in a broader spectrum of biological functions, including the IFN‐γ‐mediated induction of inducible NO synthase, guanylate binding protein, 2′,5′‐oligoadenylate synthetase (OAS), and MHC class I, the development of CD8+ T cells, induction of IL‐12 and Th1 differentiation, and NK cell development. Thus, IRF1 has crucial functions in the development and activation of various immune cells. Furthermore, IRF1 also plays a critical role in cell cycle regulation and apoptosis in response to a variety of genotoxic stresses (see below).
The expression of IRF5 is upregulated in response to type I IFN signaling and viral infection.( 70 ) In vitro experiments with overexpression assays have shown that IRF5 induces IFN‐α1 as the major subtype.( 71 , 72 ) Gene‐disruption studies have revealed that IRF5 plays a critical role in innate immune responses and is involved in TLR‐mediated induction of proinflammatory cytokines such as IL‐6, TNF‐α, and IL‐12, rather than in type I IFN induction.( 9 , 73 ) IRF4 competes with the binding of IRF5 to MyD88, an adaptor critical to TLR signaling, for the induction of these cytokine genes.( 10 ) There seems to be several differences between the properties of human IRF5 and murine IRF5: unlike human IRF5, which is expressed in multiple splice variants,( 70 ) there is only one IRF5 splice variant that is expressed at very low levels in the bone marrow of C57BL/6J mice.( 48 ) In contrast, it was reported that elevated expression of multiple unique isoforms of IRF5 by aberrant splicing and inappropriate polyadenylation, which are driven by single nucleotide polymorphisms (SNP) mutations in the IRF5 gene in humans, is an important genetic risk factor for systemic lupus erythematosus (SLE), suggesting that IRF5 is a susceptibility gene in SLE.( 74 , 75 , 76 ) IRF5 can also be induced by the tumor suppressor p53,( 77 ) and promotes cell cycle arrest and apoptosis in response to DNA damage.( 72 , 78 )
Hematopoietic IRF: IRF4 and IRF8. IRF4 and IRF8 are expressed predominantly in hematopoietic cells, including lymphocytes, macrophages, B cells, and DC.( 28 , 79 ) Both factors, whose DNA‐binding affinity is weak, can potently bind to DNA by dimerization with other transcription factors,( 80 ) including IRF1, IRF2, PU.1, and E47. IRF4 has a critical role in the maturation of B and T cells,( 81 ) and also in the development of CD4+ DC.( 82 ) It has been shown that IRF4 regulates both isotype switching and plasma cell differentiation by controlling the expression of activation‐induced cytidine deaminase and Blimp‐1.( 83 , 84 ) In human multiple myeloma cells, IRF4 was found to be overexpressed by chromosomal translocation.( 85 ) IRF4 acts as an antagonist of the IRF1‐mediated transactivation of the TRAIL promoter, and also negatively regulates IRF5‐mediated induction of proinflammatory cytokines by competitively blocking the binding of IRF5 to MyD88.( 10 ) IRF8 was originally identified as a protein that binds to the ISRE motif in the promoter region of the MHC class I gene H‐2LD.( 22 , 86 ) IRF8 null mice( 87 , 88 ) also show major defects in the differentiation of CD8+ DC and plasmacytoid DC and develop chronic myelogenous leukemia (CML)‐like syndrome. IRF8 was shown to induce IL‐12p40 and is involved in the Th1 response. Thus, this factor is critical for the regulation of both immunity and oncogenesis, as described below.
A novel member of IRF, IRF10, has been identified in chicken.( 17 ) IRF10 is most closely related to IRF4 albeit with much lower similarity (43–44%) than between avian and mammalian orthologs of IRF4 (84%), but differs in both its constitutive and inducible expression: IRF10 is expressed principally in hematopoietic cells, but at very low levels in bursal cells. IRF10 is induced by type I and type II IFN, as well as by concanavalin A, possibly through an indirect pathway. The expression of IRF10 is also upregulated by the oncogene v‐rel, the proto‐oncogene c‐rel, and IRF4 in lymphoid cells. The level of IRF10 induction in lymphoid cell lines by Rel proteins correlates with Rel transformation potential. Although it is interesting to study whether this new member participates in the regulation of human cancer development, it seems to be unlikely that a functional protein is translated as the predicted open reading frame (ORF) is incomplete.( 17 )
Morphogenic IRF: IRF6. IRF6 is structurally related to IRF5 but, in contrast to IRF5, IRF6 seems to have completely different functions, acting as a key regulator of the switch from keratinocyte proliferation to differentiation.( 89 ) Indeed, IRF6‐deficient mice have a hyperproliferative epidermis that fails to undergo terminal differentiation,( 89 ) and show an abnormality in skin, limb, and craniofacial morphogenesis.( 90 ) In humans there is an association of mutations of the IRF6 gene with Van der Woude syndrome and popliteal pterygium syndrome, which are hereditary disorders characterized by cleft lip and palate.( 91 ) In addition, it was reported recently that the IRF6 protein interacts with mammary serine protease inhibitor (maspin).( 92 ) Maspin is characterized as a tumor suppressor owing to its ability to promote apoptosis and inhibit cell invasion, and its expression is attenuated or absent in aggressive breast carcinomas. This interaction occurs via the conserved IAD and is regulated by IRF6 phosphorylation. Similarly to maspin, the IRF6 expression level correlates inversely with breast cancer invasiveness. Further investigation may clarify its role in the metastatic potential of various human cancers.
Role of IRF family members in the regulation of oncogenesis
There are numerous reports about the involvement of IRF family members in the regulation of tumor development. Some IRF members suppress tumor development, whereas some accelerate it. Consistently, there have been many reports regarding genetic abnormalities and aberrant expression of IRF genes in primary tissues derived from human cancer patients (Fig. 2b). In this section, IRF members are classified into three categories: (1) antioncogenic IRF (IRF1, IRF8, and IRF5); (2) oncogenic IRF (IRF2 and IRF4); and (3) viral oncogenic IRF. On the basis of this classification, we describe how these IRF regulate cell proliferation, DNA damage‐induced responses, and oncogenesis.
Antioncogenic IRF
IRF1. Gene‐disruption studies have revealed that IRF1 has similar but not identical functions to the p53 tumor suppressor. Similarly to p53, IRF1 is critical to cell cycle regulation and induction of apoptosis in response to stress signals.( 93 ) IRF1‐deficient MEF cannot undergo cell cycle arrest in response to DNA damage. It was subsequently found that a well‐studied cyclin‐dependent kinase (CDK) inhibitor, p21WAF/cip , is a common target gene that is induced transcriptionally by both p53 and IRF1.( 93 ) Their cooperative action was also found in the induction of apoptosis in oncogene‐expressing MEF. When MEF carrying an activated form of the c‐Ha‐ras oncogene are exposed to DNA damage such as that caused by radiation or chemotherapeutic agents, apoptosis is induced in a p53‐ and IRF1‐dependent manner.( 94 ) In thymocytes, DNA‐induced apoptosis occurs through a p53‐dependent pathway, whereas in mitogen‐activated mature T lymphocytes, this type of apoptosis is dependent on IRF1 but not p53.( 95 ) Thus, like p53, IRF1 is an essential mediator in cellular responses to DNA damage, thereby functioning as a tumor suppressor. However, IRF2 is shown to have an oncogenic feature;( 61 ) overexpression of IRF2 leads to the transformation of NIH3T3 cells. This phenotype can be reverted by the concomitant overexpression of IRF1, suggesting that IRF2 constitutively occupies the IRF‐E of putative tumor‐suppressor genes that would be otherwise activated by IRF1.( 96 ) However, the mutually antagonistic effect of IRF1 and IRF2 might vary depending on the promoter context, that is, IRF2 itself may activate the promoter of an oncogene.( 34 )
The antioncogenic function of IRF1 is not limited to only IRF2‐expressing cells, but other oncogene (e.g. c‐myc or fosB)‐transformed cells can be reverted by introduction of the IRF1 gene, suggesting the broad role of IRF1 as a tumor suppressor.( 97 , 98 ) Although it was shown that at least two oncogenes need to be activated to transform normal fibroblasts,( 99 ) IRF1‐deficient MEF can undergo transformation even when a single oncogene is activated.( 94 ) Further analysis of IRF1‐deficient mice has revealed that the IRF1 gene belongs to a class of tumor‐susceptibility genes that may indirectly suppress tumor development.( 100 , 101 , 102 ) IRF1 single knockout mice did not show any spontaneous tumor development. In contrast, IRF1 null mice crossed with mice carrying the activated c‐Ha‐ras transgene or IRF1 and p53 double‐knockout mice were more susceptible to tumor development.( 103 ) In this context, the loss of IRF1 may also contribute to the development of human cancers.
Supporting this feature of IRF1, genetic alterations in IRF1 expression have so far been reported in human hematological cancers as well as solid cancers. Defects in one or both IRF1 alleles (5q31.1) accompanied by deletion or inactivating rearrangement have been observed in human leukemia and preleukemic myelodysplasia.( 104 ) In this context, the loss of IRF1 expression by deletion or exon skipping was reported (summarized in Fig. 2b). Indeed, approximately 20% of patients with myelodysplastic syndrome or overt leukemia developing from myelodysplastic syndrome carry the exon‐skipped form of IRF1, that is, lacking exons 2 and 3, which shows neither DNA‐binding activity nor tumor‐suppressive activity.( 105 ) In addition, frequent loss of heterozygosity at the IRF1 locus has been found in patients with gastric and esophageal cancers.( 106 , 107 , 108 ) There is also a report about a gastric cancer patient with a missense point mutation in the second exon of the IRF1 gene of the residual allele, which leads to the expression of functionally impaired IRF1.( 109 ) However, protein factors that interfere with the function of IRF1 were also reported. A nuclear factor, nucleophosmin/B23/numatrin, which is overexpressed in human leukemia cells, inhibits the DNA binding and transcriptional activities of IRF1.( 110 )
What is the mechanism underlying IRF1‐mediated tumor‐suppressive activity? Various genes that exert growth‐inhibitory effects are induced by IRF1, for example 2′,5′‐oligo(A) synthetase E,( 111 ) indoleamine 2,3‐dioxigenase,( 112 ) protein kinase, RNA‐dependent (PKR),( 113 ) p21WAF/cip,( 93 ) lysyloxidase (LOX),( 114 ) angiotensin type II receptor,( 115 ) and caspase‐1.( 116 ) Among them, LOX is an extracellular matrix enzyme that catalyzes the crosslinking of collagens or elastin in the extracellular compartment, thereby regulating the integrity of tissue structure. In addition, it also functions in various intracellular processes, including cell motility and transcriptional gene regulation.( 117 ) These vital functions of LOX suggest that aberrant regulation of LOX leads to tumorigenesis and tumor progression. Indeed, there have been many reports about the loss of LOX expression and function in human basal and squamous cells, bronchogenic, colon, esophageal, gastric, head and neck squamous cells, pancreatic and prostatic carcinomas, as well as melanoma.( 117 ) This may also be closely related to identification of the LOX gene as a downregulated gene in ras‐transformed fibroblasts.( 118 ) Interestingly, persistent treatment of ras‐transformed fibroblasts with IFN‐α and IFN‐β yielded a revertant of the ras‐transformed phenotype with the reexpression of this gene.( 118 , 119 ) The promoter of the LOX gene does indeed contain an IRF1‐responsive element, and transformation of the activated c‐Ha‐ras‐expressing IRF1‐deficient MEF can be suppressed by expression of the LOX gene.( 114 ) Thus, LOX is a potential downstream mediator of the tumor‐suppressive activity of IRF1. However, the involvement of LOX in IRF1‐mediated transformation suppression is not observed under different conditions: the ras + myc‐induced transforming phenotype with respect to growth in soft agar is not altered even by the overexpression of LOX, yet it is suppressed by IRF1 expression.( 97 ) Another study also showed that inhibition of LOX transcription after transformation by the ras oncogene is not due to regulation of IRF1 and IRF2.( 120 ) Another potential candidate, p21WAF/cip, does not appear to account for the IRF1‐mediated tumor suppression either.( 97 ) Therefore, more studies will be required for further understanding of this mechanism.
IRF8. It was reported that IRF8 is expressed predominantly in hematopoietic cells, such as cells of myeloid and lymphoid lineages, and its gene expression is upregulated by IFN‐γ. Because IFN‐γ is a pivotal cytokine that is crucial for the clearance of not only virally infected cells but also cancerous cells, it can be presumed that IRF8 regulates tumor development. Of note, IRF8‐deficient mice exhibit marked expansion of granulocytes followed by a fatal blast crisis, which is quite similar to human CML,( 88 ) a disease known to be caused by the constitutive kinase activity of the BCR‐ABL (breakpoint cluster region‐Abelson murine leukemia) oncoprotein. Particularly worth noting is that the IRF8 expression level decreases markedly in CML and acute myelogenous leukemia cells from patients,( 121 ) and that a return to normal levels was observed in patients in remission following treatment with IFN‐α.
What is the underlying mechanism of IRF8 function? IRF8−/– myeloid progenitor cells have defects in both differentiation and growth. IRF8 drives their differentiation toward macrophages whereas it inhibits granulocytic differentiation.( 122 , 123 ) Moreover, IRF8 inhibits myeloid cell growth and promotes apoptosis.( 123 , 124 ) Thus, the loss of IRF8 results in the accumulation of granulocytes, and then presumably an additional genetic hit or hits in the progenitor cells causes clonal expansion of undifferentiated cells (i.e. blast crisis). Concerning the target genes of IRF8, one report shows that some of these IRF8 effects may be explained in part by an IRF8‐mediated repression of bcl‐2, a major antiapoptotic target of BCR/ABL, on a transcriptional and protein level.( 125 ) The results of another group indicate that some of the myeloleukemia suppressor activities of IRF8 are mediated through the regulation of promyelocytic leukemia (PML), which is a tumor suppressor that serves as a scaffold protein for nuclear bodies.( 126 ) In addition, IRF8 has been shown to inhibit the growth of p210 Bcr/Abl‐transformed myeloid progenitor cells. IRF8 suppresses c‐Myc expression at least in part by direct activation of B‐lymphocycte‐induced maturation protein‐1 (Blimp‐1) and mitogenic Ets transcriptional suppresor (METS), which may explain the mechanism of growth arrest induced by IRF8.( 127 ) The antagonistic role of IRF8 against Bcr/Abl is also supported by evidence that IRF8 can ameliorate Bcr/Abl‐mediated murine myeloid leukemia in vivo.( 128 ) These data indicate that the loss of IRF8 expression may be a major event leading to the development of human CML, and that the restoration of IRF8 expression can antagonize the oncogenic activity of Bcr/Abl.
In addition to the effect of IRF8 in hematopoietic tumors described above, this factor has also been shown to manifest antitumor activity even in solid tumors. IRF8 expression was found to be repressed by DNA methylation in human metastatic colon carcinoma cell lines and murine mammary carcinoma with lung metastasis in vivo.( 129 ) It has been further shown that the overexpression of IRF8 enhances apoptosis of cancer cells, whereas the disruption of IRF8 function diminishes primary tumor cell sensitivity to apoptosis and can convert a poorly metastatic tumor to a metastatic phenotype.
Interferon regulatory factor 8 appears to exert its antileukemic activity not only by the direct control of cell growth, differentiation, and apoptosis (see above) but also by modulating antitumor immunity. Indeed, the coexpression of IRF8 in Bcr/Abl‐transformed BaF3 cells causes a CD8+ cytotoxic T‐cell response to prevent the establishment of leukemia in vivo.( 130 ) Furthermore, human CML cells are sensitive to T cell‐mediated immunity.( 131 ) Given the roles of IRF8 in macrophages and DC, IRF8 may also elicit antitumor immunity through its ability to support the differentiation and function of antigen‐presenting cells.
IRF5. Along with accumulating studies of the role of IRF5 in the innate immune response,( 9 , 71 , 72 , 73 , 78 ) this factor was also reported to be a direct target of p53,( 77 ) and to undergo nuclear translocation upon DNA damage, suggesting a possible role of IRF5 in DNA‐damage responses and tumor suppression.( 78 , 132 ) This functional activity of IRF5 seems to be similar to that of IRF1. It has been reported that the effects of IRF5 on cell cycle regulation and apoptosis are independent of p53.( 133 ) In addition, the overexpression of IRF5 in B‐cell lymphomas expressing non‐functional p53 results in G2–M cell cycle arrest and cell death with the upregulation of genes involved in cell cycle regulation and apoptosis (e.g. the p21WAF/cip , caspase‐8, Bak, Bax, and DAP kinase 2 genes).( 133 ) IRF5 was also shown to sensitize tumor cells to DNA damage‐induced cell growth inhibition and apoptosis via a p53‐independent pathway.( 132 ) Intriguingly, the loss of IRF5 mRNA expression was often found in leukemia cells as well as gastrointestinal tumors (Fig. 2b),( 132 , 133 ) suggesting a putative role of IRF5 as a tumor suppressor that mediates cell cycle arrest, apoptosis, and immune activation. Consistent with these previous reports, our recent analysis of IRF5‐deficient mice revealed that IRF5 is a critical mediator of the induction of apoptosis in response to DNA damage.( 78 ) However, unlike previous reports,( 132 , 133 ) a recent study using MEF from IRF5‐deficient mice showed that IRF5 is dispensable for the induction of p21WAF/cip, and cell cycle arrest after X‐ray irradiation or adriamycin treatment normally occurs in these cells. It was shown that IRF5 is involved selectively in DNA damage‐induced apoptosis but not in cell cycle arrest.( 78 ) IRF5‐deficient MEF undergo transformation by the expression of activated c‐Ha‐Ras alone, and these cells show tumorigenic properties in nude mice. There is a similar report by another group showing that overexpression of IRF5, but not IRF7, inhibits growth of the human B‐lymphoma cells BJAB (carrying non‐functional p53) and tumor formation in nude mice.( 133 ) Interestingly, induction of known p53‐dependent proapoptotic genes, such as Puma and Noxa, is normally observed in the absence of IRF5,( 78 ) suggesting that IRF5 functions as a tumor suppressor by acting on a pathway that may be distinct from that of p53.
In light of the findings above, it will be of great interest to examine the status of the IRF5 gene and its expression in human cancers. It is interesting that the constitutive expression of IRF5 occurs primarily in lymphoid tissue, peripheral blood lymphocytes, and DC, but it has not been detected in B‐ and T‐cell leukemia cell lines,( 71 ) nor in most of the clinical samples from patients with hematological malignancies (e.g. chronic lymphocytic leukemia [CLL], acute lymphocytic leukemia [ALL], acute myelogenous leukemia [AML]) as well as gastrointestinal tumors.( 132 , 133 ) Although it remains to be investigated further whether the IRF5 gene is deleted in these tumors or silenced by hypermethylation, these data suggest that the loss of IRF5 expression in hematological malignancies may be associated with leukemogenesis.
What are the mechanisms underlying DNA damage‐induced IRF5 activation? Post‐translational modification, nuclear translocation, and the transactivating function of IRF5 are induced after virus infection and DNA damage, but the signaling pathways leading to its activation appears to be quite different.( 132 , 134 ) It was previously shown that several serine residues (Ser‐477/Ser‐480 or Ser‐427/Ser‐430) at the C terminus of human IRF5 play critical roles in the virus‐induced activation of IRF5.( 132 , 134 ) Newcastle disease virus induced phosphorylation of IRF5 at Ser‐427 and Ser‐430 and phosphorylation was not detected in cells treated with the DNA‐damaging agent irinotecan, indicating that irinotecan‐induced IRF5 phosphorylation occurs at distinct serine residues. It will be interesting to determine which kinases are involved in the phosphorylation of IRF5 for its transcriptional activity. The phosphatidylinositol‐3 kinase‐related kinases ataxia talangiectasia‐mutated (ATM), ATM and Rad3‐related (ATR), and DNA‐PK become activated in response to DNA damage and transduce signals to downstream targets, including p53 and the checkpoint kinases Chk1 and Chk2.( 135 ) Therefore, it can be speculated that these kinases may be candidate kinases involved in IRF5 phosphorylation. Indeed, database analysis shows that Thr‐272 of IRF5 may be a potential target site for phosphorylation by ATM or DNA‐PK.( 132 , 134 ) Thus, several IRF can be activated via a DNA damage‐triggered signaling pathway, and have emerged as crucial regulators of stress‐induced cellular responses. In this regard, IRF can also be considered as key transcription factors that link immunity and oncogenesis.
Regarding IRF9, the role of IRF9 in tumor suppression is implicated in the context of type I IFN‐mediated antitumor activities.( 136 , 137 ) The binding of IFN to the receptor complex leads to the activation of two main signaling pathways mediated by ISGF3 and IFN‐α‐activated factor (AAF)/IFN‐γ‐activated factor (GAF): IRF9 acts as a DNA‐binding component of the former transcriptional complex. Hundreds of cellular genes are activated transcriptionally after IFN stimulation,( 138 ) and most ISG require IRF9. In this context, IRF1 is also a downstream mediator that is induced through the AAF/GAF pathway. Most of the IFN‐inducible IRF, such as IRF5 and IRF7, are dependent on ISGF3, that is, IRF9. In general, some of these ISG were shown to encode proteins that mediate tumor‐suppressor activities directly in tumor cells, or indirectly through the activation of tumor immunity. It has been demonstrated that there is crosstalk between type I IFN‐mediated signaling and a p53‐mediated pathway, which further revealed a new regulatory mechanism for p53‐mediated responses in tumor suppression. The p53 gene was found to be induced by treatment with IFN‐α and IFN‐β, resulting in an increase in the expression level of the p53 protein.( 139 ) This induction of the p53 gene is mediated in an ISGF3‐dependent manner through the activation of two ISRE, which were found to be within the promoter and first‐intron regions of the p53 gene. Therefore, the p53 gene is not induced by IFN‐α or IFN‐β in the absence of IRF9. In this respect, IRF9 acts as a critical component of IFN‐induced p53 upregulation, which contributes to boosting the activation of the p53‐mediated proapoptotic pathway upon stimulation with DNA‐damaging agents such as radiation and chemotherapeutic agents.( 139 ) However, it was reported that the protooncogene myc directly regulates expression of the IRF9 gene.( 140 ) IRF9‐deficient cells are highly susceptible to cytotoxic chemotherapeutic agents,( 140 ) and this result suggests that IRF9 may function in the regulation of DNA damage‐induced responses.
Oncogenic IRF
IRF2. IRF2 was initially found to antagonize IRF1 in terms of transcriptional activity.( 141 ) In contrast to the antioncogenic activity of IRF1, IRF2 shows an oncogenic potential: NIH3T3 cells with overexpressed IRF2 became transformed and were more tumorigenic in nude mice, implicating IRF2 as a potential oncoprotein.( 61 ) One putative mechanism of the oncogenic activity of IRF2 is that IRF2 antagonizes the antiproliferative action of IRF1 by competing for binding sites in the promoters of several growth‐suppressing genes.( 111 , 114 , 142 ) Consistently, several studies showed that the IRF2 expression level increased in clinical samples from esophageal squamous cell cancer( 143 ) or breast cancer,( 144 ) whereas the IRF1 expression level decreased in these cancers (Fig. 2b). The ectopic expression of activated N‐ras in primary hematopoietic cells and myeloid cell lines can lead to proliferation inhibition; overexpression of the IRF2 gene in U937 myeloid leukemic cells abrogates this N‐ras‐induced growth suppression.( 62 ) Although the exact mechanism underlying this cell transformation is still unknown, it is assumed that IRF2 exerts its oncogenic activity by competing with IRF1 or other IRF‐family members for ISRE. This notion is supported by the finding that NIH 3T3 cells expressing only the DNA‐binding domain of IRF2 can undergo transformation.( 96 ) An alternative possibility for the oncogenic activity of IRF2 is that IRF2, also known as histone nuclear factor M (HiNF‐M), plays a positive role in the cell cycle regulation of the human histone H4 gene FO108.( 34 , 145 )
IRF4. There are several pieces of evidence suggesting a relationship between IRF4 and oncogenesis. The expression of IRF4 has been shown to be upregulated in v‐Rel‐expressing cells, and an increased expression level of IRF4 plays a role in the v‐Rel‐mediated transformation process.( 146 ) Similarly to IRF8 described above, there are also several reports suggesting a possible role of IRF4 in the pathogenesis of CML. The expression level of IRF4 is significantly low in CML patients.( 147 , 148 ) IRF8 and IRF4 play a cooperative role in the differentiation of B cells and DC.( 84 ) The downregulation of IRF4 may promote leukemogenesis in the myeloid cell context,( 148 ) suggesting the possibility that IRF4 may play a cooperative role with IRF8 in myeloid cell growth and differentiation as well. In this regard, one putative mechanism underlying downregulated IRF4 expression in leukemia is possibly associated with CpG‐site‐specific IRF4 promoter methylation.( 147 ) The expression of another IRF, IRF8, is also impaired in myeloid leukemias, particularly CML as mentioned above. However, in contrast to IRF4, the defective expression of IRF8 does not seem to be explained by promoter methylation.( 147 )
The IRF4 gene is a critical factor for the regulation of B‐cell proliferation and differentiation, which suggests that the deregulated expression of this gene may contribute to B‐cell malignancies. IRF4 upregulation seems to induce the growth of lymphomas or multiple myeloma.( 85 , 149 ) In human multiple myeloma cells, a chromosomal translocation t(6;14)(p25;q32) juxtaposes the IgH locus to the multiple myeloma oncogene 1/IRF4 gene, resulting in the overexpression of IRF4.( 85 ) Indeed, exogenous expression of IRF4 in Rat‐1 cells causes anchorage‐independent growth.( 85 ) However, the overexpression of IRF4 alone is not sufficient for leukemogenesis in transgenic mice overexpressing IRF4 in lymphocytes,( 150 ) suggesting that additional factors are required for the oncogenic activity of IRF4 in vivo.
The deregulation of IRF4 expression is also involved in human T cell leukemia virus (HTLV)‐1‐induced oncogenesis. IRF4 is upregulated constitutively in HTLV‐1‐infected cell lines and by overexpression of the HTLV‐1‐derived oncoprotein Tax. This Tax‐driven IRF4 expression in HTLV‐I‐infected cells suggests a role of IRF4 in reprogramming T cell gene expression.( 151 , 152 ) The constitutive expression of IRF4 in T cells results in decreased expression of the G2–M checkpoint gene encoding cyclin B1, and several DNA‐repair genes encoding Rad51, XRCC1, Yng1, RPA, and PCNA. Such a transcriptional phenotype is strikingly similar to that in HTLV‐infected T cells.( 151 , 152 , 153 ) Further studies will identify and characterize IRF4 target genes in an effort to further characterize the role of the IRF4 transcription factor in HTLV‐1‐induced leukemogenesis.
Viral IRF. Kaposi's sarcoma‐associated herpesvirus (KSHV/HHV8) has been associated etiologically with several malignancies, including Kaposi's sarcoma and primary effusion lymphoma (PEL). HHV‐8 encodes several viral IRF homologs (vIRF). One of the KSHV non‐structural regulatory lytic genes is ORF‐K9, encoding the viral interferon regulatory factor (vIRF1). vIRF1 (Fig. 1) is a 449‐amino acid protein that shares sequence homology with cellular IRF( 154 , 155 ) but does not compete with IRF1 for DNA binding, nor does it bind to or sequester IRF1.( 156 ) In contrast, several studies have shown that vIRF1 can associate with cellular proteins such as IRF1, IRF8, IRF9, CBP/p300, and p53 tumor suppressor. 157 , 158 Despite an as yet undefined mechanism, these characteristics of vIRF1 may explain the observation that vIRF1 inhibits IFN‐ and IRF‐mediated signaling (Table 2) through an as yet undefined mechanism and transforms NIH3T3. (159) Another HHV‐8‐derived protein, the replication and transcription activator (RTA), which is necessary and sufficient for the switch from viral latency to lytic replication, carries the DNA‐binding domain that is similar to those of IRF‐family members. It is speculated that HHV‐8 RTA may usurp the cellular IRF pathway, or vice versa. Indeed, it was shown that IRF7 can compete with the RTA protein for binding to the RTA response element in the ORF57 promoter to downregulate RTA‐induced gene expression.( 160 )
Table 2.
Effects of viral proteins on interferon regulatory factors (IRF) or IRF‐mediated pathways
| Virus | Viral protein | Targeted IRF | Possible mechanism |
|---|---|---|---|
| HCV | NS3/4A | (IRF3, IRF7)× † | Blockade of type I IFN production through cleavage of IPS‐1/VISA/MAVS/Cardif or TRIF/TICAM‐1 |
| EBV | LMP‐1 | IRF7↑ | Induction by LMP‐1 |
| HHV‐8 | vIRF | (IRF)× | Unknown mechanism(s) |
| RTA | IRF× | Blockade of IRF pathway via its homologous DNA‐binding domain | |
| HPV | E7 | IRF1× | Inhibition of its transcriptional activity through the interaction |
| E6 | IRF3× | Inhibition of its transcriptional activity through the interaction |
These IRF‐mediatad pathways are inactivated indirectly. Cardif, CARD adaptor inducing interferon‐β; EBV, Epstein–Barr virus; HCV, hepatitis C virus; HHV, human herpes virus; HPV, human papilloma virus; IFN, interferon; IPS, interferon‐β promoter stimulator; LMP, latent membrane protein; MAVS, mitochondrial antiviral signaling; PEL, primary effusion lymphoma; RTA, replication and transcription activator; TRIF(TICAM1), TIR domain‐containing adaptor inducing interferon‐β (TIR‐containing adaptor molecule‐1); VISA, virus‐induced signaling adaptor.
Human herpesvirus‐8‐associated PEL is a lymphoproliferative disease of B‐cell origin. PEL cells harbor a non‐B, non‐T phenotype and lack significant surface Ig expression. In PEL cells, IRF4 is reported to be expressed constitutively.( 161 ) As described above, this transcriptional factor, whose expression is restricted specifically to the lymphoid and myeloid compartments,( 28 ) interacts with PU.1 to activate genes essential for B‐cell development. However, in PEL‐derived B‐cell lines, PU.1 expression is completely abrogated. In addition, defective or markedly decreased expression levels of Oct‐2, IRF8, and BSAP/Pax5 are observed in these transformed cells. It is considered that such a disruption of the B‐cell‐specific transcriptional program may thus contribute to lymphomagenesis and to the development of the non‐B, non‐T phenotype in PEL cells.( 161 )
Viral oncoproteins inhibiting IRF function
Viruses have evolved various strategies to counteract the activity of IRF members as well as the IFN system, including type I IFN production and receptor‐mediated IFN signaling,( 162 , 163 , 164 , 165 ) so as to evade the host immune system. In particular, IRF3 and IRF7 are targeted frequently by viral factors and inactivated directly or indirectly. A complex combination of these viral strategies is considered to contribute to the persistence of viral infection, which is also considered to be one of the major risk factors for virus‐induced carcinogenesis (Table 2). A good example is the association between hepatitis C virus and hepatocellular carcinoma, although it remains controversial whether the virus plays a direct or an indirect role in the pathogenesis of hepatocellular carcinoma. To block the IFN system, hepatitis C virus encodes a serine protease, NS3/4A, which cleaves IPS‐1/VISA/MAVS/Cardif, an adaptor protein, leading to inactivation of the RIG‐I‐ or MDA5‐mediated IRF3/IRF7 pathway.( 166 ) In addition, it was reported that this protease also causes proteolysis of Toll/IL‐1 receptor (TIR) domain‐containing adaptor inducing IFN‐β (TRIF)/TIR‐containing adaptor molecule‐1 (TICAM‐1), which is the critical adaptor protein linking TLR3 to its downstream IRF3 activation pathway for dsRNA responses.( 167 )
Epstein–Barr virus (EBV) latency has also been associated with various human cancers.( 168 ) In particular, the major EBV oncoprotein latent membrane protein (LMP)‐1 is one of the key viral proteins required for the transformation of primary B cells.( 169 ) However, IRF7 was initially identified as a negative regulator of the BamHI Q promoter (Qp),( 32 ) which is used for the transcription of EBV nuclear antigen 1 (EBNA‐1) mRNA in type I latency. LMP‐1 acts as a constitutively active receptor‐like molecule, and LMP‐1‐triggered signaling induces the expression of IRF7 (Table 2).( 170 ) Thus, the induction of IRF7 by LMP‐1 may cause the silencing of Qp in EBV type III latency. It was also shown that IRF7‐expressing NIH 3T3 cells show both anchorage‐independent growth and tumorigenicity in athymic mice, and speculated that IRF7 has oncogenic properties and, along with LMP‐1, may mediate EBV transformation in the pathogenesis of EBV‐associated lymphomas.( 169 ) However, there are also several inconsistent observations that suggest that IRF7 is a tumor‐suppressor gene or that IRF7 does not have a major role in tumor suppression,( 133 ) as described above. Further studies will therefore be required to clarify this controversial issue.
Human papilloma virus (HPV) is a causative agent in the etiology of cervical dysplasia and cervical cancer.( 171 ) In contrast, low‐risk HPV‐6 and HPV‐11 are associated with benign condyloma formation, whereas high‐risk HPV‐16 and HPV‐18 are detected frequently in cervical cancer. The malignant phenotype of high‐risk types (HPV‐16 and HPV‐18) depends on the expression of two viral oncogenes, E6 and E7, both of which have been shown previously to inactivate two cellular tumor suppressor proteins: E6 binds to p53 and promotes its proteolysis, whereas E7 binds to the hypophosphorylated form of pRb and interferes with its binding to E2F. These HPV oncoproteins also target IRF‐family members, and inhibit their activities (Table 2). The E7 oncoprotein was shown to interact with IRF1 and interfere with the transactivation function of IRF1 by recruiting histone deacetylases (HDAC) to the promoter.( 172 ) Unlike pRb, IRF1 can also be inactivated by the low‐risk HPV 11 E7 oncoprotein. The functional inactivation of IRF1 by both high‐risk and low‐risk HPV E7 oncoproteins may provide another advantage to malignant and benign tumor formation in the cervix, respectively. In addition, it has also been reported that HPV‐16 E6 binds to the C‐terminal transactivation domain of IRF3, and the transcriptional activity of IRF3 toward the IFN‐β promoter decreases in the presence of E6.( 173 ) Therefore, it can be postulated that the HPV E7 and E6 oncoproteins might interfere with IRF1‐mediated antioncogenic activity and IRF3‐mediated type I IFN production, respectively, thereby overcoming the host immunity in cervical tumor development. In this respect, it can be presumed that the functional role of IRF3 in virus‐induced apoptosis( 174 ) may be another reason why IRF3 is targeted by the HPV16 E6.
Conclusion and future prospects
Only two decades has passed since the initial discovery of IRF, namely IRF1 and IRF2.( 1 ) The subsequent discovery of other IRF members and functional studies of IRF, particularly the generation and analyses of mutant mice lacking one or more of the IRF members, have provided new insights into the intricate gene‐regulation networks underlying many aspects of host defense. Recent progress in studies of innate immunity has also further accelerated advances in IRF studies. It has been revealed that IRF family members are involved crucially in many facets of cellular activity by regulating the gene transcription of type I IFN and other cytokines and chemokines, IFN‐inducible proteins, cell cycle and apoptosis regulators, and cell differentiation factors. Although each IRF member contains the conserved DNA‐binding domain, all members show various distinct regulatory effects on gene expression. Thus, a given IRF member may positively or negatively regulate its target genes depending on the promoter context or its association with other transcription factors. In order to determine the mechanism underlying these differential activities of IRF, it will be interesting to search for new IRF‐interacting molecules. In relation to this issue, there are several interesting studies demonstrating that that IRF3 associates with a subunit of NF‐κB and functions as a coactivator of gene transcription.( 38 , 39 ) These studies would provide further insights into the complex network of gene‐induction pathways, which underlie the diversified activities of IRF.
In the present review, we described the ‘IRF world’ by focusing mainly on the intrinsic aspects of IRF activity in the regulation of oncogenesis. Although not touched upon in this review, there are numerous studies demonstrating the role of IRF in antitumor immunity. IRF1 is an essential regulator of NK cell differentiation,( 5 ) and it is also critical to Th1 differentiation of CD4+ T cells through induction of IL‐12 and IL‐12 receptor subunit β1, the critical signaling components for Th1 differentiation.( 175 ) In addition, recently accumulated evidence has revealed that almost all members of the IRF family play crucial roles in the differentiation, maturation, and activation of DC, which are powerful sensors of cancer cells as well as pathogens.( 87 ) Thus, we envisage that our knowledge about the role of IRF in the regulation of oncogenesis will expand further, particularly in the context of human cancers. Furthermore, understanding IRF biology will have an important impact on the clinical field because various aspects of host defense are regulated by the IRF and IFN systems. It will also provide a molecular basis for cancer therapy.
Acknowledgments
The authors would like to thank Drs Honda, Ohba, Yanai, Negishi, and Hayakawa, Miss Chen, and other colleagues for their continuous support of our work described in this review, and also Miss Satoh and Mrs Yoshida for their assistance with this manuscript. This research program is supported in part by a grant (KAKENHI) for Advanced Research on Cancer and a Grant‐In‐Aid for Scientific Research on Priority Areas, and for Scientific Research, from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Miyamoto M, Fujita T, Kimura Y et al . Regulated expression of a gene encoding a nuclear factor, IRF‐1, that specifically binds to IFN‐β gene regulatory elements. Cell 1988; 54: 903–13. [DOI] [PubMed] [Google Scholar]
- 2. Kamijo R, Harada H, Matsuyama T et al . Requirement for transcription factor IRF‐1 in NO synthase induction in macrophages. Science 1994; 263: 1612–15. [DOI] [PubMed] [Google Scholar]
- 3. Kimura T, Nakayama K, Penninger J et al . Involvement of the IRF‐1 transcription factor in antiviral responses to interferons. Science 1994; 264: 1921–4. [DOI] [PubMed] [Google Scholar]
- 4. Lohoff M, Ferrick D, Mittrucker HW et al . Interferon regulatory factor‐1 is required for a T helper 1 immune response in vivo . Immunity 1997; 6: 681–9. [DOI] [PubMed] [Google Scholar]
- 5. Ogasawara K, Hida S, Azimi N et al . Requirement for IRF‐1 in the microenvironment supporting development of natural killer cells. Nature 1998; 391: 700–3. [DOI] [PubMed] [Google Scholar]
- 6. Taki S, Sato T, Ogasawara K et al . Multistage regulation of Th1‐type immune responses by the transcription factor IRF‐1. Immunity 1997; 6: 673–9. [DOI] [PubMed] [Google Scholar]
- 7. Takaoka A, Wang Z, Choi MK et al . DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007; 448: 501–5. [DOI] [PubMed] [Google Scholar]
- 8. Yoneyama M, Kikuchi M, Natsukawa T et al . The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 2004; 5: 730–7. [DOI] [PubMed] [Google Scholar]
- 9. Takaoka A, Yanai H, Kondo S et al . Integral role of IRF‐5 in the gene induction programme activated by Toll‐like receptors. Nature 2005; 434: 243–9. [DOI] [PubMed] [Google Scholar]
- 10. Negishi H, Ohba Y, Yanai H et al . Negative regulation of Toll‐like‐receptor signaling by IRF‐4. Proc Natl Acad Sci USA 2005; 102: 15 989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Negishi H, Fujita Y, Yanai H et al . Evidence for licensing of IFN‐γ‐induced IFN regulatory factor 1 transcription factor by MyD88 in Toll‐like receptor‐dependent gene induction program. Proc Natl Acad Sci USA 2006; 103: 15 136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moore PS, Chang Y. Kaposi's sarcoma‐associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu Rev Microbiol 2003; 57: 609–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Offermann MK. Kaposi sarcoma herpesvirus‐encoded interferon regulator factors. Curr Top Microbiol Immunol 2007; 312: 185–209. [DOI] [PubMed] [Google Scholar]
- 14. Barnes B, Lubyova B, Pitha PM. On the role of IRF in host defense. J Interferon Cytokine Res 2002; 22: 59–71. [DOI] [PubMed] [Google Scholar]
- 15. Mamane Y, Heylbroeck C, Genin P et al . Interferon regulatory factors: the next generation. Gene 1999; 237: 1–14. [DOI] [PubMed] [Google Scholar]
- 16. Taniguchi T, Ogasawara K, Takaoka A et al . IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 2001; 19: 623–55. [DOI] [PubMed] [Google Scholar]
- 17. Nehyba J, Hrdlickova R, Burnside J et al . A novel interferon regulatory factor (IRF), IRF‐10, has a unique role in immune defense and is induced by the v‐Rel oncoprotein. Mol Cell Biol 2002; 22: 3942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Escalante CR, Yie J, Thanos D et al . Structure of IRF‐1 with bound DNA reveals determinants of interferon regulation. Nature 1998; 391: 103–6. [DOI] [PubMed] [Google Scholar]
- 19. Fujii Y, Shimizu T, Kusumoto M et al . Crystal structure of an IRF–DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J 1999; 18: 5028–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka N, Kawakami T, Taniguchi T. Recognition DNA sequences of interferon regulatory factor 1 (IRF‐1) and IRF‐2, regulators of cell growth and the interferon system. Mol Cell Biol 1993; 13: 4531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Darnell JJ, Kerr IM, Stark GR. Jak–STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264: 1415–21. [DOI] [PubMed] [Google Scholar]
- 22. Driggers PH, Ennist DL, Gleason SL et al . An interferon γ‐regulated protein that binds the interferon‐inducible enhancer element of major histocompatibility complex class I genes. Proc Natl Acad Sci USA 1990; 87: 3743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugita K, Miyazaki J, Appella E et al . Interferons increase transcription of a major histocompatibility class I gene via a 5′ interferon consensus sequence. Mol Cell Biol 1987; 7: 2625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharf R, Meraro D, Azriel A et al . Phosphorylation events modulate the ability of interferon consensus sequence binding protein to interact with interferon regulatory factors and to bind DNA. J Biol Chem 1997; 272: 9785–92. [DOI] [PubMed] [Google Scholar]
- 25. Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol 2006; 8: 907–22. [DOI] [PubMed] [Google Scholar]
- 26. Bovolenta C, Driggers PH, Marks MS et al . Molecular interactions between interferon consensus sequence binding protein and members of the interferon regulatory factor family. Proc Natl Acad Sci USA 1994; 91: 5046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brass AL, Kehrli E, Eisenbeis CF et al . Pip, a lymphoid‐restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev 1996; 10: 2335–47. [DOI] [PubMed] [Google Scholar]
- 28. Eisenbeis CF, Singh H, Storb U. Pip, a novel IRF family member, is a lymphoid‐specific, PU.1‐dependent transcriptional activator. Genes Dev 1995; 9: 1377–87. [DOI] [PubMed] [Google Scholar]
- 29. Harada H, Willison K, Sakakibara J et al . Absence of the type I IFN system in EC cells: transcriptional activator (IRF‐1) and repressor (IRF‐2) genes are developmentally regulated. Cell 1990; 63: 303–12. [DOI] [PubMed] [Google Scholar]
- 30. Nelson N, Marks MS, Driggers PH et al . Interferon consensus sequence‐binding protein, a member of the interferon regulatory factor family, suppresses interferon‐induced gene transcription. Mol Cell Biol 1993; 13: 588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamagata T, Nishida J, Tanaka S et al . A novel interferon regulatory factor family transcription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of interferon‐regulated genes. Mol Cell Biol 1996; 16: 1283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang L, Pagano JS. IRF‐7, a new interferon regulatory factor associated with Epstein–Barr virus latency. Mol Cell Biol 1997; 17: 5748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masumi A, Wang IM, Lefebvre B et al . The histone acetylase PCAF is a phorbol‐ester‐inducible coactivator of the IRF family that confers enhanced interferon responsiveness. Mol Cell Biol 1999; 19: 1810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaughan PS, Aziz F, Van Wijnen AJ et al . Activation of a cell‐cycle‐regulated histone gene by the oncogenic transcription factor IRF‐2. Nature 1995; 377: 362–5. [DOI] [PubMed] [Google Scholar]
- 35. Imai KS, Hino K, Yagi K et al . Gene expression profiles of transcription factors and signaling molecules in the ascidian embryo: towards a comprehensive understanding of gene networks. Development 2004; 131: 4047–58. [DOI] [PubMed] [Google Scholar]
- 36. Krause CD, Pestka S. Evolution of the class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol Ther 2005; 106: 299–346. [DOI] [PubMed] [Google Scholar]
- 37. Leung TH, Hoffmann A, Baltimore D. One nucleotide in a κB site can determine cofactor specificity for NF‐κB dimers. Cell 2004; 118: 453–64. [DOI] [PubMed] [Google Scholar]
- 38. Ogawa S, Lozach J, Benner C et al . Molecular determinants of crosstalk between nuclear receptors and toll‐like receptors. Cell 2005; 122: 707–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wietek C, Miggin SM, Jefferies CA et al . Interferon regulatory factor‐3‐mediated activation of the interferon‐sensitive response element by Toll‐like receptor (TLR) 4 but not TLR3 requires the p65 subunit of NF‐κB. J Biol Chem 2003; 278: 50 923–31. [DOI] [PubMed] [Google Scholar]
- 40. Taniguchi T, Takaoka A. A weak signal for strong responses: interferon‐α/β revisited. Nat Rev Mol Cell Biol 2001; 2: 378–86. [DOI] [PubMed] [Google Scholar]
- 41. Hida S, Ogasawara K, Sato K et al . CD8+ T cell‐mediated skin disease in mice lacking IRF‐2, the transcriptional attenuator of interferon‐α/β signaling. Immunity 2000; 13: 643–55. [DOI] [PubMed] [Google Scholar]
- 42. Honda K, Yanai H, Negishi H et al . IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 2005; 434: 772–7. [DOI] [PubMed] [Google Scholar]
- 43. Takayanagi H, Kim S, Matsuo K et al . RANKL maintains bone homeostasis through c‐Fos‐dependent induction of interferon‐β. Nature 2002; 416: 744–9. [DOI] [PubMed] [Google Scholar]
- 44. Hiscott J. Convergence of the NF‐κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev 2007; 18: 483–90. [DOI] [PubMed] [Google Scholar]
- 45. Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006; 25: 349–60. [DOI] [PubMed] [Google Scholar]
- 46. Honda K, Taniguchi T. IRF: master regulators of signalling by Toll‐like receptors and cytosolic pattern‐recognition receptors. Nat Rev Immunol 2006; 6: 644–58. [DOI] [PubMed] [Google Scholar]
- 47. Ozato K, Tailor P, Kubota T. The interferon regulatory factor family in host defense: mechanism of action. J Biol Chem 2007; 282: 20065–9. [DOI] [PubMed] [Google Scholar]
- 48. Paun A, Pitha PM. The IRF family, revisited. Biochimie 2007; 89: 744–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sato M, Suemori H, Hata N et al . Distinct and essential roles of transcription factors IRF‐3 and IRF‐7 in response to viruses for IFN‐α/β gene induction. Immunity 2000; 13: 539–48. [DOI] [PubMed] [Google Scholar]
- 50. Karpova AY, Trost M, Murray JM et al . Interferon regulatory factor‐3 is an in vivo target of DNA‐PK. Proc Natl Acad Sci USA 2002; 99: 2818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim TK, Kim T, Kim TY et al . Chemotherapeutic DNA‐damaging drugs activate interferon regulatory factor‐7 by the mitogen‐activated protein kinase kinase‐4‐cJun NH2‐terminal kinase pathway. Cancer Res 2000; 60: 1153–6. [PubMed] [Google Scholar]
- 52. Lu R, Au WC, Yeow WS et al . Regulation of the promoter activity of interferon regulatory factor‐7 gene. Activation by interferon snd silencing by hypermethylation. J Biol Chem 2000; 275: 31805–12. [DOI] [PubMed] [Google Scholar]
- 53. Yu J, Zhang H, Gu J et al . Methylation profiles of thirty four promoter‐CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer 2004; 4: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Andrews HN, Mullan PB, McWilliams S et al . BRCA1 regulates the interferon γ‐mediated apoptotic response. J Biol Chem 2002; 277: 26225–32. [DOI] [PubMed] [Google Scholar]
- 55. Buckley NE, Hosey AM, Gorski JJ et al . BRCA1 regulates IFN‐γ signaling through a mechanism involving the type I IFNs. Mol Cancer Res 2007; 5: 261–70. [DOI] [PubMed] [Google Scholar]
- 56. Scully R, Livingston DM. In search of the tumour‐suppressor functions of BRCA1 and BRCA2. Nature 2000; 408: 429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108: 171–82. [DOI] [PubMed] [Google Scholar]
- 58. Osterlund PI, Pietila TE, Veckman V et al . IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN‐λ) genes. J Immunol 2007; 179: 3434–42. [DOI] [PubMed] [Google Scholar]
- 59. Jesse TL, LaChance R, Iademarco MF et al . Interferon regulatory factor‐2 is a transcriptional activator in muscle where it regulates expression of vascular cell adhesion molecule‐1. J Cell Biol 1998; 140: 1265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rouyez MC, Lestingi M, Charon M et al . IFN regulatory factor‐2 cooperates with STAT1 to regulate transporter associated with antigen processing‐1 promoter activity. J Immunol 2005; 174: 3948–58. [DOI] [PubMed] [Google Scholar]
- 61. Harada H, Kitagawa M, Tanaka N et al . Anti‐oncogenic and oncogenic potentials of interferon regulatory factors‐1 and ‐2. Science 1993; 259: 971–4. [DOI] [PubMed] [Google Scholar]
- 62. Passioura T, Shen S, Symonds G et al . A retroviral library genetic screen identifies IRF‐2 as an inhibitor of N‐ras‐induced growth suppression in leukemic cells. Oncogene 2005; 24: 7327–36. [DOI] [PubMed] [Google Scholar]
- 63. Taki S, Nakajima S, Ichikawa E et al . IFN regulatory factor‐2 deficiency revealed a novel checkpoint critical for the generation of peripheral NK cells. J Immunol 2005; 174: 6005–12. [DOI] [PubMed] [Google Scholar]
- 64. Honda K, Mizutani T, Taniguchi T. Negative regulation of IFN‐α/β signaling by IFN regulatory factor 2 for homeostatic development of dendritic cells. Proc Natl Acad Sci USA 2004; 101: 2416–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ichikawa E, Hida S, Omatsu Y et al . Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor‐2. Proc Natl Acad Sci USA 2004; 101: 3909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Darnell JE Jr, Kerr IM, Stark GR. Jak‐STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264: 1415–21. [DOI] [PubMed] [Google Scholar]
- 67. Majumder S, Zhou LZ, Chaturvedi P et al . p48/STAT‐1α‐containing complexes play a predominant role in induction of IFN‐γ‐inducible protein, 10 kDa (IP‐10) by IFN‐γ alone or in synergy with TNF‐α. J Immunol 1998; 161: 4736–44. [PubMed] [Google Scholar]
- 68. Lau JF, Parisien JP, Horvath CM. Interferon regulatory factor subcellular localization is determined by a bipartite nuclear localization signal in the DNA‐binding domain and interaction with cytoplasmic retention factors. Proc Natl Acad Sci USA 2000; 97: 7278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luker KE, Pica CM, Schreiber RD et al . Overexpression of IRF9 confers resistance to antimicrotubule agents in breast cancer cells. Cancer Res 2001; 61: 6540–7. [PubMed] [Google Scholar]
- 70. Mancl ME, Hu G, Sangster‐Guity N et al . Two discrete promoters regulate the alternatively spliced human interferon regulatory factor‐5 isoforms. Multiple isoforms with distinct cell type‐specific expression, localization, regulation, and function. J Biol Chem 2005; 280: 21078–90. [DOI] [PubMed] [Google Scholar]
- 71. Barnes BJ, Moore PA, Pitha PM. Virus‐specific activation of a novel interferon regulatory factor, IRF‐5, results in the induction of distinct interferon α genes. J Biol Chem 2001; 276: 23382–90. [DOI] [PubMed] [Google Scholar]
- 72. Barnes BJ, Field AE, Pitha‐Rowe PM. Virus‐induced heterodimer formation between IRF‐5 and IRF‐7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem 2003; 278: 16630–41. [DOI] [PubMed] [Google Scholar]
- 73. Barnes BJ, Richards J, Mancl M et al . Global and distinct targets of IRF‐5 and IRF‐7 during innate response to viral infection. J Biol Chem 2004; 279: 45 194–207. [DOI] [PubMed] [Google Scholar]
- 74. Demirci FY, Manzi S, Ramsey‐Goldman R et al . Association of a common interferon regulatory factor 5 (IRF5) variant with increased risk of systemic lupus erythematosus (SLE). Ann Hum Genet 2007; 71: 308–11. [DOI] [PubMed] [Google Scholar]
- 75. Graham RR, Kozyrev SV, Baechler EC et al . A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 2006; 38: 550–5. [DOI] [PubMed] [Google Scholar]
- 76. Sigurdsson S, Nordmark G, Goring HH et al . Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet 2005; 76: 528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mori T, Anazawa Y, Iiizumi M et al . Identification of the interferon regulatory factor 5 gene (IRF‐5) as a direct target for p53. Oncogene 2002; 21: 2914–18. [DOI] [PubMed] [Google Scholar]
- 78. Yanai H, Chen HM, Inuzuka T et al . Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci USA 2007; 104: 3402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Politis AD, Sivo J, Driggers PH et al . Modulation of interferon consensus sequence binding protein mRNA in murine peritoneal macrophages. Induction by IFN‐γ and down‐regulation by IFN‐α, dexamethasone, and protein kinase inhibitors. J Immunol 1992; 148: 801–7. [PubMed] [Google Scholar]
- 80. Tailor P, Tamura T, Ozato K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res 2006; 16: 134–40. [DOI] [PubMed] [Google Scholar]
- 81. Lu R, Medina KL, Lancki DW et al . IRF‐4,8 orchestrate the pre‐B‐to‐B transition in lymphocyte development. Genes Dev 2003; 17: 1703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tamura T, Tailor P, Yamaoka K et al . IFN regulatory factor‐4 and ‐8 govern dendritic cell subset development and their functional diversity. J Immunol 2005; 174: 2573–81. [DOI] [PubMed] [Google Scholar]
- 83. Klein U, Casola S, Cattoretti G et al . Transcription factor IRF4 controls plasma cell differentiation and class‐switch recombination. Nat Immunol 2006; 7: 773–82. [DOI] [PubMed] [Google Scholar]
- 84. Sciammas R, Shaffer AL, Schatz JH et al . Graded expression of interferon regulatory factor‐4 coordinates isotype switching with plasma cell differentiation. Immunity 2006; 25: 225–36. [DOI] [PubMed] [Google Scholar]
- 85. Iida S, Rao PH, Butler M et al . Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet 1997; 17: 226–30. [DOI] [PubMed] [Google Scholar]
- 86. Weisz A, Marx P, Sharf R et al . Human interferon consensus sequence binding protein is a negative regulator of enhancer elements common to interferon‐inducible genes. J Biol Chem 1992; 267: 25589–96. [PubMed] [Google Scholar]
- 87. Gabriele L, Ozato K. The role of the interferon regulatory factor (IRF) family in dendritic cell development and function. Cytokine Growth Factor Rev 2007; 18: 503–10. [DOI] [PubMed] [Google Scholar]
- 88. Holtschke T, Lohler J, Kanno Y et al . Immunodeficiency and chronic myelogenous leukemia‐like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 1996; 87: 307–17. [DOI] [PubMed] [Google Scholar]
- 89. Richardson RJ, Dixon J, Malhotra S et al . Irf6 is a key determinant of the keratinocyte proliferation–differentiation switch. Nat Genet 2006; 38: 1329–34. [DOI] [PubMed] [Google Scholar]
- 90. Ingraham CR, Kinoshita A, Kondo S et al . Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet 2006; 38: 1335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kondo S, Schutte BC, Richardson RJ et al . Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet 2002; 32: 285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bailey CM, Khalkhali‐Ellis Z, Kondo S et al . Mammary serine protease inhibitor (Maspin) binds directly to interferon regulatory factor 6: identification of a novel serpin partnership. J Biol Chem 2005; 280: 34210–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tanaka N, Ishihara M, Lamphier MS et al . Cooperation of the tumour suppressors IRF‐1 and p53 in response to DNA damage. Nature 1996; 382: 816–18. [DOI] [PubMed] [Google Scholar]
- 94. Tanaka N, Ishihara M, Kitagawa M et al . Cellular commitment to oncogene‐induced transformation or apoptosis is dependent on the transcription factor IRF‐1. Cell 1994; 77: 829–39. [DOI] [PubMed] [Google Scholar]
- 95. Tamura T, Ishihara M, Lamphier MS et al . An IRF‐1‐dependent pathway of DNA damage‐induced apoptosis in mitogen‐activated T lymphocytes. Nature 1995; 376: 596–9. [DOI] [PubMed] [Google Scholar]
- 96. Nguyen H, Mustafa A, Hiscott J et al . Transcription factor IRF‐2 exerts its oncogenic phenotype through the DNA binding/transcription repression domain. Oncogene 1995; 11: 537–44. [PubMed] [Google Scholar]
- 97. Kroger A, Dallugge A, Kirchhoff S et al . IRF‐1 reverts the transformed phenotype of oncogenically transformed cells in vitro and in vivo . Oncogene 2003; 22: 1045–56. [DOI] [PubMed] [Google Scholar]
- 98. Tanaka N, Ishihara M, Taniguchi T. Suppression of c‐myc or fosB‐induced cell transformation by the transcription factor IRF‐1. Cancer Lett 1994; 83: 191–6. [DOI] [PubMed] [Google Scholar]
- 99. Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res 1989; 49: 3713–21. [PubMed] [Google Scholar]
- 100. Demant P. Genetic resolution of susceptibility to cancer – new perspectives. Semin Cancer Biol 1992; 3: 159–66. [PubMed] [Google Scholar]
- 101. Ghebranious N, Donehower LA. Mouse models in tumor suppression. Oncogene 1998; 17: 3385–400. [DOI] [PubMed] [Google Scholar]
- 102. Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science 1998; 280: 1036–7. [DOI] [PubMed] [Google Scholar]
- 103. Nozawa H, Oda E, Nakao K et al . Loss of transcription factor IRF‐1 affects tumor susceptibility in mice carrying the Ha‐ras transgene or nullizygosity for p53. Genes Dev 1999; 13: 1240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Willman CL, Sever CE, Pallavicini MG et al . Deletion of IRF‐1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science 1993; 259: 968–71. [DOI] [PubMed] [Google Scholar]
- 105. Harada H, Kondo T, Ogawa S et al . Accelerated exon skipping of IRF‐1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene 1994; 9: 3313–20. [PubMed] [Google Scholar]
- 106. Ogasawara S, Tamura G, Maesawa C et al . Common deleted region on the long arm of chromosome 5 in esophageal carcinoma. Gastroenterology 1996; 110: 52–7. [DOI] [PubMed] [Google Scholar]
- 107. Peralta RC, Casson AG, Wang RN et al . Distinct regions of frequent loss of heterozygosity of chromosome 5p and 5q in human esophageal cancer. Int J Cancer 1998; 78: 600–5. [DOI] [PubMed] [Google Scholar]
- 108. Tamura G, Ogasawara S, Nishizuka S et al . Two distinct regions of deletion on the long arm of chromosome 5 in differentiated adenocarcinomas of the stomach. Cancer Res 1996; 56: 612–15. [PubMed] [Google Scholar]
- 109. Nozawa H, Oda E, Ueda S et al . Functionally inactivating point mutation in the tumor‐suppressor IRF‐1 gene identified in human gastric cancer. Int J Cancer 1998; 77: 522–7. [DOI] [PubMed] [Google Scholar]
- 110. Kondo T, Minamino N, Nagamura‐Inoue T et al . Identification and characterization of nucleophosmin/B23/numatrin which binds the anti‐oncogenic transcription factor IRF‐1 and manifests oncogenic activity. Oncogene 1997; 15: 1275–81. [DOI] [PubMed] [Google Scholar]
- 111. Benech P, Vigneron M, Peretz D et al . Interferon‐responsive regulatory elements in the promoter of the human 2′,5′‐oligo(A)synthetase gene. Mol Cell Biol 1987; 7: 4498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Takikawa O, Kuroiwa T, Yamazaki F et al . Mechanism of interferon‐γ action. Characterization of indoleamine 2,3‐dioxygenase in cultured human cells induced by interferon‐γ and evaluation of the enzyme‐mediated tryptophan degradation in its anticellular activity. J Biol Chem 1988; 263: 2041–8. [PubMed] [Google Scholar]
- 113. Kirchhoff S, Koromilas AE, Schaper F et al . IRF‐1 induced cell growth inhibition and interferon induction requires the activity of the protein kinase PKR. Oncogene 1995; 11: 439–45. [PubMed] [Google Scholar]
- 114. Tan RS, Taniguchi T, Harada H. Identification of the lysyl oxidase gene as target of the antioncogenic transcription factor, IRF‐1, and its possible role in tumor suppression. Cancer Res 1996; 56: 2417–21. [PubMed] [Google Scholar]
- 115. Horiuchi M, Yamada T, Hayashida W et al . Interferon regulatory factor‐1 up‐regulates angiotensin II type 2 receptor and induces apoptosis. J Biol Chem 1997; 272: 11952–8. [DOI] [PubMed] [Google Scholar]
- 116. Chin YE, Kitagawa M, Kuida K et al . Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol Cell Biol 1997; 17: 5328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Payne SL, Hendrix MJ, Kirschmann DA. Paradoxical roles for lysyl oxidases in cancer – a prospect. J Cell Biochem 2007; 101: 1338–54. [DOI] [PubMed] [Google Scholar]
- 118. Contente S, Kenyon K, Rimoldi D et al . Expression of gene rrg is associated with reversion of NIH 3T3 transformed by LTR‐c‐H‐ras. Science 1990; 249: 796–8. [DOI] [PubMed] [Google Scholar]
- 119. Kenyon K, Contente S, Trackman PC et al . Lysyl oxidase and rrg messenger RNA. Science 1991; 253: 802. [DOI] [PubMed] [Google Scholar]
- 120. Contente S, Kenyon K, Sriraman P et al . Epigenetic inhibition of lysyl oxidase transcription after transformation by ras oncogene. Mol Cell Biochem 1999; 194: 79–91. [DOI] [PubMed] [Google Scholar]
- 121. Schmidt M, Nagel S, Proba J et al . Lack of interferon consensus sequence binding protein (ICSBP) transcripts in human myeloid leukemias. Blood 1998; 91: 22–9. [PubMed] [Google Scholar]
- 122. Scheller M, Foerster J, Heyworth CM et al . Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood 1999; 94: 3764–71. [PubMed] [Google Scholar]
- 123. Tamura T, Nagamura‐Inoue T, Shmeltzer Z et al . ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 2000; 13: 155–65. [DOI] [PubMed] [Google Scholar]
- 124. Gabriele L, Phung J, Fukumoto J et al . Regulation of apoptosis in myeloid cells by interferon consensus sequence‐binding protein. J Exp Med 1999; 190: 411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Burchert A, Cai D, Hofbauer LC et al . Interferon consensus sequence binding protein (ICSBP; IRF‐8) antagonizes BCR/ABL and down‐regulates bcl‐2. Blood 2004; 103: 3480–9. [DOI] [PubMed] [Google Scholar]
- 126. Dror N, Rave‐Harel N, Burchert A et al . Interferon regulatory factor‐8 is indispensable for the expression of promyelocytic leukemia and the formation of nuclear bodies in myeloid cells. J Biol Chem 2007; 282: 5633–40. [DOI] [PubMed] [Google Scholar]
- 127. Tamura T, Kong HJ, Tunyaplin C et al . ICSBP/IRF‐8 inhibits mitogenic activity of p210 Bcr/Abl in differentiating myeloid progenitor cells. Blood 2003; 102: 4547–54. [DOI] [PubMed] [Google Scholar]
- 128. Hao SX, Ren R. Expression of interferon consensus sequence binding protein (ICSBP) is downregulated in Bcr‐Abl‐induced murine chronic myelogenous leukemia‐like disease, and forced coexpression of ICSBP inhibits Bcr‐Abl‐induced myeloproliferative disorder. Mol Cell Biol 2000; 20: 1149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yang D, Thangaraju M, Greeneltch K et al . Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res 2007; 67: 3301–9. [DOI] [PubMed] [Google Scholar]
- 130. Deng M, Daley GQ. Expression of interferon consensus sequence binding protein induces potent immunity against BCR/ABL‐induced leukemia. Blood 2001; 97: 3491–7. [DOI] [PubMed] [Google Scholar]
- 131. Lim SH, Coleman S. Chronic myeloid leukemia as an immunological target. Am J Hematol 1997; 54: 61–7. [DOI] [PubMed] [Google Scholar]
- 132. Hu G, Mancl ME, Barnes BJ. Signaling through IFN regulatory factor‐5 sensitizes p53‐deficient tumors to DNA damage‐induced apoptosis and cell death. Cancer Res 2005; 65: 7403–12. [DOI] [PubMed] [Google Scholar]
- 133. Barnes BJ, Kellum MJ, Pinder KE et al . Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res 2003; 63: 6424–31. [PubMed] [Google Scholar]
- 134. Barnes BJ, Kellum MJ, Field AE et al . Multiple regulatory domains of IRF‐5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol Cell Biol 2002; 22: 5721–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sancar A, Lindsey‐Boltz LA, Unsal‐Kacmaz K et al . Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85. [DOI] [PubMed] [Google Scholar]
- 136. Belardelli F, Ferrantini M, Proietti E et al . Interferon‐α in tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13: 119–34. [DOI] [PubMed] [Google Scholar]
- 137. Strander H, Einhorn S. Interferons and the tumor cell. Biotherapy 1996; 8: 213–18. [DOI] [PubMed] [Google Scholar]
- 138. Der SD, Zhou A, Williams BR et al . Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci USA 1998; 95: 15 623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Takaoka A, Hayakawa S, Yanai H et al . Integration of interferon‐α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003; 424: 516–23. [DOI] [PubMed] [Google Scholar]
- 140. Weihua X, Lindner DJ, Kalvakolanu DV. The interferon‐inducible murine p48 (ISGF3γ) gene is regulated by protooncogene c‐myc. Proc Natl Acad Sci USA 1997; 94: 7227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Harada H, Fujita T, Miyamoto M et al . Structurally similar but functionally distinct factors, IRF‐1 and IRF‐2, bind to the same regulatory elements of IFN and IFN‐inducible genes. Cell 1989; 58: 729–39. [DOI] [PubMed] [Google Scholar]
- 142. Tanaka H, Samuel CE. Mechanism of interferon action: structure of the mouse PKR gene encoding the interferon‐inducible RNA‐dependent protein kinase. Proc Natl Acad Sci USA 1994; 91: 7995–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wang Y, Liu DP, Chen PP et al . Involvement of IFN regulatory factor (IRF)‐1 and IRF‐2 in the formation and progression of human esophageal cancers. Cancer Res 2007; 67: 2535–43. [DOI] [PubMed] [Google Scholar]
- 144. Doherty GM, Boucher L, Sorenson K et al . Interferon regulatory factor expression in human breast cancer. Ann Surg 2001; 233: 623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Vaughan PS, Van Der Meijden CM, Aziz F et al . Cell cycle regulation of histone H4 gene transcription requires the oncogenic factor IRF‐2. J Biol Chem 1998; 273: 194–9. [DOI] [PubMed] [Google Scholar]
- 146. Hrdlickova R, Nehyba J, Bose HR Jr. Interferon regulatory factor 4 contributes to transformation of v‐Rel‐expressing fibroblasts. Mol Cell Biol 2001; 21: 6369–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ortmann CA, Burchert A, Holzle K et al . Down‐regulation of interferon regulatory factor 4 gene expression in leukemic cells due to hypermethylation of CpG motifs in the promoter region. Nucleic Acids Res 2005; 33: 6895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Schmidt M, Hochhaus A, Konig‐Merediz SA et al . Expression of interferon regulatory factor 4 in chronic myeloid leukemia: correlation with response to interferon α therapy. J Clin Oncol 2000; 18: 3331–8. [DOI] [PubMed] [Google Scholar]
- 149. Tsuboi K, Iida S, Inagaki H et al . MUM1/IRF4 expression as a frequent event in mature lymphoid malignancies. Leukemia 2000; 14: 449–56. [DOI] [PubMed] [Google Scholar]
- 150. Saito T, Yamagata T, Takahashi T et al . ICSAT overexpression is not sufficient to cause adult T‐cell leukemia or multiple myeloma. Biochem Biophys Res Commun 1999; 260: 329–31. [DOI] [PubMed] [Google Scholar]
- 151. Mamane Y, Grandvaux N, Hernandez E et al . Repression of IRF‐4 target genes in human T cell leukemia virus‐1 infection. Oncogene 2002; 21: 6751–65. [DOI] [PubMed] [Google Scholar]
- 152. Mamane Y, Loignon M, Palmer J et al . Repression of DNA repair mechanisms in IRF‐4‐expressing and HTLV‐I‐infected T lymphocytes. J Interferon Cytokine Res 2005; 25: 43–51. [DOI] [PubMed] [Google Scholar]
- 153. Mamane Y, Sharma S, Grandvaux N et al . IRF‐4 activities in HTLV‐I‐induced T cell leukemogenesis. J Interferon Cytokine Res 2002; 22: 135–43. [DOI] [PubMed] [Google Scholar]
- 154. Moore PS, Boshoff C, Weiss RA et al . Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996; 274: 1739–44. [DOI] [PubMed] [Google Scholar]
- 155. Russo JJ, Bohenzky RA, Chien MC et al . Nucleotide sequence of the Kaposi sarcoma‐associated herpesvirus (HHV8). Proc Natl Acad Sci USA 1996; 93: 14 862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zimring JC, Goodbourn S, Offermann MK. Human herpesvirus 8 encodes an interferon regulatory factor (IRF) homolog that represses IRF‐1‐mediated transcription. J Virol 1998; 72: 701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Burysek L, Yeow WS, Lubyová B et al . Functional analysis of human herpesvirus 8‐encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J Virol 1999; 73: 7334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Seo T, Park J, Lee D et al . Viral interferon regulatory factor 1 of Kaposi's sarcoma‐associated herpesvirus binds to p53 and represses p53‐dependent transcription and apoptosis. J Virol 2001; 75: 6193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Gao SJ, Boshoff C, Jayachandra S et al . KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 1997; 15: 1979–85. [DOI] [PubMed] [Google Scholar]
- 160. Wang J, Zhang J, Zhang L et al . Modulation of human herpesvirus 8/Kaposi's sarcoma‐associated herpesvirus replication and transcription activator transactivation by interferon regulatory factor 7. J Virol 2005; 79: 2420–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Arguello M, Sgarbanti M, Hernandez E et al . Disruption of the B‐cell specific transcriptional program in HHV‐8 associated primary effusion lymphoma cell lines. Oncogene 2003; 22: 964–73. [DOI] [PubMed] [Google Scholar]
- 162. Garcia‐Sastre A. Mechanisms of inhibition of the host interferon α/β‐mediated antiviral responses by viruses. Microbes Infect 2002; 4: 647–55. [DOI] [PubMed] [Google Scholar]
- 163. Goodbourn S, Didcock L, Randall RE. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J General Virol 2000; 81: 2341–64. [DOI] [PubMed] [Google Scholar]
- 164. Katze MG, He Y, Gale M Jr. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2002; 2: 675–87. [DOI] [PubMed] [Google Scholar]
- 165. Levy DE, Garcia‐Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev 2001; 12: 143–56. [DOI] [PubMed] [Google Scholar]
- 166. Meylan E, Curran J, Hofmann K et al . Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437: 1167–72. [DOI] [PubMed] [Google Scholar]
- 167. Li K, Foy E, Ferreon JC et al . Immune evasion by hepatitis C virus NS3/4A protease‐mediated cleavage of the Toll‐like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA 2005; 102: 2992–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Pagano JS. Epstein–Barr virus: the first human tumor virus and its role in cancer. Proc Assoc Am Physicians 1999; 111: 573–80. [DOI] [PubMed] [Google Scholar]
- 169. Zhang L, Zhang J, Lambert Q et al . Interferon regulatory factor 7 is associated with Epstein–Barr virus‐transformed central nervous system lymphoma and has oncogenic properties. J Virol 2004; 78: 12 987–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zhang L, Pagano JS. Interferon regulatory factor 7 is induced by Epstein–Barr virus latent membrane protein 1. J Virol 2000; 74: 1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2002; 2: 342–50. [DOI] [PubMed] [Google Scholar]
- 172. Park JS, Kim EJ, Kwon HJ et al . Inactivation of interferon regulatory factor‐1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7‐mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem 2000; 275: 6764–9. [DOI] [PubMed] [Google Scholar]
- 173. Ronco LV, Karpova AY, Vidal M et al . Human papillomavirus 16, E6 oncoprotein binds to interferon regulatory factor‐3 and inhibits its transcriptional activity. Genes Dev 1998; 12: 2061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Heylbroeck C, Balachandran S, Servant MJ et al . The IRF‐3 transcription factor mediates Sendai virus‐induced apoptosis. J Virol 2000; 74: 3781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Langowski JL, Zhang X, Wu L et al . IL‐23 promotes tumour incidence and growth. Nature 2006; 442: 461–5. [DOI] [PubMed] [Google Scholar]