

Abstract
We previously performed gene expression profile analyses of 20 intestinal‐type gastric cancers, and identified a set of genes whose expression levels were elevated in cancer tissues compared to their corresponding non‐cancerous tissues. In the present study we focused on the immunoglobulin superfamily 11 gene (IGSF11). Its expression was also elevated in colorectal cancers and hepatocellular carcinomas as well as intestinal‐type gastric cancers. Northern blot analysis showed that it was expressed abundantly in testis and ovary. These data suggest that IGSF11 is a good candidate of cancer‐testis antigen. Furthermore, suppression of IGSF11 by siRNA retarded the growth of gastric cancer cells. To investigate the possibility of clinical application of peptide vaccine to IGSF11, we synthesized candidate epitope peptides for IGSF11 and tested whether the peptides elicit IGSF11‐specific CTL. As a result, we successfully established oligo‐clonal CTL by stimulation with IGSF11‐9‐207 (ALSSGLYQC). In addition, we also established additional CTL using IGSF11‐9V (ALSSGLYQV), anchor‐modified peptides of IGSF11‐9‐207. These peptides showed IGSF11‐specific cytotoxic activity in an HLA‐A*0201‐restricted fashion, suggesting that these peptides may be applicable for cancer immunotherapy. These findings have provided a novel insight into carcinogenesis of the stomach, colon and liver, and will be helpful for the development of novel therapeutic strategies to a wide range of human cancers. (Cancer Sci 2005; 96: 498 –506)
Abbreviations
- APC
antigen presenting cells
- CTA
cancer‐testis antigen
- CTL
cytotoxic T lymphocyte
- CXADR
coxsackie adenovirus receptor
- DC
dendritic cell
- ESAM
endothelial cell‐selective adhesion molecule
- EST
expressed sequence tag
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HCC
hepatocellular carcinoma
- IGSF11
immunoglobulin superfamily 11
- IL
interleukin
- MAGE
melanoma antigen
- MHC
major histocompatibility complex
- NK
natural killer
- PBMC
peripheral blood mononuclear cell
- PDZ
postsynaptic density protein 95/drosophila disks large/zona occludens‐1
- RP‐HPLC
reverse phase high‐performance liquid chromatography
- RT‐PCR
reverse transcription–polymerase chain reaction
- SEREX
serological analysis of recombinant cDNA expression cloning
- siRNA
short interfering RNA
- TAA
tumor‐associated antigen
- TCR
T‐cell receptor.
Gastric cancer is one of the most frequent malignancies worldwide, accounting for 10.4% of cancer deaths in 2000.( 1 ) Recent progress in the diagnosis of gastric cancer using endoscopy has enabled the detection of gastric tumors at relatively early stages, which improves the cure rate by surgical resection. However, the prognosis of patients with advanced gastric cancer still remains poor, because other therapeutic modalities, such as chemotherapy, remain ineffective; the overall 5‐year survival rate of advanced tumors ranges from 5 to 15%.( 2 ) Hence, the development of novel therapeutic strategies is urgently required for the treatment of patients with advanced cancer.
The effective induction of CTL by TAA has provided hope for the success of cancer immunotherapy.( 3 ) Utilization of CTL elicited by TAA is an ideal therapeutic approach, if they specifically attack tumor cells expressing the antigen and reveal no or little adverse effect on normal cells. The identification of TAA immunodominant epitopes has highlighted the utilization of these epitopes as a promising therapeutic tool for immunotherapy. In addition, the understanding of mechanisms of antigen presentation to T lymphocytes in association with MHC has underscored the concept of cancer vaccines as a therapeutic option. More than 50 TAA have been identified to date, including MAGE‐1, MAGE‐2, MAGE‐3, MAGE‐12, BAGE, GAGE, PAGE, XAGE, NY‐ESO‐1, SSX, HER‐2/neu, SPANX and TRAG‐3.( 4 ) Clinical trials using their epitope peptides alone or loaded on to DC have been carried out not only with malignant melanoma( 5 , 6 , 7 , 8 ) but also with epithelial malignancies.( 9 , 10 , 11 ) Recently, it was reported that four reactive peptide vaccines lead to prolonged survival in gastric cancer patients, and this was associated with cellular and humoral immunoresponses.( 12 ) In addition, epitope peptides of immediate early response gene X‐1 induced CTL toward gastric cancer in an HLA‐A33‐restricted manner.( 13 ) However, a greater number of TAA are required to exert effective immunotherapy to cancer cells that are prone to escape from immunity.
Cancer‐testis antigens are one category of TAA that are expressed in tumors as well as in germ cells within the testis, and in some cases also in the ovary.( 14 ) The gonads themselves do not express molecules of MHC; therefore, when tumor‐specific T‐cell‐mediated immune systems are activated, cytotoxic T cells elicited by CTA will attack CTA in cancer cells and have no effect on germ cells.
The immunoglobulin superfamily 11 gene (IGSF11) was identified as a gene expressed in the brain and testis.( 15 ) The predicted protein comprises V‐type and C2‐type immunoglobulin domains, a C‐terminal PDZ‐binding domain and a transmembrane domain, and was classified as belonging to a novel immunoglobulin superfamily. A search for expressed sequence tags in nucleotide databases identified two forms of IGSF11 transcripts that were composed of different exons in their 5′ terminal regions.( 16 ) Homology searches revealed that both forms of the predicted IGSF11 proteins share similarity in amino acid sequence with CXADR and ESAM.
To disclose mechanisms of gastric carcinogenesis and discover target molecules for their diagnosis and treatment, we had analyzed global expression profiles of 20 gastric cancers by means of a cDNA microarray representing 23 040 genes.( 17 ) Among the genes commonly upregulated in cancer tissues, we focused, in this study, on IGSF11, a type‐1 transmembrane protein. Its expression was also elevated in six of 11 colon cancers, and 12 of 20 hepatocellular carcinomas in our microarray data. In the present study, we demonstrate novel insights into carcinogenesis of the stomach, colon and liver, and document antigenic epitope peptides of IGSF11 for the treatment of human cancers as a vaccine strategy.
Materials and Methods
Cell lines
Human gastric cancer cell lines MKN1, MKN28 and MKN45 (expressing HLA‐A*2402), MKN74 (HLA‐A*3101/), Kato III (HLA‐A*0201/0207), St‐4 (HLA‐A*0201/1101), a human colon cancer cell line SNU‐C4 (HLA‐A*0201/2402), a mouse fibroblast line NIH3T3, a monkey kidney cell line COS‐7, and a human hybrid between B and T lymphoblastic cell line T2 (HLA‐A*0201) were purchased from the American Type Culture Collection (ATCC). A human hepatocellular carcinoma cell line SNU475 (HLA‐A*0201/1101) was obtained from Korea Cell‐Line Bank (KCLB, Seoul National University, Seoul, Korea). An Epstein–Barr virus‐transformed B‐lymphoblastoid cell line A3LCL (HLA‐A*0301/) was generously provided by Takara Shuzo Co. Although abundant HLA‐A*0201 protein expression was observed in both SNU‐C4 and SNU475 cells, low level and no expression of HLA‐A*0201 protein was detected in St‐4 and MKN74, respectively. All of these cells were cultured in appropriate media: Dulbecco's modified Eagle's medium (COS7, NIH3T3) and RPMI‐1640 medium (T2, A3LCL, MKN1, MKN28, MKN45, MKN74, Kato III, St‐4, SNU‐C4 and SNU475), supplemented with 10% fetal bovine serum (Cansera International) and 1% antibiotic/antimycotic solution (Sigma‐Aldrich). Cells were maintained at 37°C in an atmosphere of humidified air with 5% CO2. Cancerous tissues and corresponding non‐cancerous tissues were excised from patients during surgery, after informed consent had been obtained.
RNA preparation and RT‐PCR analysis
Total RNA was extracted using Trizol reagent (Life Technologies) according to the manufacturer's instructions. Total RNA (10 µg) was reverse transcribed for single‐stranded cDNA using the poly dT12‐18 primer (Amersham Pharmacia Biotech) with Superscript II reverse transcriptase (Life Technologies). Each single‐stranded cDNA was then diluted for subsequent PCR amplification by monitoring GAPDH as a quantitative control. All of the reactions involved initial denaturation at 94°C for 4 min followed by 20 cycles at 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s (for GAPDH), or 30 cycles at 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s (for IGSF11). Primer sequences were as follows: GAPDH forward, 5′‐ACAACAGCCTCAAGATCATCAG‐3′; GAPDH reverse, 5′‐GGTCCACCACTGACACGTTG‐3′; IGSF11 forward, 5′‐ACCTGTCTTCTGGATCTCCAG‐3′; and IGSF11 reverse, 5′‐GTGTGAAATGCTTTGGCAGAAG‐3′.
Multiple tissue northern blot analysis
Human multiple‐tissue blots (Clontech) were hybridized with 32P‐labeled IGSF11 cDNA as a probe. Prehybridization, hybridization and washing were carried out according to the manufacturer's recommendations. The blots were then autoradiographed with intensifying screens at −80°C for 120 h.
Colony formation assay of IGSF‐11 in NIH3T3 cells
To prepare plasmids expressing IGSF11‐2 (pcDNA3.1myc/His‐IGSF11), we amplified the coding region of IGSF11‐2 with a set of primers (5′‐AGTTAAGCTTGCCGGGATGACTTCTCAGCGTTCCCCTCTGG‐3′ and 5′‐ATCTCGAGTACCAAGGACCCGGCCCGACTCTG‐3′) by RT‐PCR using cDNA from gastric cancer tissue as a template. The PCR products were cloned into an appropriate cloning site in the pcDNA3.1‐Myc/His vector (Invitrogen). NIH3T3 cells were plated at a density of 1 × 105 cells/100 mm dish. After 24 h, the cells were transfected by plasmid vector using FuGENE6 reagent, and were cultured with an appropriate concentration of geneticin for 2 weeks. Cells were fixed with 100% methanol and stained using Giemsa solution.
Construction of IGSF‐11 siRNA plasmids
We prepared psiH1BX3.0 vector plasmids that express siRNA under the control of the H1RNA promoter.( 18 ) Control plasmid, psi‐EGFP, was prepared by cloning double‐stranded oligonucleotides of 5′‐TCCCGAAGCAGCACGACTTCTTCTTCAAGAGAGAAGAAGTCGTGCTGCTTC‐3′ and 5′‐ AAAAGAAGCAGCACGACTTCTTCTCTCTTGAAGAAGAAGTCGTGCTGCTTC‐3′ into the BbsI site in the psiH1BX3.0 vector. Plasmids expressing IGSF11 siRNA were prepared by cloning double‐stranded oligonucleotides into the psiH1BX3.0 vector. The oligonucleotides used for IGSF11 siRNA were 5′‐TCCCCCTTCCAGACATAGGGGGCTTCAAGAGAGCCCCCTATGTCTGGAAGG‐3′ and 5′‐AAAACCTTCCAGACATAGGGGGCTCTCTTGAAGCCCCCTATGTCTGGAAGG‐3′ (psi‐IGSF11‐12). The plasmid psi‐IGSF11‐12 was transfected into St‐4 cells using FuGENE6 reagent (Roche) or Nucleofector reagent (Alexa) according to the suppliers’ recommendations. Total RNA was extracted from the cells 48 h after the transfection. Cells were cultured in the presence of 0.3 mg/mL geneticin for 14 days and stained with Giemsa solution (Merck) as described elsewhere.( 18 ) To determine cell viability in response to siRNA to either IGSF11 or EGFP (a control gene), St‐4 cells were plated in six‐well plates at a density of 1 × 105 cells/well. On the next day, the cells were transfected in triplicate with psi‐IGSF11 or psi‐EGFP, and maintained in media containing 0.3 mg/mL geneticin. After 7 days of culture, cell viability was measured using the MTT assay with cell counting kit‐8 (DOJINDO).
Peptide prediction and synthesis
Candidate peptide sequences of nine and 10 amino acids in length were predicted using the HLA Peptide Binding Predictions program,( 19 ) available at http://bimas.dcrt.nih.gov/cgi‐bin/molbio/ken_parker_comboform in the NIH Bioinformatics and Molecular Analysis Section (BIMAS). Among the predicted sequences, we synthesized 40 different peptides that were ranked according to highest binding affinity for the MHC class I molecule HLA‐A*0201, the most common HLA‐allele in the world (Table 1). The peptides synthesized revealed over 90% purity by RP‐HPLC, and their structures were verified using mass spectrometry. CMVpp65495−503 (NLVPMVATV), an HLA‐A*0201‐restricted epitope peptide derived from cytomegalovirus protein pp65, was used as a control for the cytotoxicity assay.( 20 )
Table 1.
Candidate peptides derived from IGSF11 and their predicted binding affinities to HLA‐A*0201
| Position † | Sequence (9‐mer) | Score ‡ | Position † | Sequence (10‐mer) | Score ‡ |
|---|---|---|---|---|---|
| 176 | YLWEKLDNT | 1314.6 | 176 | YLWEKLDNTL | 3344.0 |
| 11 | LLLLSLHGV | 1006.2 | 52 | LINLNVIWMV | 280.4 |
| 53 | INLNVIWMV | 49.2 (N.S.) § | 207 | ALSSGLYQCV | 104.3 |
| 59 | WMVTPLSNA | 37.9 | 51 | ALININVIWM | 62.8 (N.S.) § |
| 120 | CLVNNLPDI | 23.9 | 162 | ILLCSSEEGI | 32.1 |
| 15 | SLHGVAASL | 21.3 | 41 | VLPCTFTTSA | 32.0 |
| 252 | VIIIFCIAL | 18.9 (N.S.) § | 12 | LLLSIHGVAA | 31.2 |
| 52 | LINLNVIWM | 14.6 (N.S.) § | 356 | SIYANGTHLV | 30.6 |
| 40 | AVLPCTFTT | 13.9 | 111 | QLSDTGTYQC | 20.3 |
| 207 | ALSSGLYQC | 11.4 | 211 | GLYQCVASNA | 15.8 |
| 384 | VMSRSNGSV | 11.1 | 10 | PLLLISLHGV | 13.0 |
| 104 | SIFINNTQL | 10.8 | 32 | IQVARGQPAV | 11.9 |
| 327 | KVHRNTDSV | 10.4 | 106 | FINNTQLSDT | 10.8 |
| 413 | RIGAVPVMV | 9.5 | 364 | LVPGQHKTLV | 10.3 |
| 132 | NIGVTGLTV | 9.5 | 124 | NLPDIGGRNI | 8.5 |
| 356 | SIYANGTHL | 9.3 | 140 | VLVPPSAPHC | 8.4 |
| 163 | LLCSSEEGI | 8.6 (N.S.) § | 251 | AVIIIFCIAL | 7.1 |
| 13 | LLSLHGVAA | 8.4 | 252 | VIIIFCIALI | 5.6 (N.S.) § |
| 254 | IIFCIALIL | 7.5 | 261 | ILGAFFYWRS | 5.4 |
| 97 | TMPATNVSI | 7.5 | 137 | GLTVIVPPSA | 4.9 |
Amino acid sequences of candidate peptides are listed in the order of binding affinities to HLA‐A*0201 molecules. †The amino acid sequence number for position 1 in the candidate peptide, counting from the N‐terminus of IGSF11. ‡The estimated binding affinity of each peptide to HLA‐A*0201 molecules using Bioinformatics and Molecular Analysis Section software. §Not synthesized due to the hydrophobicity of amino acid sequences.
CTL induction by candidate peptides in vitro
We prepared DC of HLA‐A*0201 healthy volunteers for the use of APC as described previously.( 21 , 22 ) Subsequently, the DC were pulsed with 20 µg/mL of the candidate peptides over 4 h at room temperature. The cells were then irradiated (50 Gy) and incubated with CD8‐positive T lymphocytes in the presence of IL‐7 at a concentration of 10 U/mL (Genzyme). We added IL‐2 (BD Biosciences) into the culture medium at a final concentration of 20 U/mL at day 2. Two additional weekly stimulations with autologous peptide‐loaded DC using the same procedure were carried out on days 7 and 14. The CTL activity was examined by cytotoxicity assay at day 21 as described elsewhere.( 21 )
Cytotoxicity assay
We examined the cytotoxic activity of the stimulated T cells by means of a 4‐h 51Cr‐release assay. T2 cells (HLA‐A*0201‐positive) were pulsed with or without 20 µM peptide for 16 h, and tumor cells expressing IGSF11 were removed with phosphate‐buffered saline containing 0.53 mM ethylenediaminetetracetic acid. Each target was subsequently incubated with 100 µCi of 51Cr‐labeled sodium chromate for an additional hour at 37°C. The labeled cells (target) were then washed three times with phosphate‐buffered saline and mixed with the pretreated T cells (effectors) at various effector‐to‐target (E/T) ratios in 200 µL of medium in 96‐well plates. Half of the culture medium was collected from each well after 4 h of incubation, and its radioactivity was counted in a scintillation counter. Background activity was measured without effector cells, while maximum 51Cr‐releasing activity was examined by incubating target cells with 1 M hydrochloric acid. The percentage of cytotoxicity (%cytotoxicity) was calculated using the following formula:
| [(51Cr‐releasing activity of each experiment − background activity)/(maximum 51Cr‐releasing activity − background activity)] × 100(%). |
Establishment of oligo‐clonal CTL
After testing the cytotoxicities, the peptide‐pulsed CTL that responded with increased peptide‐specific killing either were expanded to establish CTL lines or underwent limiting dilution and expansion to establish oligo‐clonal CTL. To establish CTL lines, a total of 5 × 104 CTL were resuspended in 25 mL of AIM‐V/2%AS, along with 2.5 × 107 irradiated (33 Gy) allogenic PBMC and 5 × 106 irradiated (80 Gy) A3LCL cells, in the presence of 30 ng/mL of anti‐CD3 monoclonal antibody, as reported by Walter et al.( 23 ) The following day, IL‐2 was added to the cultures to a final concentration of 120 U/mL. The cultures were fed with fresh AIM‐V/2% autologous serum containing 30 U/mL of IL‐2 on days 5, 8, 11 and 14. To establish oligo‐clonal CTL, we carried out limiting dilution of CTL that showed increased cytotoxicity to the peptide‐pulsed target cells in our 51Cr‐release assay, before expansion.
Cold target‐inhibition assay
For the cold target‐inhibition assay, SNU475 cells expressing both HLA‐A*0201 and IGSF11 were labeled with 51Cr (hot target) and subsequently mixed with unlabeled T2 cells (cold target) pulsed with IGSF11‐9‐207 or control (CMVpp65495−503) peptides at various cold target/hot target (C/H) ratios. After preparation of target cells, CTL (effectors) were added and reacted for 4 h. The cytotoxicity of each C/H ratio was measured using the 51Cr‐release assay. The percentage of specific inhibition (%inhibition) was calculated using the following formula:
| {[%cytotoxicity (C/H ratio 0) − %cytotoxicity (each experiment)]/%cytotoxicity (C/H ratio 0)} × 100. |
Antibody blocking assay
SNU475 cells pretreated with anti‐HLA‐A/B/C monoclonal antibody or anti‐HLA‐DR/DB monoclonal antibody for 1 h were co‐cultured with oligo‐clonal CTL that were pretreated with anti‐CD4 monoclonal antibody or anti‐CD8 monoclonal antibody for 1 h, at an E/T ratio of 20 (all monoclonal antibodies were purchased from Dako, Carpinteria, CA, USA). We used antimouse IgG2a monoclonal antibody for anti‐HLA‐A/B/C monoclonal antibody and antimouse IgG1 monoclonal antibody for the other, as isotype antibodies. The percentage of specific inhibition (%inhibition) was calculated using the following formula:
| {[%cytotoxicity (Isotype) − %cytotoxicity (experimental)]/%cytotoxicity (Isotype)} × 100. |
Results
Identification of IGSF11, a human gene commonly upregulated in gastric cancer
Using a genome‐wide cDNA microarray containing 23 040 genes, we had previously examined the expression profiles of 20 gastric cancers and their corresponding normal gastric mucosae.( 17 ) Among the genes commonly upregulated in the cancer cells, we focused on a gene corresponding to an EST, Hs.112873 of a UniGene cluster (http://www.ncbi.nlm.nih.gov/UniGene/), which later turned out to be IGSF11 on chromosomal band 3q13 (GenBank accession no. AC068984). The signal intensity ratios of cancer‐to‐non‐cancerous gastric tissues in the microarray data ranged between 4.09 and 48.6. Elevated IGSF11 expression was subsequently confirmed in seven out of eight additional gastric cancer tissues (data not shown). Interestingly, its expression was also elevated in six of 11 colon cancers, and in 12 of 20 hepatocellular carcinomas in our microarray data. Enhanced expression was confirmed in five out of eight hepatocellular carcinomas by semiquantitative RT‐PCR (data not shown). Because two forms of IGSF11 transcripts had been reported,( 16 ) we prepared PCR primers specific to each transcript and carried out RT‐PCR experiments to examine the transcriptional levels of the two transcripts in gastric cancer. Although an isoform (IGSF11‐2) containing exon 1b was successfully amplified using cDNA from gastric cancer tissues as a template, the other form (IGSF11‐1) containing exons 1a, 2a and 3a could not be amplified with various sets of primers, suggesting that IGSF11‐2 was the isoform upregulated in gastric cancer tissues. Hence, we further investigated this form of transcript (GenBank accession no. AB071618) in the present study.
Expression and characterization of IGSF11
We further analyzed IGSF11 expression in normal adult organs by multiple‐tissue northern‐blot analysis, using two 3′ regions of IGSF11 cDNA (which were conserved between the two isoforms) as probes, and revealed that the 3.5‐kb transcript was abundantly expressed in testis and ovary, and expressed in brain, kidney and skeletal muscle at lower levels (Fig. 1a). Semi‐quantitative RT‐PCR analysis revealed that MKN‐1, MKN‐7, MKN‐28, MKN‐74 and St‐4 gastric cancer cells, and Alexander, Huh7 and SNU475 hepatoma cells expressed IGSF11, but not MKN‐45 and Kato‐III gastric cancer cells or HepG2 hepatoma cells (Fig. 1b).
Figure 1.
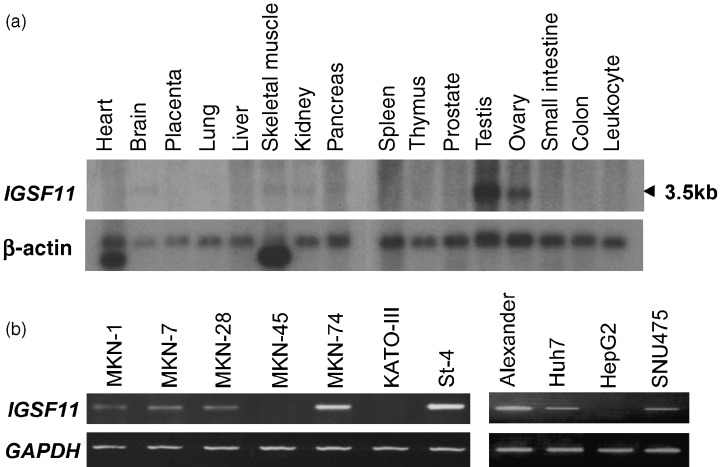
(a) Multiple tissue northern‐blot analyses of IGSF11. The transcript of IGSF11 is approximately 3.5 kb in size. Expression of β‐actin served as a control. (b) IGSF11 expression in gastric cancer and hepatoma cell lines.
Effect of IGSF11 on cell growth
To analyze the effect of IGSF11‐2 on cell growth, we carried out a colony‐formation assay by transfecting NIH3T3 cells with pcDNA3.1myc/His‐IGSF11. Expression of exogenous Myc‐tagged IGSF11 was confirmed by Western blot analysis using anti‐Myc antibody (Fig. 2a). Compared with a control plasmid (pcDNA3.1myc/His‐LacZ), pcDNA3.1myc/His‐IGSF11 induced markedly more colonies in NIH3T3 cells (Fig. 2b). This result was confirmed in three independent experiments.
Figure 2.
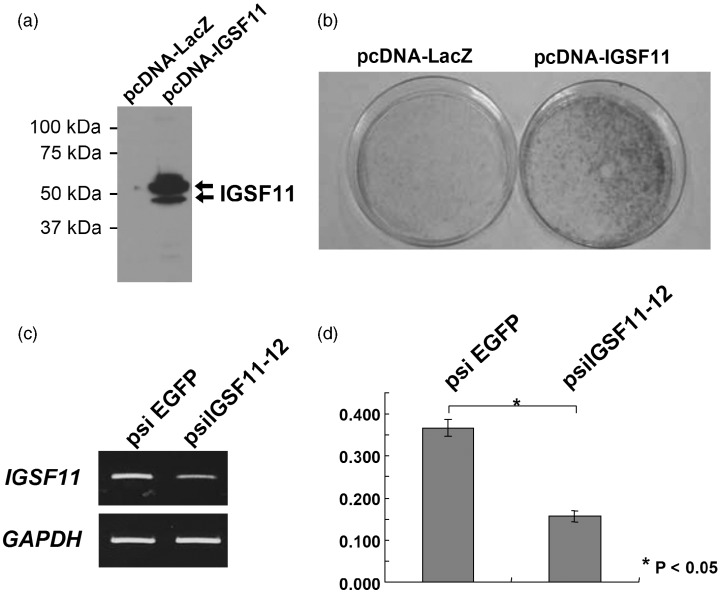
(a) Expression of Myc‐tagged IGSF11 in NIH3T3 cells transfected with pcDNA3.1myc/His‐IGSF11 was examined by Western blot analysis using anti‐Myc antibody. (b) Colony formation activity of exogenous IGSF11 in NIH3T3 cells. (c) Gene‐knockdown effect of IGSF11‐specific siRNA. (d) Significant growth‐inhibitory effect of the IGSF11‐specific siRNA in St‐4 cells (P < 0.05).
Effect of the siRNA to IGSF11 on the growth of gastric cancer cells
To test whether the suppression of IGSF11 may reduce the growth of cancer cells or not, we constructed plasmids expressing IGSF11 siRNA (psiIGSF11‐12) and those expressing siRNA to a control gene (psiEGFP), and transfected them into St‐4 gastric cancer cells that constitutively express IGSF11. As a result, transfection with psiIGSF11‐12 reduced IGSF11 expression (Fig. 2c) and decreased the number of transfected St‐4 cells compared to psiEGFP plasmids (Fig. 2d), suggesting that the elevated expression of IGSF11 may be essential for the growth of cancer cells.
Identification of peptides that induce CTL to cells expressing IGSF11
Using the HLA Peptide Binding Predictions in the NIH BIMAS, we synthesized a total of 40 different candidate 9‐ or 10 amino‐acid peptides that were expected to have the highest binding affinity to HLA‐A*0201, the most common HLA‐allele (Table 1). Each of the 40 peptides was tested as to whether they could induce peptide‐reactive CTL or not by means of the 51Cr‐release assay. As a result, IGSF11‐9‐207 (ALSSGLYQC) and IGSF11‐10‐207 (ALSSGLYQCV) were shown to induce peptide‐reactive CTL (data not shown). Notably, these CTL did not show cytotoxic activity to T2 cells that were pulsed with the control peptides. We further carried out limiting dilutions to establish oligo‐clonal CTL from both CTL lines.
Establishment of oligo‐clonal CTL
Among the oligo‐clonal CTL established from the CTL line against IGSF11‐9‐207, CTL207‐11 cells showed higher cytotoxic activity against T2 cells (HLA‐A*0201 positive) pulsed with the peptides compared to the parental cells, while the cells did not show cytotoxic activity against T2 cells without IGSF11‐9‐207‐stimulation (Fig. 3a). Furthermore, SNU475 human hepatocellular carcinoma cells that express both HLA‐A*0201 and IGSF11 (Fig. 1b) were killed by CTL207‐11 cells (Fig. 3b). On the other hand, CTL207‐11 cells did not reveal any cytotoxity to SNU‐C4 hepatocellular carcinoma cells that express HLA‐A*0201 but do not express IGSF11 (Fig. 3b). These data confirm the IGSF11‐specific cytotoxic activity of CTL207‐11 cells. To further examine HLA‐dependence of the cytotoxity, we analyzed their cytotoxic activity using MKN74 human gastric cancer cells, which express IGSF11 but do not express HLA‐A*0201 (Fig. 3b). As expected, we observed no target killing, recapitulating IGSF11‐specific and HLA‐A*0201‐restricted cytotoxity elicited by the peptide. Although we analyzed oligo‐clonal CTL induced by IGSF11‐10‐207 (ALSSGLYQCV), the cells failed to display any (or displayed a low level of) cytotoxity against SNU475 cells (data not shown).
Figure 3.
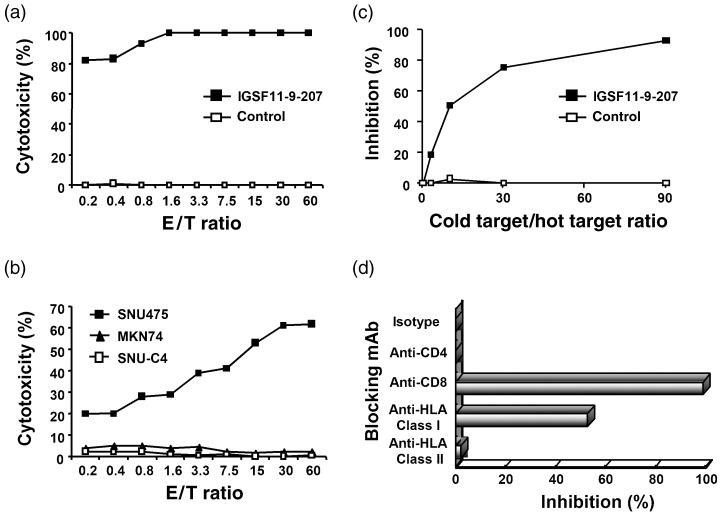
Cytotoxic activities of CTL207‐11 oligo‐clonal CTL induced with IGSF11‐9‐207 peptide. Cytotoxic activities of CTL207‐11 cells were examined using a 4‐h 51Cr‐release assay. (a) Cytotoxic activities to T2 cells (HLA‐A*0201) pulsed with IGSF11‐9‐207 (▪) or control (□) peptides. (b) Cytotoxic activities to SNU475 (both IGSF11 and HLA‐A*0201 positive), SNU‐C4 (HLA‐A*0201 positive and IGSF11 negative) and MKN74 (IGSF11 positive and HLA‐A*0201 negative). CTL207‐11 showed potent cytotoxic activities against not only T2 cells pulsed with IGSF11‐9‐207 but also SNU475 cells. There was no significant cytotoxic activity against SNU‐C4 or MKN74, both of which do not express HLA‐A*0201 and IGSF11 simultaneously. (c) A cold target inhibition assay. 51Cr‐labeled SNU475 cells were used as hot targets at an E/T ratio of 20. T2 cells pulsed with IGSF11‐9‐207 (▪) or control (□) peptides were used as a cold target. We added T2 cells to SNU475 cells in each microculture at various cold target/hot target ratios, and examined cytotoxic activity. T2 cells inhibited cytotoxic activity against SNU475 cells only when pulsed with IGSF11‐9‐207 peptides. (d) An antibody‐blocking assay. Cytotoxic activity against SNU475 cells was inhibited by anti‐CD8 or anti‐HLA‐class I monoclonal antibody. *E/T ratio: 20.
Specificity of established oligo‐clonal CTL
To assess the specificity of CTL207‐11 cells, we carried out a cold target inhibition assay. We incubated SNU475 cells with various numbers of the cold target T2 cells that were pulsed with IGSF11‐9‐207 or control peptides. As a consequence, the killing of 51Cr‐labeled SNU475 cells was dramatically reduced when they were mixed with T2 cells pulsed with IGSF11‐9‐207 peptides, but not suppressed when they were incubated with T2 cells that were pulsed with control peptides (Fig. 3c). To confirm the HLA‐dependent cytotoxity, we carried out 51Cr‐releasing assays with monoclonal antibody to MHC class I of the target cells, or cytotoxic T‐cell‐specific antibody to the effector cells. The cytotoxic activity was decreased significantly either when SNU475 cells were pretreated with anti‐HLA‐ABC monoclonal antibody, or when the CTL207‐11 cells were preincubated with anti‐CD8 monoclonal antibody (Fig. 3d). However, the cytotoxic activity was decreased by approximately 50% in the presence of anti‐HLA monoclonal antibody. Therefore, other effector cells, such as CD8‐positive NK cells, may contribute to the cytotoxicity. Taken together, the CTL consist of CD8‐positive cells that recognize IGSF11‐9‐207 in an MHC class I‐restricted manner, and IGSF11‐9‐207 peptide is likely to be an epitope peptide restricted to HLA‐A*0201.
CTL by anchor‐modified peptides
According to the HLA‐A*0201 antigen motif previously reported by Smith et al.,( 24 ) leucine and isoleucine are the ideal anchor residues at position two that enhance the binding affinity of a peptide to HLA‐A*0201, and valine and leucine are the preferred amino acids at position nine for nonamer peptides. The newly defined epitope IGSF11‐9‐207 was ranked at a relatively low binding score by the prediction, and it did not consist of valine or leucine at the ninth position. Hence, we synthesized two different anchor‐modified peptides termed IGSF11‐9V (ALSSGLYQV) and IGSF11‐9L (ALSSGLYQL) and examined a possible enhancement of immunogenicity by these modifications (Table 2). The cysteine at position nine in IGSF11‐9‐207 was replaced by valine in IGSF11‐9V and leucine in IGSF11‐9L. Both of the two altered peptides were predicted to have higher HLA‐A*0201‐binding scores compared to the wild‐type IGSF11‐9‐207 peptide by BIMAS's epitope prediction algorithm. We also prepared CTL pulsed with IGSF11‐9V or IGSF11‐9L peptides, and successfully established oligo‐clonal CTL with IGSF11‐9V (CTL9V‐69) (Fig. 4a). CTL9V‐69 cells killed not only T2 cells pulsed with IGSF11‐9‐207 peptide but also SNU475 cells expressing IGSF11, indicating that IGSF11‐9V is also capable of inducing CTL activity against IGSF11 (Fig. 4b). Notably, although we only managed to induce CTL in one individual out of four healthy HLA‐A*0201‐positive volunteers using IGSF11‐9‐207, this number was increased to three individuals out of the four by using IGSF11‐9V peptides.
Table 2.
Anchor‐modified peptides for IGSF11‐9‐207
| Peptide | Type | Sequence | Score † |
|---|---|---|---|
| IGSF11‐9‐207 | Wild type | ALSSGLYQC | 11.4 |
| IGSF11‐9V | Anchor‐modified‐1 | ALSSGLYQV | 49.0 |
| IGSF11‐9L | Anchor‐modified‐2 | ALSSGLYQL | 159.8 |
The estimated binding affinity of each peptide to HLA‐A*0201 molecules using Bioinformatics and Molecular Analysis Section software.
Figure 4.
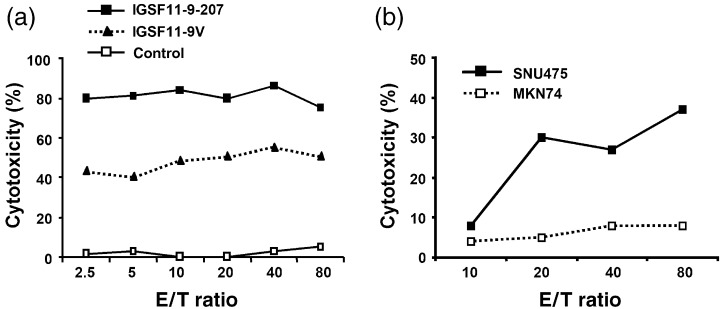
Cytotoxic activity of CTL induced with the anchor‐modified altered peptide IGSF11‐9V. Examination of the cytotoxic activity of CTL9V‐69 oligo‐clonal CTL against (a) peptide‐pulsed T2 cells (HLA‐A*0201) and (b) tumor cell lines using 4‐h 51Cr‐release assay. CTL9V‐69 cells recognized not only IGSF11‐9V (▴) but also the parental peptide IGSF11‐9‐207 (▪). The cells killed SNU475 cells expressing both IGSF11and the HLA‐A*0201 molecule.
A homology search of protein databases revealed that IGSF11‐9‐207 and IGSF11‐9V have no homologous peptide sequences derived from any other gene products, indicating that these peptides are likely to elicit IGSF11‐specific cytotoxity.
Discussion
In the present study, we have shown that IGSF11 is a promising therapeutic target for human gastric and hepatocellular carcinomas, and that peptides corresponding to a part of IGSF11 [IGSF11‐9‐207 (ALSSGLYQC) or its anchor‐modified form IGSF‐9V (ALSSGLYQV)] elicit CTL towards human cancer cells. IGSF11 was identified as an immunoglobulin superfamily gene that showed highest expression in testis and ovary, and lower expression levels in brain, kidney and adrenal gland.( 15 ) IGSF11 expression was barely detectable in other organs except for testis and ovary, brain, kidney and skeletal muscle in our study, suggesting that targeting IGSF11 was highly unlikely to give rise to life‐threatening adverse events. Our microarray data also showed that IGSF11 expression was augmented in six of 11 colon cancers, 12 of 20 hepatocellular carcinomas and none of 20 diffuse‐type gastric cancers. Hence, targeting IGSF11 should be applicable for the treatment of colorectal cancer and hepatocellular carcinoma in addition to intestinal‐type gastric cancer, but not for that of diffuse‐type gastric cancer. Although we revealed frequent enhancement of IGSF11 in a wide range of tumors, the mechanisms of its upregulation remain unresolved. Frequent gain of 3q was reported in diffuse‐type gastric cancer,( 25 ) but the amplification has not been reported so far in intestinal‐type cancers, colorectal cancer or hepatocellular carcinomas. Thus, other mechanisms, such as transcriptional activation, may be involved in its upregulation. Notably, two forms of IGSF11 transcripts were predicted, and our experiment revealed that gastric cancer cells abundantly express IGSF11‐2 transcripts but not IGSF11‐1 transcripts. Because the peptide sequence of IGSF11‐9‐207 (ALSSGLYQC) is conserved in both forms of IGSF11 protein, immunotherapy targeting this peptide sequence may have an effect on cells expressing IGSF11‐1 as well as those expressing IGSF11‐2. Further studies of the function and physiological roles of IGSF11‐1 and IGSF11‐2 are necessary.
The IGSF11‐2 protein shared 37% homology with human CXADR, 34% with Xenopus CTX homolog‐like (CTXL), and 30% with ESAM.( 16 ) CXADR serves as the cell‐surface receptor for group B coxsackie viruses and most adenoviruses;( 26 ) CTXL is a human homolog of Xenopus thymocyte receptor and is expressed in stomach, colon, prostate, trachea and thyroid gland;( 27 ) ESAM plays a role in adhesion and vascular development.( 28 ) These proteins comprise a signal peptide, an extracellular V‐type immunoglobulin‐like domain followed by a C2‐type immunoglobulin‐like domain, a transmembrane region and a cytoplasmic tail. The predicted IGSF11‐2 protein also contains a hydrophobic signal sequence, two immunoglobulin domains, a transmembrane domain and a cytoplasmic region with a PZD‐binding domain, suggesting that IGSF11‐2 is a type‐I transmembrane protein. In line with this prediction, we detected subcellular localization of exogenous IGSF11‐2 protein in the cytoplasmic membrane when we transfected NIH‐3T3 cells with plasmids expressing Flag‐tagged IGSF11‐2 (data not shown). Therefore, IGSF11 may mediate external signals as a receptor, play a role in cell–cell interaction, or enhance cellular proliferation and motility through the regulation of other receptors or adhesion molecules in a manner similar to Nectin‐like molecule‐5.( 29 ) An antibody against the extracellular domain of IGSF11‐2 is therefore an alternative approach for the treatment of gastric cancer. Because its elevated expression is essential for the growth of cancer cells, small compounds that inhibit IGSF11‐2 or signals mediated by IGSF11‐2 may also be a potential therapeutic option.
A recent report summarized the objective response rate of immunotherapy in clinical trials to be as low as 2.6%.( 30 ) This inefficacy may result from decreased expression of MHC class I antigen or co‐stimulatory molecules, low levels of circulating immune cells, induction of immune suppressive mechanisms, insufficient observation periods or heterogeneous expression of TAA. Although cancer‐testis antigens are expressed in a variety of cancers, the expression levels among cancer cells are very heterogeneous.( 31 , 32 ) Moreover, peptides suitable for use in vaccination are different from patient to patient, mainly due to large diversity of TCR repertories.( 33 , 34 ) Therefore, identification of further TAA is urgently needed to overcome the low efficacy. In particular, antigens that are homogenously expressed in all cancer cells are ideal target molecules. From this point of view, we are attempting to identify additional TAA and epitope peptides for cancer immunotherapy. As IGSF11 expression is essential for the proliferation of cancer cells, all cancer cells should express IGSF11 to some extent. Together with its low expression levels in important organs essential for life, IGSF11 should be an ideal therapeutic target.
The majority of TAA were identified using one of three approaches: (i) cDNA expression cloning of tumor infiltrating lymphocytes;( 35 , 36 ) (ii) SEREX with antibodies from patients’ sera;( 14 , 37 ) and (iii) reverse immunology using previously defined oncogene products.( 38 , 39 ) In addition to these conventional methods, advances in expression profile analysis have accelerated the identification of candidate TAA.( 40 , 41 , 42 , 43 , 44 ) Although a number of TAA have been identified to date,( 4 , 45 ) TAA of gastric cancer have not been intensively studied; only the expression of a few known TAA, such as MAGE‐1, MAGE‐2 and NY‐ESO‐1, have been investigated in gastric tumors.( 46 , 47 ) To our knowledge, IGSF11 is the first TAA for immunotherapy to gastric cancer identified by expression profile analysis. Recent studies have identified an increasing number of antigenic peptides, some of which are under clinical trials.( 3 ) However, data of clinically useful epitope peptides towards treatment of gastric cancer is limited. Several candidate epitope peptides were determined for peptide vaccines. For example, an‐HLA‐A31(A*31012)‐restricted natural antigenic peptide of gastric signet ring cell carcinoma cells was identified by RP‐HPLC.( 48 ) Peptides of IEX‐1 were recently identified as TAA for gastric cancer expressing HLA‐A33 by cDNA expression cloning.( 13 ) Another group tested a total of 30 antigenic peptides on HLA‐A24 or HLA‐A2 of 13 advanced gastric patients, which were identified by means of reverse immunology.( 49 ) We added two epitope peptide sequences for HLA‐A*0201‐restricted immunotherapy to intestinal‐type gastric cancer. Notably, HLA‐A*0201 accounts for 30–40% of Caucasian and approximately 20% of Japanese populations.( 50 ) Our data showed that IGSF11 is highly expressed in a wide range of human tumors. Taken together, the two IGSF11‐derived peptides should be applicable for the treatment of a number of patients with cancer. Although cytotoxic activity was elicited by the anchor‐modified peptides, IGSF11‐9V was approximately half of that by IGSF11‐9‐207, binding affinity of IGSF11‐9V peptides to HLA molecules was higher, and the frequency of induction of CTL with IGSF11‐9V increased compared to IGSF11‐9‐207. Therefore, IGSF11‐9V peptides may have an advantage for clinical use.
In summary, we have identified IGSF11 as a novel therapeutic target for gastric cancer, and further discovered two peptide sequences of IGSF11, both of which elicited CTL to gastric cancer cells expressing IGSF11 in an HLA‐A*0201‐restricted manner. These data may be useful not only for the gain of additional insights into human carcinogenesis but also for clinical applications of immunotherapy.
Acknowledgments
This work was supported by a ‘Research for the Future’ Program Grant (00L01402) from the Japan Society for the Promotion of Science. We thank Ms Yumi Nakajima for her technical assistance, and Ryuji Hamamoto, Ryuichiro Yagyu and Takashi Shimokawa for helpful discussions.
References
- 1. Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol 2001; 9: 533–43. [DOI] [PubMed] [Google Scholar]
- 2. Peddanna N, Holt S, Verma RS. Genetics of gastric cancer. Anticancer Res 1995; 15: 2055–64. [PubMed] [Google Scholar]
- 3. Stevanovic S. Identification of tumor associated T‐cell epitopes for vaccine development. Nature Rev 2002; 2: 1–7. [DOI] [PubMed] [Google Scholar]
- 4. Renkvist N, Castelli C, Robbins PF, Parmiani G. A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 2001; 50: 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawakami Y, Eliyahu SC, Jennings K, et al. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor‐infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol 1995; 154: 3961–8. [PubMed] [Google Scholar]
- 6. Rosenberg SA, Yang JC, Schwartentruber DJ, et al. Immunologic and therapeutic evaluation of synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 1998; 4: 321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide‐ or tumor lysate‐pulsed dendritic cells. Nat Med 1998; 4: 328–32. [DOI] [PubMed] [Google Scholar]
- 8. Panelli MC, Wunderlich J, Jeffries J, et al. Phase I study in patients with metastatic melanoma of immunization with dendritic cells presenting epitopes derived from the melanoma‐associated antigens MART‐1 and gp100. J Immunother 2000; 23: 487–98. [DOI] [PubMed] [Google Scholar]
- 9. Sadanaga N, Nagashima H, Mashino K, et al. Dendritic cell vaccination with MAGE peptide is a novel therapeutic approach for gastrointestinal carcinomas. Clin Cancer Res 2001; 7: 2277–84. [PubMed] [Google Scholar]
- 10. Tanaka H, Tsunoda T, Nukaya I, et al. Mapping the HLA‐A24‐restricted T‐cell epitope peptide from a tumor‐associated antigen HER2/neu: Possible immunotherapy for colorectal carcinomas. Br J Cancer 2001; 84: 94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rentzsch C, Kayser S, Stumm S, et al. Evaluation of pre‐existent immunity in patients with primary breast cancer: molecular and cellular assays to quantify antigen‐specific T lymphocytes in peripheral blood mononuclear cells. Clin Cancer Res 2003; 9: 4376–86. [PubMed] [Google Scholar]
- 12. Kono K, Takahashi A, Sugai H, et al. Dendritic cells pulsed with HER‐2/neu‐derived peptide can induce specific T‐cell responses in patients with gastric cancer. Clin Cancer Res 2002; 8: 3394–400. [PubMed] [Google Scholar]
- 13. Sasada T, Takedatsu H, Azuma K, et al. Immediate early response Gene X‐1, a stress‐inducible anti‐apoptotic gene, encodes cytotoxic T‐lymphocyte (CTL) epitopes capable of inducing Human Leukocyte Antigen‐A33‐restricted and tumor‐reactive CTLs in gastric cancer patients. Cancer Res 2004; 64: 2882–8. [DOI] [PubMed] [Google Scholar]
- 14. Chen YT, Scanlan MJ, Sahin U, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA 1997; 94: 1914–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suzu S, Hayashi Y, Harumi T, et al. Molecular cloning of a novel immunoglobulin superfamily gene preferentially expressed by brain and testis. Biochem Biophys Res Commun 2002; 296: 1215–21. [DOI] [PubMed] [Google Scholar]
- 16. Katoh M, Katoh M. Identification and characterization of human TMEM25 and mouse Tmem25 genes in silico . Oncol Rep 2004; 12: 429–33. [PubMed] [Google Scholar]
- 17. Hasegawa S, Furukawa Y, Li M, et al. Genome‐wide analysis of gene expression in intestinal‐type gastric cancers using a complementary DNA microarray representing 23 040 genes. Cancer Res 2002; 62: 7012–17. [PubMed] [Google Scholar]
- 18. Shimokawa T, Furukawa Y, Sakai M, et al. Involvement of the FGF18 gene in colorectal carcinogenesis, as a novel downstream target of the beta‐catenin/T‐cell factor complex. Cancer Res 2003; 63: 6116–20. [PubMed] [Google Scholar]
- 19. Lu J, Celis E. Use of predictive algorithms of the world wide web for the identification of tumor‐reactive T‐cell epitopes. Cancer Res 2000; 60: 5223–7. [PubMed] [Google Scholar]
- 20. Solache A, Morgan CL, Dodi AI, et al. Identification of three HLA‐A*0201‐restricted cytotoxic T cell epitopes in the cytomegalovirus protein pp65 that are conserved between eight strains of the virus. J Immunol 1999; 163: 5512–18. [PubMed] [Google Scholar]
- 21. Tsai V, Southwood S, Sidney J, et al. Identification of subdominant CTL epitopes of the GP100 melanoma‐associated tumor antigen by primary in vitro immunization with peptide‐pulsed dendritic cells. J Immunol 1997; 158: 1796–802. [PubMed] [Google Scholar]
- 22. Nakahara S, Tsunoda T, Baba T, Asabe S, Tahara H. Dendritic cells stimulated with a bacterial product, OK‐432, efficiently induce cytotoxic T lymphocytes specific to tumor rejection peptide. Cancer Res 2003; 63: 4112–18. [PubMed] [Google Scholar]
- 23. Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogenic bone marrow by transfer of T‐cell clones from the donor. N Engl J Med 1995; 333: 1038–44. [DOI] [PubMed] [Google Scholar]
- 24. Smith MH, Nuara AA, Egen JG, Shirjani DB, Lam KS, Grimes WJ. Baculoviral‐expressed HLA‐class I heavy chains used to screen a synthetic peptide library for allele‐specific peptide binding motifs. Mol Immunol 1998; 35: 1033–43. [DOI] [PubMed] [Google Scholar]
- 25. Peng DF, Sugihara H, Mukaisho K, Tsubosa Y, Hattori T. Alterations of chromosomal copy number during progression of diffuse‐type gastric carcinomas: metaphase‐ and array‐based comparative genomic hybridization analyses of multiple samples from individual tumours. J Pathol 2003; 201: 439–50. [DOI] [PubMed] [Google Scholar]
- 26. Honda T, Saitoh H, Masuko M, et al. The coxsackievirus–adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res Mol Brain Res 2000; 77: 19–28. [DOI] [PubMed] [Google Scholar]
- 27. Chretien I, Marcuz A, Courtet M, et al. CTX, a xenopus thymocyte receptor, defines a molecular family conserved throughout vertebrates. Eur J Immunol 1998; 28: 4094–104. [DOI] [PubMed] [Google Scholar]
- 28. Hirata Ki, Ishida T, Penta K, et al. Cloning of an immunoglobulin family adhesion molecule selectively expressed by endothelial cells. J Biol Chem 2001; 276: 16 223–31. [DOI] [PubMed] [Google Scholar]
- 29. Kakunaga S, Ikeda W, Shingai T, et al. Enhancement of serum‐ and platelet‐derived growth factor‐induced cell proliferation by Necl‐5/Tage4/poliovirus receptor/CD155 through Ras‐Raf‐MEK‐ERK signaling. J Biol Chem 2004; 279: 36 419–25. [DOI] [PubMed] [Google Scholar]
- 30. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Neumann E, Engelsberg A, Decker J, et al. Heterogeneous expression of the tumor‐associated antigens RAGE‐1, PRAME, and glycoprotein 75 in human renal cell carcinoma: candidates for T‐cell‐based immunotherapies? Cancer Res 1998; 58: 4090–5. [PubMed] [Google Scholar]
- 32. Jungbluth AA, Antonescu CR, Busam KJ, et al. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY‐ESO‐1 but not MAGE‐A1 or CT7. Int J Cancer 2001; 94: 252–6. [DOI] [PubMed] [Google Scholar]
- 33. Noguchi M, Kobayashi K, Suetsugu N, et al. Induction of cellular and humoral immune responses to tumor cells and peptides in HLA‐A24‐positive hormone‐refractory prostate cancer patients by peptide vaccination. Prostate 2003; 57: 80–92. [DOI] [PubMed] [Google Scholar]
- 34. Sato M, Takayama T, Tanaka H, et al. Generation of mature dendritic cells fully capable of T helper type 1 polarization using OK‐432 combined with prostaglandin E(2). Cancer Sci 2003; 94: 1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254: 1643–7. [DOI] [PubMed] [Google Scholar]
- 36. Boon T. Tumor antigens recognized by cytolytic T lymphocytes: present perspectives for specific immunotherapy. Int J Cancer 1993; 54: 177–80. [DOI] [PubMed] [Google Scholar]
- 37. Sahin U, Tureci O, Schmitt H, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci USA 1995; 92: 11 810–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang S, Cordon‐Cardo C, Zhang HS, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer 1997; 73: 42–9. [DOI] [PubMed] [Google Scholar]
- 39. Zhang S, Zhang HS, Cordon‐Cardo C, Ragupathi G, Livingston PO. Selection of tumor antigens as targets for immune attack using immunohistochemistry: protein antigens. Clin Cancer Res 1998; 4: 2669–76. [PubMed] [Google Scholar]
- 40. Mathiassen S, Lauemoller SL, Ruhwald M, Claesson MH, Buus S. Tumor‐associated antigens identified by mRNA expression profiling induce protective anti‐tumor immunity. Eur J Immunol 2001; 31: 1239–46. [DOI] [PubMed] [Google Scholar]
- 41. Boer JM, Huber WK, Sultmann H, et al. Identification and classification of differentially expressed genes in renal cell carcinoma by expression profiling on a global human 31 500‐element cDNA array. Genome Res 2001; 11: 1861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmidt SM, Schag K, Muller MR et al. Induction of adipophilin‐specific cytotoxic T lymphocytes using a novel HLA‐A2‐binding peptide that mediates tumor cell lysis. Cancer Res 2004; 64: 1164–70. [DOI] [PubMed] [Google Scholar]
- 43. Yoshitake Y, Nakatsura T, Monji M, et al. Proliferation potential‐related protein, an ideal esophageal cancer antigen for immunotherapy, identified using complementary DNA microarray analysis. Clin Cancer Res 2004; 10: 6437–48. [DOI] [PubMed] [Google Scholar]
- 44. Uchida N, Tsunoda T, Wada S, Furukawa Y, Nakamura Y, Tahara H. Ring finger protein 43 as a new target for cancer immunotherapy. Clin Cancer Res 2004; 10: 8577–86. [DOI] [PubMed] [Google Scholar]
- 45. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 2002; 188: 22–32. [DOI] [PubMed] [Google Scholar]
- 46. Granelli P, Siardi C, Zennaro F, et al. Melanoma antigen genes 1 and 2 are differentially expressed in human gastric and cardial carcinomas. Scand J Gastroenterol 2000; 35: 528–33. [DOI] [PubMed] [Google Scholar]
- 47. Wang Y, Wu XJ, Zhao AL, et al. Cancer/testis antigen expression and autologous humoral immunity to NY‐ESO‐1 in gastric cancer. Cancer Immun 2004; 4: 11–17. [PubMed] [Google Scholar]
- 48. Suzuki K, Sahara H, Okada Y, et al. Identification of natural antigenic peptides of a human gastric signet ring cell carcinoma recognized by HLA‐A31‐restricted cytotoxic T lymphocytes. J Immunol 1999; 163: 2783–91. [PubMed] [Google Scholar]
- 49. Sato Y, Shomura H, Maeda Y, et al. Immunological evaluation of peptide vaccination for patients with gastric cancer based on pre‐existing cellular response to peptide. Cancer Sci 2003; 94: 802–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Date Y, Kimura A, Kato H, Sasazuki T. DNA typing of the HLA‐A gene: population study and identification of four new alleles in Japanese. Tissue Antigens 1996; 47: 93–101. [DOI] [PubMed] [Google Scholar]