

Abstract
Accumulated understanding of the molecular pathways regulating cancer progression has led to the development of novel targeted therapies. Hepatocellular carcinoma (HCC) remains a highly lethal disease that is resistant to conventional cytotoxic chemotherapy and radiotherapy. Unlike conventional chemotherapy, molecular‐targeted agents offer the potential advantages of a relatively high therapeutic window and use in combination with other anticancer strategies without overlapping toxicity. It is hoped that these drugs will become valuable therapeutic tools within the multimodal approach to treating cancer. A recent clinical trial revealed an oral multikinase inhibitor, sorafenib, as the first agent that has demonstrated improved overall survival in patients with advanced HCC. The present review summarizes molecular abnormalities of HCC with a focus on clinical studies, and current status as well as problems of the targeted strategies for HCC. (Cancer Sci 2009; 100: 1–8)
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide accounting for 500 000–600 000 deaths per year,( 1 , 2 ) and the incidence is still increasing.( 3 ) Although the primary curative treatment for HCC is surgical resection, there has been limited improvement in the availability of alternative treatments in the last decade.( 4 ) A major obstacle for the treatment of HCC is the high frequency of tumor recurrence after curative resection. In fact, effective palliative treatment is hindered by the fact that HCC is frequently resistant to conventional chemotherapy and radiotherapy.( 4 ) Moreover, the existing conventional chemotherapeutics are more or less non‐selective cytotoxic drugs with significant systemic side effects. Importantly, as most patients with HCC have compromised liver function aggressive medical therapy regimens can not be applied. Thus, usually no effective therapy can be offered to these patients.( 2 , 4 ) There is an urgent need to develop novel treatments for recurrent and advanced HCC.
Numerous studies on molecular abnormalities in HCC progression have revealed the crucial roles of such molecules in cell proliferation, as well as survival not only of cancer cells but also angiogenic or stromal cells.( 5 , 6 ) Among the key pathways in the pathogenesis of HCC, this review focuses on the pathological processes including vascular endothelial growth factor (VEGF)‐dependent tumor angiogenesis, epidermal growth factor receptor (EGFR), and insulin‐like growth factor (IGF)‐dependent tumor cell proliferation and survival in HCC (Fig. 1). Furthermore, several intracellular factors essential for hepatocarcinogenesis are demonstrated (2, 3), coupled with molecularly targeted agents in clinical trials (Table 1).
Figure 1.

Molecular targets in the (a) epidermal growth factor (EGF) and EGF receptor (EGFR) family, (b) vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR) family, and (c) insulin‐like growth factor (IGF) and IGF receptor (IGFR). Targeted agents are indicated by arrows.
Figure 2.
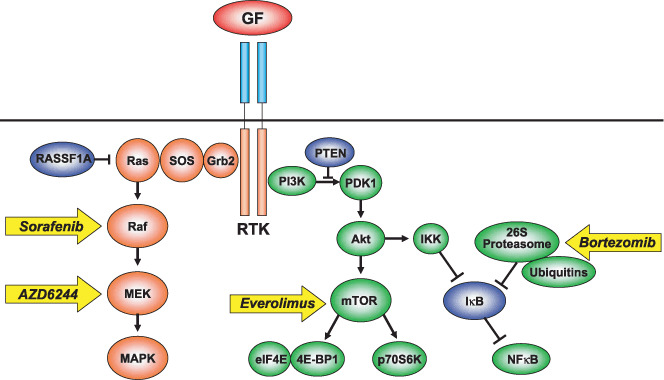
Molecular targets in mitogen‐activated protein kinase (MAPK) and phosphatidylinositol‐3 kinase (PI3K) signal transduction pathways stimulated by receptor tyrosine kinases (RTK). Targeted agents are indicated by arrows. GF, growth factor; SOS, sun of sevenless; MEK, MAPK/extracellular regulated kinase kinase; PTEN, phospharase and tensin homologue; PDK, phosphoinositide‐dependent kinase; IKK, IκB kinase; mTOR, mammalian target of rapamycin.
Figure 3.
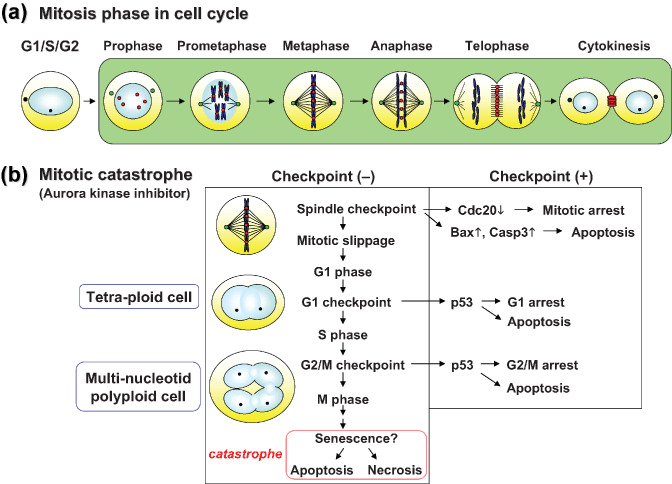
(a) The mitosis phase of the cell cycle. Localization of Aurora kinases is shown. Green spots, the centrosome protein Aurora kinase A; red spots, the chromosomal passenger Aurora kinase B. (b) The concept of mitotic catastrophe, induced by inhibition of Aurora kinases in cancer cells. Under abnormalities in each checkpoint system, p53‐independent death is induced as senescence‐like polyploidy without successfully completing mitosis.
Table 1.
Molecularly targeted agents for hepatocellular carcinoma
| Agent | Classification | Function | Clinical trial |
|---|---|---|---|
| Sorafenib (Nexabar, BAY43‐9006) | Small molecule compound | VEGFR2, VEGFR3, PDGFR‐β tyrosine kinase, Raf serine/threonine kinase inhibitor | Phase III |
| Sunitinib (Sutent, SU11248) | Small molecule compound | VEGFR1, VEGFR2, PDGFR, Flt‐3, c‐KIT tyrosine kinase inhibitor | Phase II |
| SU6688 (TSU‐68) | Small molecule compound | VEGFR2, PDGF R‐β, FGFR tyrosine kinase inhibitor | Phase II |
| Vatalanib (PTK787/ZK222584) | Small molecule compound | VEGFR1, VEGFR2, VEGFR3, PDGFR‐β, c‐KIT tyrosine kinase inhibitor | Phase II |
| Cediranib (AZD2171) | Small molecule compound | VEGFR1, VEGFR2, VEGFR3, PDGFR, c‐KIT tyrosine kinase inhibitor | Phase II |
| Bevacizumab (Avastin) | Monoclonal antibody | VEGF‐A neutralization | Phase II |
| Gefintinib (Iressa, ZD1839) | Small molecule compound | EGFR/ErbB1/Her1 tyrosine kinase inhibitor | Phase II |
| Erlotinib (Tarceva, OSI774) | Small molecule compound | EGFR/ErbB1/Her1 tyrosine kinase inhibitor | Phase II |
| Lapatinib (Tykerb, GW572016) | Small molecule compound | EGFR/ErbB1/Her1 and ErbB2/Her2/Neu tyrosine kinase inhibitor | Phase II |
| BMS‐599626 | Small molecule compound | EGFR/ErbB1/Her1 and ErbB2/Her2/Neu tyrosine kinase inhibitor | Phase II |
| Cetuximab (Erbitux, GW572016) | Monoclonal antibody | EGFR/ErbB1/Her1 neutralization | Phase II |
| AZD6244 (ARRY‐142886) | Small molecule compound | MEK serine‐threonine tyrosine kinase inhibitor | Phase II |
| IMC‐A12 | Monoclonal antibody | IGF‐IR neutralization | Phase II |
| Everolimus (RAD001) | Small molecule compound | mTOR serine‐threonine kinase inhibitor | Phase I and II |
| Sirolimus (Rapamune) | Small molecule compound | mTOR serine‐threonine kinase inhibitor | Phase I |
| Bortezomib (Velcade) | Small molecule compound | Proteasome inhibitor | Phase I and II |
| PXD101 (Belinostat) | Small molecule compound | HDAC inhibitor | Phase I and II |
| PI‐88 | Small molecule compound | Heparanase inhibitor | Phase III |
VEGF and VEGF receptor family
In vivo tumor progression requires various host factors as well as tumor factors; in particular, neovascularization is one of the most important host factors.( 7 ) HCC is well known as one of the tumors to present with typical neovascularization. A dramatic alteration in the arterial hypervascularity is observed in moderately to poorly differentiated‐type HCC, but the new blood vessels are so irregular that the flow is often stagnated.( 7 , 8 ) VEGF is one of the most potent growth factors of the vascular endothelial cells, as well as one of the critical effectors on progenitor cells.( 9 ) The VEGF ligand family consists of VEGF‐A, VEGF‐B, VEGF‐C, VEGF‐D, PIGF‐1, PIGF‐2, and VEGF‐E derived from a virus gene (Fig. 1a).( 10 ) VEGF‐A and VEGF‐B have spliced variants. The currently known VEGF genes and polypeptides belong to a family of structurally and functionally related growth factors, which also includes the platelet‐derived growth factors (PDGF) that mainly function in the vascular mural cells (pericytes). In Drosophila, PEGF and VEGF‐like factors share a single receptor. The human VEGF receptor (VEGFR) family consists of VEGFR‐1/Flt‐1, VEGFR‐2/KDR/Flk‐1, and VEGFR‐3/Flt‐4. Although either VEGFR‐1 or VEGFR‐2 regulates angiogenesis and vasculogenesis, VEGFR‐3 is mainly related to lymphoangiogenesis.( 11 ) We first found a close relationship between VEGF expression and the vascularity of HCC tumors compared that of non‐cancerous liver tissue from a clinical specimen.( 12 ) The expression of VEGF protein was found to correlate with clinicopathological factors such as proliferation, vascular invasion, and tumor multiplicity.( 13 ) VEGF expression was reported to associate with not only invasion and metastasis of HCC,( 14 ) but also postoperative recurrence.( 15 )
Expression of VEGF is regulated by micro‐environmental and genetic alterations in cancer cells. Hypoxia is a key micro‐environmental factor of angiogenesis, and hypoxia‐inducible factors (HIF) are known to stimulate VEGF expression.( 16 , 17 ) The upregulation of VEGF in HCC is controlled by transcriptional levels as well as the mRNA stability of VEGF.( 18 ) In addition, the p53 tumor‐suppressor and HBx genes might regulate VEGF expression in HCC.( 19 , 20 ) Furthermore, we previously identified angiopoietin‐2–Tie2 signaling as another angiogenic pathway essential for HCC progression.( 9 , 21 ) These angiopoietin‐2 signals also require VEGF activation in the angiogenic switch.( 22 )
It should be mentioned that the receptors VEGFR‐1/Flt‐1 and VEGFR‐2/KDR/Flk‐1 have been identified on HCC cells.( 23 ) These findings suggest that VEGF might function in the migration of endothelial cells, as well as in HCC cells per se, indicating a possibly novel mechanism for HCC progression.( 24 ) Recent studies revealed critical roles of VEGFR‐1‐expressing hematopoietic cells in formation of the premetastatic niche.( 25 , 26 ) VEGF signaling functions not only in angiogenesis but also in cancer invasion and metastasis.( 27 ) Given that the VEGF and VEGFR pathways are required for the pathogenesis and progression of HCC, it is possible that inhibitors of VEGF signaling are promising therapeutic agents for HCC treatment.
Most of these compounds can be broadly classified into two main categories: small‐molecule kinase inhibitors and monoclonal antibodies. Sorafenib (Nexavar, BAY43‐9006) is a unique multitargeting small molecule that inhibits the receptor tyrosine kinases (RTK) VEGFR‐2, VEGFR‐3, Flt‐3, PDGF receptor (PDGFR), and fibroblast growth factor receptor (FGFR)‐1, as well as Raf serine‐threonine kinase in the signal transduction pathway Ras–Raf–mitogen‐activated protein kinase/extracellular regulated kinase (MEK) –mitogen‐activated protein kinase (MAPK)( 28 ) (1, 2). In a recent phase III trial, the Sorafenib HCC Assessment Randomized Protocol (SHARP), 602 patients with advanced HCC who had received no prior systemic therapy were evaluated and randomized to receive either sorafenib (n = 299) or placebo (n = 303).( 1 ) The median overall survival was 10.7 months in sorafenib‐treated patients compared with 7.9 months in patients who received placebo, indicating a 44% increase in overall survival (hazard ratio, 0.69; P < 0.0001). The median time to radiological progression was 5.5 months in sorafenib‐treated patients compared with 2.8 months in patients who received placebo (hazard ratio, 0.58; P < 0.0001). The overall incidence of treatment‐related adverse events was 80% in the sorafenib group and 52% in the placebo group. Grade 3 drug‐related adverse events included diarrhea (8% in the sorafenib group vs 2% in the placebo group, P < 0.001), and hand or foot skin reaction (8% in the sorafenib group vs <1% in the placebo group, P < 0.001). Grade 3 or 4 laboratory abnormalities occurred with grade 3 hypophosphatemia (11% in the sorafenib group vs 2% in the placebo group, P < 0.001) and grade 3 or 4 thrombocytopenia (4% in the sorafenib group vs <1% in the placebo group, P = 0.006).
Sunitinib (Sutent, SU11248) is another oral inhibitor that targets RTK of the split‐kinase domain family, including VEGFR‐1, VEGFR‐2, PDGFR‐α, PDGFR‐β, c‐Kit, Flt‐3, and RET.( 29 ) The phase II studies have examined the tolerability and efficacy of sunitinib in patients with advanced HCC.( 30 ) Of the 37 patients enrolled, one patient had a confirmed partial response (PR), and 39% patients had stable disease (SD) as their best response. Grade 3 and 4 toxicities included thrombocytopenia (43%), neutropenia (24%), central nervous system symptoms (24%), asthenia (22%), and hemorrhage (14%). Dose reductions were required in 27% patients. Four patients developed grade 5 events, including ascites, edema, bleeding, drowsiness, and hepatic encephalopathy.
Vatalanib (PTK787/ZK222584) is an oral pan‐VEGFR inhibitor with activity against PDGFR‐β and c‐Kit (Fig. 1a).( 31 ) Preclinical studies suggested anti‐angiogenic and angiogenesis‐independent effects on HCC growth arrest.( 32 ) In a phase I study of vatalanib in 18 patients with unresectable HCC, nine patients had a best response of SD, and nine patients had progressive disease (PD).( 33 ) Cediranib (AZD2171) is another potent pan‐VEGFR inhibitor with activity against PDGFR and c‐Kit.( 34 ) According to a phase II study of cediranib in patients with advanced HCC, 28 patients were accrued, and 19 patients were evaluable for toxicity.( 35 ) Of these, 16 patients (84%) developed grade 3 toxicity. Fatigue, hypertension, and anorexia accounted for the majority of adverse events. A high rate of refusal of further treatment was encountered and apparently was related to the high rate of grade 3 fatigue. A dose reduction of cediranib was planned in an ongoing study. SU6668 (TSU‐68) is a potent RTK inhibitor against VEGFR‐2, PDGFR‐β, and FGFR.( 36 ) According to preliminary data from a Japanese phase I and II study in 15 HCC patients, one patient had PR, seven patients had SD (two patients for over 12 months), and seven patients had PD.( 37 ) Tumor necrosis was observed in eight patients. The adverse events were hypoalbuminemia, diarrhea, abdominal pain, fever, and aspartate aminotransferase (AST) and alanine aminotransferase (ALT) elevation.
Bevacizumab (Avastin), a recombinant, humanized monoclonal antibody that targets VEGF, has emerged as an important therapeutic agent in several malignancies.( 38 ) In addition to its direct antiangiogenic effects, bevacizumab may enhance chemotherapy administration by normalizing tumor vasculature.( 39 ) Several studies have explored the use of bevacizumab either as a single agent or in combination with cytotoxic or molecularly targeted agents in patients with HCC. In a phase I study in 25 patients using single‐agent bevacizumab, two patients had a PR, and 18 patients had SD. The median time to progression was 6.5 months.( 40 ) In a phase II study using single‐agent bevacizumab,( 41 ) among the 24 patients who were evaluable for efficacy, three patients had a PR and seven patients had SD that lasted at least 16 weeks. The combination of bevacizumab with cytotoxic agents also was evaluated. In a recent phase II study, bevacizumab in combination with gemcitabine and oxaliplatin (GEMOX) was used as treatment for patients with advanced HCC.( 42 ) This regimen had moderate antitumor activity in HCC with an overall response rate of 20% in evaluable patients. An additional 27% patients had SD with a median duration of 9 months. The median overall survival was 9.6 months, the median progression‐free survival was 5.3 months, and the progression‐free survival rate at 3 and 6 months approached 70 and 48%, respectively. Bevacizumab‐related side effects, including hypertension, bleeding, and proteinuria, were generally manageable. The encouraging results from that early study should be confirmed cautiously by an independent study in the future.
EGFR/ErbB family
EGFR/ErbB1/Her1 is a member of a RTK family that also includes ErbB2/Her2/Neu, ErbB3/Her3, and ErbB4/Her4 (Fig. 1b).( 43 ) EGFR/ErbB1 binds not only epidermal growth factor (EGF) but also transforming growth factor (TGF)‐α, amphiregulin, HB‐EGF, β‐cellulin, and epiregulin. Although heregulins, epiregulin, and NRG have been identified as ligands of ErbB3/Her3, and heregulins, epiregulin, HB‐EGF, β‐cellulin, and NRG have been identified as ligands of ErbB4/Her4, no ligands have been identified for ErbB2/Her2/Neu. Either EEGFR/ErbB1 or ErbB2/Her2/Neu is overexpressed in various cancers. The binding of ligand to EGFR/ErbB1 leads to homodimerization or heterodimerization with ErbB2/Her2/Neu or ErbB3/Her3, resulting in tyrosine kinase activation and self‐phosphorylation (not in ErbB2/Her3). Activation of RTK transduces the MAPK and phosphatidylinositol‐3 kinase (PI3K) signaling pathways. The MAPK signals mainly stimulate cell proliferation, and the PI3K signals activate Akt anti‐apoptotic pathways. Overexpression of TGF‐α has been observed in the early stages of hepatocarcinogenesis, and is associated with upregulation of VEGF.( 44 )
The crucial role of EGFR in HCC proliferation has provided the rationale for targeting and interrupting this key signaling network. Gefinitib (Iressa, ZD1839) is an oral tyrosine kinase inhibitor that selectively suppresses EGFR and not other tyrosine kinases such as ErbB2 or VEGFR (Fig. 1).( 45 ) According to the clinical trials for non‐small lung cancers, the effects of gefitinib correlate with EGFR mutations in cancer cells.( 46 , 47 ) In a phase II clinical trial of 31 patients with HCC, one patient had PR and seven patients had SD, but there were no clear effects using single administration as the median overall survival was 6.5 months and the median progression‐free survival was 2.8 months.( 48 ) The criterion for second‐stage accrual was not met, and the authors concluded that gefitinib as a single agent was not active in patients with advanced HCC. Erlotinib (Tarceva, OSI774) is another oral EGFR‐selective inhibitor, and two phase II clinical studies have evaluated its safety and efficacy in patients with advanced HCC.( 49 ) In the study by Philip et al., 3 of 38 patients had PR and 12 patients had progression‐free survival at 6 months.( 50 ) The median overall survival for this cohort was 13 months. In another report by Thomas et al. 17 of 40 patients achieved progression‐free survival at 16 weeks and the progression‐free survival rate at 24 weeks was 28%.( 51 ) No PR or complete response (CR) was observed in that study, but the median overall survival was 25 weeks. Lapatinib (Tykerb, GW572016), a selective dual inhibitor of both EGFR and ErbB2 tyrosine kinases, also demonstrated modest activity in HCC in a preliminary report.( 52 ) Among the first 17 patients with advanced HCC, two patients had a confirmed PR and an additional eight patients had SD. However, the progression‐free survival was only 2.3 months in this cohort. For another dual inhibitor, BMS‐599626, a phase II clinical trial is ongoing in patients with HCC.
Cetuximab (Erbitux, IMC‐C225), a chimeric monoclonal antibody against EGFR, has attracted attention in colorectal cancer as a K‐Ras biomarker.( 53 , 54 ) Cetuximab was also tested in two phase II studies in patients with advanced HCC. Zhu et al. reported five patients with SD in 30 patients with advanced HCC.( 55 ) The median overall survival was 9.6 months, and the median progression‐free survival was 1.4 months. Gruenwald et al. reported their preliminary experience of cetuximab in a similarly designed study in patients with HCC.( 56 ) Of the 32 patients who were enrolled, 27 patients were evaluable for efficacy. No responses were observed, and the median time to progression for all patients was 8 weeks. The combination of cetuximab with GEMOX was evaluated in a phase II study.( 57 ) Of the 43 patients who were enrolled, 35 patients were evaluable for efficacy, with a response rate of 23%. Given the known antitumor activity of GEMOX in prior phase II studies and the lack of activity of cetuximab as a single agent, the relative contribution of cetuximab to this regimen remains to be defined. The axis of TGF‐α–EGFR signaling might be an attractive therapeutic target as frequent mutations have not been found in downstream molecules, such as Ras and Raf family members, in HCC.
Insulin‐like growth factor and IGF receptor family
There is compelling evidence that both of the insulin‐like growth factors IGF‐I and IGF‐II and their receptor IGF‐1R are involved in the development and progression of cancer( 58 ) (Fig. 1c). Interaction of IGF‐I and IGF‐II with IGF‐1R plays a pivotal role in the tumorigenesis, proliferation, and spread of many cancers by promoting cell‐cycle progression, preventing apoptosis, and regulating and maintaining the tumorigenic phenotype. A wide variety of tumors (including HCC) show abnormal or enhanced expression of IGF and IGF‐1R, which has been correlated with disease stage, reduced survival, development of metastases, and tumor dedifferentiation.( 59 ) We have previously identified insulin receptor substrate (IRS)‐1, the main substrate of IGF‐1R/insulin receptor (IR),( 60 , 61 ) as an overexpressed molecule with significant roles in hepatocarcinogenesis.( 62 , 63 ) Interestingly, serine phosphorylated IRS‐1 protein by TNF‐α is converted into an inhibitor of IGF‐1R/IR.( 64 ) Obesity and diabetes are clearly associated with an increased risk of HCC, and this seems to be due to alterations in the IGF signaling systems.
Several approaches have demonstrated the therapeutic potential of interfering with IGF‐1R‐mediated signaling in vitro and in vivo. Specific IGF receptor (IGFR) antibodies have also been shown to suppress prostate and breast cancer cell growth in a recent preclinical study.( 65 ) The most advanced clinical anti‐IGFR antibody is IMC‐A12, which is currently being tested in a phase II trial for HCC. Importantly, IGFR inhibition appears to be well tolerated in the preliminary clinical studies conducted so far.( 66 ) Safety is important, as IGFR‐based inhibition has long been regarded as a high‐risk intervention because of the high homology of IGF‐1R with the related IR, and there is a fear that IGF‐1R‐tyrosine kinase inhibitors in particular might also block IR, which could lead to insulin resistance and overt diabetes.( 67 ) However, the current in vivo studies did not confirm this apprehension, resulting in growing interest in anti‐IGFR‐based therapies. OSI‐906, an IGF‐1R tyrosine kinase inhibitor, is currently being tested in phase I trials for solid tumors including pancreatic cancer. The most promising IGF‐ and IGFR‐targeted agents are currently under intense investigation in preclinical and early clinical trials.( 68 )
Intracellular signaling pathways
Activated RTK stimulate several intracellular signal transduction pathways, including Ras–Raf–MEK–MAPK and PI3K–Akt–mammalian target of rapamycin (mTOR) (Fig. 2).( 69 ) In a series of specific phosphorylation events, the adaptor protein Grb2 stimulates sun of sevenless (SOS), leading to the activation of Ras, which is farnesylated and localized under the cell membrane. Farnesyl transferase inhibitors have been used in clinical trials of pancreatic cancer treatments, usually with mutations in the K‐ras oncogene.( 70 ) In spite of the low incidence of Ras gene mutations in HCC, silencing of the RASSF1A gene (a member of the Ras inhibitor family) with DNA methylation was found frequently in human HCC.( 71 ) Inactivation of the Ras inhibitor might result in persistent activation of the downstream pathway during hepatocarcinogenesis. The activated form of Ras then stimulates Raf serine‐threonine kinase. As mentioned above, Raf kinase is one of the targets of the multikinase inhibitor sorafenib.( 1 , 28 ) Activated Raf kinase phosphorylates MEK kinases, which activate the extracellular regulated kinases Erk1/2 of the MAPK family. A MEK kinase inhibitor, AZD6244 (ARRY‐142886), has been evaluated for HCC treatment in a phase II clinical trial. Once activated, Erk1/2 translocates to the nucleus where it acts as a regulator of gene expression, including those for proteins involved in cell cycle progression, apoptosis resistance, and cellular motility.( 72 )
The PI3K–Akt–mTOR pathway has emerged as a contributor to hepatocarcinogenesis.( 73 ) PI3K consists of p85 adaptor and p110 kinase subunits. After association with the intracellular domain of several RTK or specific substrates such as IRS‐1, PI3K phosphorylates phosphatidylinositol 3,4,5‐trisphosphate (PIP3) to generate phosphatidylinositol 4,5‐bisphosphate (PIP2), which transduces phosphoinositide‐dependent kinase (PDK), which in turn activates the serine‐threonine kinase Akt.( 72 ) PIP3 is dephosphorylated by phospharase and tensin homologue (PTEN), a tumor suppressor, which reverses this pathway. Once activated, Akt regulates multiple cellular target proteins, including mTOR, IκB kinase (IKK), Bad, and Gsk3. The mTOR protein regulates phosphorylation of the p70 S6 serine‐threonine kinase and the translational repressor protein 4E‐BP1.( 74 ) Both proteins regulate the translation of proliferative and angiogenic factors, such as c‐myc, cyclin‐D1, and HIF1‐α, and are indirectly involved in the expression of VEGF.( 75 ) The mTOR inhibitors temsirolimus (CCI‐779) and everolimus (RAD001) have been developed as rapamycin derivates.( 76 , 77 ) A phase I clinical trial using rapamycin and bevacizumab and a phase I and II clinical trial using everolimus are ongoing in patients with HCC.( 76 )
Another downstream protein of Akt, IKK, provokes subsequent activation of the transcription factor nuclear factor (NF)‐κB.( 78 ) NF‐κB promotes cell survival by activating transcription of normally repressed target genes by binding the specific inhibitor IκB, which sequesters the NF‐κB p50–p65 heterodimer in the cytoplasm.( 79 ) Inhibition is reversed in response to several intracellular stimuli, resulting in targeted, ubiquitin–proteasome‐mediated degradation of IκB.( 80 ) The proteasome is a 26S multiprotein complex that consists of a 19S regulatory subunit and a 20S catalytic subunit. Ubiquitinated proteins are recognized by the 19S unit, which results in the liberation of ubiquitin chains that are recycled and the formation of a denatured protein that is transferred to the outer ring or the 20S core unit. Bortezomib (Velcade, PS‐341) is a potent and selective inhibitor of the 20S proteasome.( 81 ) The actions of bortezomib are pleiotropic and include inhibition of NF‐κB activation by preventing IκB degradation. 26S proteasome‐mediated protein degradation is a central metabolic and regulatory process in cell physiology. Apart from its role in scavenging damaged proteins, the 26S complex is an important regulator of cell life and fate. For instance, specific ubiquitination of key proteins, such as cyclins A, B, D, and E, eEF2‐kinase, c‐Myc, Notch, c‐Jun, p21WAF/CIP1, p27, p53, topoisomerases I and II, the apoptosis modulators XIAP, Bik/NBK, Bad and Bid, the transcription coactivator β‐catenin, and the NF‐κB regulator IκB, targets these proteins toward proteasome degradation.( 80 ) Through its regulation of protein turnover, the 26S proteasome is thus involved in cell‐cycle progression, apoptosis, and other processes like angiogenesis and cell motility that are important in cancer progression. With the unique and independent anti‐tumor effects of bortezomib, combination therapy with other cytotoxic or molecular‐targeting agents is expected. Bortezomib received approval for second‐line therapy of patients with progressive multiple myeloma,( 80 ) and a phase I and II study for HCC is ongoing.( 82 ) Because abnormalities of intracellular signal transduction pathways should play essential roles in carcinogenesis and cancer progression, further studies should be carried out.
Aurora kinases and mitotic catastrophe
Cell‐cycle checkpoints are pivotal mechanisms safeguarding genomic stability. Cells that harbor defects in checkpoints are predisposed to genomic instability and neoplastic transformation. Of all the different checkpoint controls, the most important one is the mitotic spindle checkpoint, which is considered the primary defense against aneuploidy and ensures accurate chromosome segregation to produce genetically identical daughter cells (Fig. 3a).( 83 ) Among spindle checkpoint kinases, the Aurora family of serine‐threonine kinases has recently emerged as a key mitotic regulator required for genomic stability.( 84 ) Aberrant expression of the Aurora kinase family has been reported in a variety of solid tumors. In mammals, the Aurora family consists of three members: A, B, and C. Aurora kinase A plays important roles in centrosome maturation and separation, and acentrosomal and centrosomal spindle assembly. Recent studies revealed that Aurora kinase A also controls spindle axis orientation during asymmetric division of stem cells. Abnormalities of asymmetric division were detected in Drosophila that had a mutation in the mitotic kinase Aurora A, resulting in massive overproliferation in tumors.( 85 ) Thus, the cell fate of cancer stemness might be regulated by Aurora kinase.
Aurora kinase B, another member of the family, is a chromosomal passenger protein that regulates accurate chromosomal segregation, cytokinesis, protein localization to the centromere and kinetochore, correct microtubule–kinetochore attachments, and regulation of the mitotic checkpoint.( 86 ) Recently, we identified overexpression of Aurora kinase B as the only independent factor predictive of aggressive recurrence of HCC, based on analysis of genome‐wide microarray profiling on clinical samples.( 87 , 88 ) It is of interest that Aurora kinase B overexpression is closely correlated with genetic instability of HCC. Several small‐molecule inhibitors of Aurora kinases have been developed as potential anticancer agents, including ZM447439,( 89 ) hesperadin,( 90 ) VX‐680,( 91 ) PHA‐680632,( 92 ) and MLN 8054.( 93 ) Aurora kinase inhibitors such as AT9283 and AZD1152( 94 ) are currently undergoing phase I clinical evaluation as treatments for malignancies.( 95 )
Aurora kinase B, in particular, may be a suitable anticancer target as its inhibition rapidly results in catastrophic mitosis with senescence.( 95 , 96 ) During mitotic karyokinesis under downregulation of Aurora kinases, a process termed micronucleation occurs in the cancer cells. Thus, unable to maintain G2 arrest, they enter mitosis and after being arrested for several hours at metaphase, they eventually die without successfully completing mitosis. This process is known as p53‐independent cell death or mitotic catastrophe (Fig. 3b).( 96 ) In our studies, a selective Aurora kinase B inhibitor induced in vitro polyploidy of human HCC cells, resulting in mitotic catastrophe. Our preclinical studies using the Aurora kinase B inhibitor revealed remarkable anti‐tumor effects on HCC models in vivo. The inhibitor was well tolerated within the dose range required to elicit a potent and durable effect in mice. Specific inhibition of Aurora kinases is a promising novel therapeutic approach for the treatment of HCC. Further studies and clinical trials of Aurora inhibitors will confirm their significance in HCC therapeutics.
Conclusion
The concept of targeted therapies that specifically inhibit molecular abnormalities has emerged as a promising approach for the innovative and effective medical treatment of various cancers, including HCC. In this regard, sorafenib must throw new light and impact on studies of molecularly targeted agents in HCC.( 6 ) However, although the SHARP study revealed positive and landmark results, the benefits of sorafenib were reported to be relatively modest in patients with HCC.( 1 ) Furthermore, biomarkers to predict its effects are poorly understood. Future research should continue to unravel the mechanism of hepatocarcinogenesis and to identify key relevant molecular targets for therapeutic intervention. The advantages of molecular targeting are being explored in combination treatments as well as adjuvant or neoadjuvant therapies with surgical resection, liver transplantation, radiofrequency and transarterial chemoembolization.( 4 ) We are only now at the beginning of the history of finding novel treatments for HCC.
References
- 1. Llovet JM, Ricci S, Mazzaferro V et al . Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378–90. [DOI] [PubMed] [Google Scholar]
- 2. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006; 6: 674–87. [DOI] [PubMed] [Google Scholar]
- 3. Ince N, Wands JR. The increasing incidence of hepatocellular carcinoma. N Engl J Med 1999; 340: 798–9. [DOI] [PubMed] [Google Scholar]
- 4. Arii S, Yamaoka Y, Futagawa S et al . Results of surgical and nonsurgical treatment for small‐sized hepatocellular carcinomas: a retrospective and nationwide survey in Japan. The Liver Cancer Study Group of Japan. Hepatology 2000; 32: 1224–9. [DOI] [PubMed] [Google Scholar]
- 5. Tanaka S, Sugimachi K, Maehara S et al . Oncogenic signal transduction and therapeutic strategy for hepatocellular carcinoma. Surgery 2002; 131: S142–7. [DOI] [PubMed] [Google Scholar]
- 6. Zhu AX. Development of sorafenib and other molecularly targeted agents in hepatocellular carcinoma. Cancer 2008; 112: 250–9. [DOI] [PubMed] [Google Scholar]
- 7. Tanaka S, Arii S. Current status of perspective of antiangiogenic therapy for cancer: hepatocellular carcinoma. Int J Clin Oncol 2006; 11: 82–9. [DOI] [PubMed] [Google Scholar]
- 8. Hopfner M, Schuppan D, Scherubl H. Growth factor receptors and related signalling pathways as targets for novel treatment strategies of hepatocellular cancer. World J Gastroenterol 2008; 14: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanaka S, Sugimachi K, Yamashita Yi et al . Tie2 vascular endothelial receptor expression and function in hepatocellular carcinoma. Hepatology 2002; 35: 861–7. [DOI] [PubMed] [Google Scholar]
- 10. Shibuya M, Claesson‐Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res 2006; 312: 549–60. [DOI] [PubMed] [Google Scholar]
- 11. Tammela T, Enholm B, Alitalo K, Paavonen K. The biology of vascular endothelial growth factors. Cardiovasc Res 2005; 65: 550–63. [DOI] [PubMed] [Google Scholar]
- 12. Mise M, Arii S, Higashituji H et al . Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996; 23: 455–64. [DOI] [PubMed] [Google Scholar]
- 13. Ng IO, Poon RT, Lee JM, Fan ST, Ng M, Tso WK. Microvessel density, vascular endothelial growth factor and its receptors Flt‐1 and Flk‐1/KDR in hepatocellular carcinoma. Am J Clin Pathol 2001; 116: 838–45. [DOI] [PubMed] [Google Scholar]
- 14. Li XM, Tang ZY, Zhou G, Liu YK, Ye SL. Significance of vascular endothelial growth factor mRNA expression in invasion and metastasis of hepatocellular carcinoma. J Exp Clin Cancer Res 1998; 17: 13–17. [PubMed] [Google Scholar]
- 15. El Assal ON, Yamanoi A, Soda Y et al . Clinical significance of microvessel density and vascular endothelial growth factor expression in hepatocellular carcinoma and surrounding liver: possible involvement of vascular endothelial growth factor in the angiogenesis of cirrhotic liver. Hepatology 1998; 27: 1554–62. [DOI] [PubMed] [Google Scholar]
- 16. Sugimachi K, Tanaka S, Taguchi K, Aishima S, Shimada M, Tsuneyoshi M. Angiopoietin switching regulates angiogenesis and progression of human hepatocellular carcinoma. J Clin Pathol 2003; 56: 854–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yasuda S, Arii S, Mori A et al . Hexokinase II and VEGF expression in liver tumors: correlation with hypoxia‐inducible factor 1 alpha and its significance. J Hepatol 2004; 40: 117–23. [DOI] [PubMed] [Google Scholar]
- 18. Von Marschall Z, Cramer T, Hocker M, Finkenzeller G, Wiedenmann B, Rosewicz S. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut 2001; 48: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsukamoto A, Kaneko Y, Yoshida T, Ichinose M, Kimura S. Regulation of angiogenesis in human hepatomas: possible involvement of p53‐inducible inhibitor of vascular endothelial cell proliferation. Cancer Lett 1999; 141: 79–84. [DOI] [PubMed] [Google Scholar]
- 20. Lee SW, Lee YM, Bae SK, Murakami S, Yun Y, Kim KW. Human hepatitis B virus X protein is a possible mediator of hypoxia‐induced angiogenesis in hepatocarcinogenesis. Biochem Biophys Res Commun 2000; 268: 456–61. [DOI] [PubMed] [Google Scholar]
- 21. Tanaka S, Mori M, Sakamoto Y, Makuuchi M, Sugimachi K, Wands JR. Biologic significance of angiopoietin‐2 expression in human hepatocellular carcinoma. J Clin Invest 1999; 103: 341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holash J, Maisonpierre PC, Compton D et al . Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999; 284: 1994–8. [DOI] [PubMed] [Google Scholar]
- 23. Schmitt M, Horbach A, Kubitz R, Frilling A, Haussinger D. Disruption of hepatocellular tight junctions by vascular endothelial growth factor (VEGF): a novel mechanism for tumor invasion. J Hepatol 2004; 41: 274–83. [DOI] [PubMed] [Google Scholar]
- 24. Arii S. Role of vascular endothelial growth factor on the invasive potential of hepatocellular carcinoma. J Hepatol 2004; 41: 333–5. [DOI] [PubMed] [Google Scholar]
- 25. Kaplan RN, Riba RD, Zacharoulis S et al . VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 2005; 438: 820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour‐mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 2006; 8: 1369–75. [DOI] [PubMed] [Google Scholar]
- 27. Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 2008; 319: 195–8. [DOI] [PubMed] [Google Scholar]
- 28. Wilhelm SM, Carter C, Tang L et al . BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64: 7099–109. [DOI] [PubMed] [Google Scholar]
- 29. Mendel DB, Laird AD, Xin X et al . In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003; 9: 327–37. [PubMed] [Google Scholar]
- 30. Faivre SJ, Raymond E, Douillard JBE et al . Assessment of safety and drug‐induced tumor necrosis with sunitinib in patients (pts) with unresectable hepatocellular carcinoma (HCC). J Clin Oncol 2007; 25 (18S): 3546. [Google Scholar]
- 31. Wood JM, Bold G, Buchdunger E et al . PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor‐induced responses and tumor growth after oral administration. Cancer Res 2000; 60: 2178–89. [PubMed] [Google Scholar]
- 32. Liu Y, Poon RT, Li Q, Kok TW, Lau C, Fan ST. Both antiangiogenesis‐ and angiogenesis‐independent effects are responsible for hepatocellular carcinoma growth arrest by tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res 2005; 65: 3691–9. [DOI] [PubMed] [Google Scholar]
- 33. Koch I, Baron A, Roberts S et al . Influence of hepatic dysfunction on safety, tolerability, and pharmacokinetics of PTK787/ZK 222584 in patients with unresectable hepatocellular carcinoma (HCC). J Clin Oncol 2005; 23 (16S): 4134. [Google Scholar]
- 34. Wedge SR, Kendrew J, Hennequin LF et al . AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor‐2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res 2005; 65: 4389–400. [DOI] [PubMed] [Google Scholar]
- 35. Alberts SR, Morlan BW, Kim GP et al . NCCTG phase II trial (N044J) of AZD2171 for patients with hepatocellular carcinoma (HCC)‐interim review of toxicity. 2007. Gastrointestinal Cancer Symposium; 19–21 Jan 2007, Orlando, FL, USA. Abstract 186.
- 36. Laird AD, Vajkoczy P, Shawver LK et al . SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res 2000; 60: 4152–60. [PubMed] [Google Scholar]
- 37. Kanai F, Yoshida H, Teratani T et al . New feasibility study design with hepatocellular carcinoma: A phase I/II study of TSU‐68, an oral angiogenesis inhibitor. J Clin Oncol 2006; 24 (18S): 4145. [Google Scholar]
- 38. Hurwitz H, Fehrenbacher L, Novotny W et al . Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 39. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307: 58–62. [DOI] [PubMed] [Google Scholar]
- 40. Schwartz JD, Schwartz M, Lehrer D et al . Bevacizumab in unresectable hepatocellular carcinoma (HCC) for patients without metastasis and without invasion of the portal vein. J Clin Oncol 2006; 24 (18S): 4144. [Google Scholar]
- 41. Malka D, Dromain C, Farace F et al . Bevacizumab in patients (pts) with advanced hepatocellular carcinoma (HCC): preliminary results of a phase II study with circulating endothelial cell (CEC) monitoring. J Clin Oncol 2007; 25 (18S): 4570. [Google Scholar]
- 42. Zhu AX, Blaszkowsky LS, Ryan DP et al . Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006; 24: 1898–903. [DOI] [PubMed] [Google Scholar]
- 43. Normanno N, De Luca A, Bianco C et al . Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006; 366: 2–16. [DOI] [PubMed] [Google Scholar]
- 44. Yamaguchi R, Yano H, Iemura A, Ogasawara S, Haramaki M, Kojiro M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology 1998; 28: 68–77. [DOI] [PubMed] [Google Scholar]
- 45. Couzin J. Cancer drugs. Smart weapons prove tough to design. Science 2002; 298: 522–5. [DOI] [PubMed] [Google Scholar]
- 46. Paez JG, Jänne PA, Lee JC et al . EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–5. [DOI] [PubMed] [Google Scholar]
- 47. Lynch TJ, Bell DW, Sordella R et al . Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 48. O'Dwyer PJ, Giantonio BJ, Levy DE, Kauh JS, Fitzgerald DB, Benson AB III. Gefitinib in advanced unresectable hepatocellular carcinoma: results from the Eastern Cooperative Oncology Group's Study E1203. J Clin Oncol 2006; 24 (18S): 4143. 16896003 [Google Scholar]
- 49. Moyer JD, Barbacci EG, Iwata KK et al . Induction of apoptosis and cell cycle arrest by CP‐358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res 1997; 57: 4838–48. [PubMed] [Google Scholar]
- 50. Philip PA, Mahoney MR, Allmer C et al . Phase II study of Erlotinib (OSI‐774) in patients with advanced hepatocellular cancer. J Clin Oncol 2005; 23: 6657–63. [DOI] [PubMed] [Google Scholar]
- 51. Thomas MB, Chadha R, Glover K et al . Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer 2007; 110: 1059–67. [DOI] [PubMed] [Google Scholar]
- 52. Ramanathan RK, Belani CP, Singh DA et al . Phase II study of lapatinib, a dual inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase 1 and 2 (Her2/Neu) in patients (pts) with advanced biliary tree cancer (BTC) or hepatocellular cancer (HCC). A California Consortium (CCC‐P) Trial. J Clin Oncol 2006; 24 (18S): 4010. [Google Scholar]
- 53. Prewett M, Rockwell P, Rockwell RF et al . The biologic effects of C225, a chimeric monoclonal antibody to the EGFR, on human prostate carcinoma. J Immunother Emphasis Tumor Immunol 1996; 19: 419–27. [DOI] [PubMed] [Google Scholar]
- 54. Lièvre A, Bachet JB, Boige V et al . KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26: 374–9. [DOI] [PubMed] [Google Scholar]
- 55. Zhu AX, Stuart K, Blaszkowsky LS et al . Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer 2007; 110: 581–589. [DOI] [PubMed] [Google Scholar]
- 56. Gruenwald V, Wilkens L, Gebel M et al . A phase II open‐label study of cetuximab in unresectable hepatocellular carcinoma: final results. J Clin Oncol 2007; 25 (18S): 4598. [Google Scholar]
- 57. Louafi S, Hebbar M, Rosmorduc O et al . Gemcitabine, oxaliplatin (GEMOX) and cetuximab for treatment of hepatocellular carcinoma (HCC): results of the phase II study ERGO. J Clin Oncol 2007; 25 (18S): 4594. [Google Scholar]
- 58. Sachdev D, Yee D. Disrupting insulin‐like growth factor signaling as a potential cancer therapy. Mol Cancer Ther 2007; 6: 1–12. [DOI] [PubMed] [Google Scholar]
- 59. Scharf JG, Braulke T. The role of the IGF axis in hepatocarcinogenesis. Horm Metab Res 2003; 35: 685–93. [DOI] [PubMed] [Google Scholar]
- 60. Tanaka S, Ito T, Wands JR. Neoplastic transformation induced by insulin receptor substrate‐1 overexpression requires an interaction with Grb2 and Syp signaling molecules. J Biol Chem 1996; 271: 14 610–16. [DOI] [PubMed] [Google Scholar]
- 61. Tanaka S, Smidt EV, Mohr L, Sugimachi K, Wands JR. Biological effects of human insulin receptor substrate‐1 overexpression in hepatocytes. Hepatology 1997; 26: 598–604. [DOI] [PubMed] [Google Scholar]
- 62. Tanaka S, Wands JR. A carboxy‐terminal truncated IRS‐1 dominant negative protein reverses the human hepatocellular carcinoma malignant phenotype. J Clin Invest 1996; 98: 2100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tanaka S, Wands JR. Insulin receptor substrate‐1 overexpression in human hepatocellular carcinoma cells prevents transforming growth factor‐β1 induced apoptosis. Cancer Res 1996; 56: 3391–4. [PubMed] [Google Scholar]
- 64. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS‐1‐mediated inhibition of insulin RTK activity in TNF‐α‐ and obesity‐induced insulin resistance. Science 1996; 271: 665–8. [DOI] [PubMed] [Google Scholar]
- 65. Feng Y, Zhu Z, Xiao X, Choudhry V, Barrett JC, Dimitrov DS. Novel human monoclonal antibodies to insulin‐like growth factor (IGF)‐II that potently inhibit the IGF receptor type I signal transduction function. Mol Cancer Ther 2006; 5: 114–20. [DOI] [PubMed] [Google Scholar]
- 66. Burtrum D, Zhu Z, Lu D et al . A fully human monoclonal antibody to the insulin‐like growth factor I receptor blocks ligand‐dependent signaling and inhibits human tumor growth in vivo . Cancer Res 2003; 63: 8912–21. [PubMed] [Google Scholar]
- 67. Garber K. IGF‐1: old growth factor shines as new drug target. J Natl Cancer Inst 2005; 97: 790–2. [DOI] [PubMed] [Google Scholar]
- 68. Desbois‐Mouthon C, Cacheux W, Blivet‐Van Eggelpoel MJ et al . Impact of IGF‐1R/EGFR cross‐talks on hepatoma cell sensitivity to gefitinib. Int J Cancer 2006; 119: 2557–66. [DOI] [PubMed] [Google Scholar]
- 69. Pawson T. Specificity in signal transduction: from phosphotyrosine‐SH2 domain interactions to complex cellular systems. Cell 2004; 116: 191–203. [DOI] [PubMed] [Google Scholar]
- 70. Macdonald JS, McCoy S, Whitehead RP et al . A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs 2005; 23: 485–7. [DOI] [PubMed] [Google Scholar]
- 71. Schagdarsurengin U, Wilkens L, Steinemann D et al . Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene 2003; 22: 1866–71. [DOI] [PubMed] [Google Scholar]
- 72. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006; 6: 674–87. [DOI] [PubMed] [Google Scholar]
- 73. Luo J, Manning BD, Cantley LC. Targeting the PI3K–Akt pathway in human cancer: rationale and promise. Cancer Cell 2003; 4: 257–62. [DOI] [PubMed] [Google Scholar]
- 74. Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med 2007; 13: 433–42. [DOI] [PubMed] [Google Scholar]
- 75. Frost P, Shi Y, Hoang B, Lichtenstein A. AKT activity regulates the ability of mTOR inhibitors to prevent angiogenesis and VEGF expression in multiple myeloma cells. Oncogene 2007; 26: 2255–62. [DOI] [PubMed] [Google Scholar]
- 76. Raymond E, Alexandre J, Faivre S et al . Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI‐779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol 2004; 22: 2336–47. [DOI] [PubMed] [Google Scholar]
- 77. Beuvink I, Boulay A, Fumagalli S et al . The mTOR inhibitor RAD001 sensitizes tumor cells to DNA‐damaged induced apoptosis through inhibition of p21 translation. Cell 2005; 120: 747–59. [DOI] [PubMed] [Google Scholar]
- 78. Luo JL, Kamata H, Karin M. IKK/NF‐κB signaling: balancing life and death – a new approach to cancer therapy. J Clin Invest 2005; 115: 2625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKβ couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005; 121: 977–90. [DOI] [PubMed] [Google Scholar]
- 80. Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol 2005; 23: 630–9. [DOI] [PubMed] [Google Scholar]
- 81. Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS‐341 in cancer therapy. Clin Cancer Res 1999; 5: 2638–45. [PubMed] [Google Scholar]
- 82. Hegewisch‐Becker S, Sterneck M, Schubert U et al . Phase I/II trial of botrezomib in patients with unresectable hepatocellular carcinoma. J Clin Oncol 2004; 22 (15S): 4089. [Google Scholar]
- 83. Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev 2007; 17: 60–5. [DOI] [PubMed] [Google Scholar]
- 84. Keen N, Taylor S. Aurora‐kinase inhibitors as anticancer agents. Nat Rev Cancer 2004; 4: 927–36. [DOI] [PubMed] [Google Scholar]
- 85. Lee CY, Andersen RO, Cabernard C et al . Drosophila Aurora‐A kinase inhibits neuroblast self‐renewal by regulating aPKC/Numb cortical polarity and spindle orientation. Genes Dev 2006; 20: 3464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 2007; 8: 798–812. [DOI] [PubMed] [Google Scholar]
- 87. Tanaka S, Noguchi N, Ochiai T et al . Outcomes and recurrence of initially resection of hepatocellular carcinoma meeting Milan criteria: Rationale for partial hepatectomy as first strategy. J Am Coll Surg 2007; 204: 1–6. [DOI] [PubMed] [Google Scholar]
- 88. Tanaka S, Arii S, Yasen M et al . Aurora kinase B is a predictive factor for the aggressive recurrence of hepatocellular carcinoma after curative hepatectomy. Br J Surg 2008; 95: 611–19. [DOI] [PubMed] [Google Scholar]
- 89. Gadea BB, Ruderman JV. Aurora kinase inhibitor ZM447439 blocks chromosome‐induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol Biol Cell 2005; 16: 1305–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hauf S, Cole RW, LaTerra S et al . The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 2003; 161: 281–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Harrington EA, Bebbington D, Moore J et al . VX‐680, a potent and selective small‐molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo . Nat Med 2004; 10: 262–7. [DOI] [PubMed] [Google Scholar]
- 92. Soncini C, Carpinelli P, Gianellini L et al . PHA‐680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res 2006; 12: 4080–9. [DOI] [PubMed] [Google Scholar]
- 93. Hoar K, Chakravarty A, Rabino C et al . MLN8054, a small‐molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol 2007; 27: 4513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wilkinson RW, Odedra R, Heaton SP et al . AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res 2007; 13: 3682–8. [DOI] [PubMed] [Google Scholar]
- 95. Girdler F, Gascoigne KE, Eyers PA et al . Validating Aurora B as an anti‐cancer drug target. J Cell Sci 2006; 119: 3664–75. [DOI] [PubMed] [Google Scholar]
- 96. Vakifahmetoglu V, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ 2008; 15: 1153–62. [DOI] [PubMed] [Google Scholar]