

Abstract
E7070 is a novel sulfonamide anticancer agent that disrupts the G1/S phase of the cell cycle. The objectives of this phase I study of E7070 were to estismate the maximal tolerated dose (MTD), to determine the recommended dose for phase II, and to clarify the pharmacokinetic profile of E7070 and its relation to polymorphisms of CYP2C9 (*2, *3) and CYP2C19 (*2, *3) in Japanese patients. Patients received 1–2‐h i.v. infusions of E7070 (400, 600, 700, 800 or 900 mg/m2) on day 1 of a 21‐day cycle. Twenty‐one patients received between one and eight cycles of E7070. The dose‐limiting toxicities (DLT) comprised leukopenia, neutropenia, thrombocytopenia, elevation of aspartate aminotransferase, colitis, and ileus. The mean area under the plasma concentration–time curve (AUC) for successive dose levels increased in a non‐dose‐proportional manner. Two patients were heterozygous for the CYP2C9 mutation. For CYP2C19, eight patients were wild type and the remainder had heterozygous (n = 8) or homozygous mutations (n = 5). Regarding the CYP2C19 genotype, the AUC of patients with mutant alleles were higher than those of patients with wild type at a dose of 600 mg/m2 or more. The severity of toxic effects, such as myelosuppression, seemed to depend on the AUC. No partial responses were observed. One patient treated at a dose of 700 mg/m2 experienced a maximum tumor volume reduction of 22.5%. The MTD was estimated to be 900 mg/m2. A dose of 800 mg/m2 is recommended for further phase II studies. The pharmacokinetic/pharmacodynamic properties of E7070 seemed to be influenced by CYP2C19 genotype. The observed safety profile and preliminary evidence of antitumor activity warrant further investigation of this drug in monotherapy or in combination chemotherapy. (Cancer Sci 2005; 96: 721 –728)
E7070, N‐(3‐chloro‐7‐indolyl)‐1,4‐benzenedisulfonamide, has been reported to exhibit a potent antitumor activity by blocking cell cycle progression. In vitro studies indicate that the drug disrupts the G1/S phase, thereby inducing cell cycle arrest and apoptosis.( 1 ) Although E7070 is not a direct inhibitor of cyclin‐dependent kinases (CDK), it causes depletion of cyclin E, with a reduction in CDK2 catalytic activity.( 2 ) The exact mechanism of cyclin E/CDK2 inactivation is unclear. Transcriptional repression of cyclin H in a p53‐independent manner also occurs in response to E7070.( 3 ) The reduction in G1 CDK activity induces arrest at the G1/S boundary, accompanied by hypophosphorylation of retinoblastoma (Rb) protein and decreases in CDK2, cyclin A, and cyclin B proteins.( 1 ) E7070 activity is associated with upregulation of p53 and p21, which may contribute to the reduced Rb phosphorylation, as well as subsequent apoptosis. In preclinical models, E7070 was cytotoxic to human HCT116 colon carcinoma and LX‐1. E7070 exhibits more potent in vivo antitumor effects than 5‐fluorouracil, mitomycin C, and irinotecan.( 4 )
E7070 has been the subject of four clinical phase I studies. In the first trial, E7070 was administered once every 21 days at doses between 50 and 1000 mg/m2,( 5 ) and in the second trial E7070 was administered five times per day once every 3 weeks at doses between 10 and 200 mg/m2 per day.( 6 ) Other schedules used were a weekly infusion given over 4 consecutive weeks repeated every 6 weeks,( 7 ) and a continuous intravenous infusion for 5 days every 3 weeks.( 8 ) In the single‐dose every 3 weeks study, neutropenia and thrombocytopenia were dose‐limiting at 700 and 800 mg/m2.( 5 ) In the second study, neutropenia and thrombocytopenia were dose‐limiting at 160 and 200 mg/m2.( 6 ) In the study that used a weekly dose schedule, neutropenia and thrombocytopenia were also dose‐limiting toxicities (DLT) and other DLT included stomatitis, diarrhea, nausea, and fatigue.( 7 ) Partial responses were observed in patients with breast and endometrial cancer.( 6 , 7 )
During a phase I trial, three patients receiving prophylactic daily oral maintenance therapy with acenocoumarol developed a hemorrhagic tendency and/or a prolonged prothrombin time following treatment with 700 and 800 mg/m2 of E7070.( 5 ) The major metabolic enzyme for acenocoumarol is cytochrome P450 (CYP)2C9.( 9 ) In vitro studies have shown that E7070 has the potential to inhibit CYP2C9 and CYP2C19.( 10 ) The pharmacokinetic drug–drug interaction study indicated that primary interaction of the two drugs could occur via inhibition by E7070 of acenocoumarol metabolism.
Based on these results from the previous phase I and pharmacokinetic trials, the present phase I study was designed to evaluate ascending doses of E7070 administered as a single dose by 1–2‐h i.v. infusion every 21 days. The objectives of the study were to determine the maximum tolerated dose (MTD) and the dose to be recommended for use in future phase II studies, as well as to assess the safety, pharmacokinetic profile and preliminary antitumor activity of the drug. We also evaluated the influence of genetic polymorphisms of CYP2C9 and CYP2C19 on the pharmacokinetics of E7070.
Materials and Methods
Patient selection
Japanese patients with histologically or cytologically confirmed malignant solid tumors refractory to conventional chemotherapy, or tumors for which no effective therapy was available, were candidates for this study. Eligibility criteria included the following: age between 20 and 75 years; World Health Organization (WHO) performance status 0–1, life expectancy ≥ 3 months, absolute leukocyte count ≥ 4000/µL and < 12 000/µL, absolute neutrophil count ≥ 2000/µL, hemoglobin level ≥ 9 g/dL, platelet count ≥ 100 000/µL, serum creatinine level <1.5 mg/dL or creatinine clearance ≥ 50 mL/min, and arterial partial pressure of oxygen of 65 torr or more. Additional entry criteria were serum bilirubin ≤ 1.5 mg/dL, and serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤ 100 IU/L. Before study entry, a 6‐week interval was required for patients previously treated with mitomycin C or nitrosoureas, a 4‐week interval was required for other chemotherapy, endocrinotherapy, surgery, radiation therapy, immunotherapy treatments or other investigational agents, and a 2‐week interval after blood transfusion or administration of granulocyte‐colony stimulating factor (G‐CSF). Patients were ineligible for the study if they had symptomatic central nervous system metastases, active infection, or other non‐malignant disease, which was considered to be incompatible with the protocol. Patients who were receiving corticosteroids or coumarin anticoagulants less than 2 weeks prior to administration of E7070 were excluded from the study. The protocol was approved by the institutional review boards of the National Cancer Center, and all patients gave written informed consent prior to study entry.
Dosage and dose escalation
E7070 was provided as a lyophilized powder in 500‐mg vials by Eisai Co. (Tokyo, Japan). The starting dose of E7070 was set at 400 mg/m2 because only mild to moderate grade 1 to grade 2 toxicity was observed at doses of 600 mg/m2 or lower in the previous phase I study.( 5 ) Subsequent doses were to be escalated to 600, 700, 800 and 900 mg/m2. E7070 was dissolved in 20 mL of distilled water, then added to 500 mL of normal saline for injection, and this solution was administered by intravenous infusion over 1 h at doses of 400 and 600 mg/m2. Injection site reaction was observed in one of three patients at 400 mg/m2 and three of three patients at 600 mg/m2. Therefore, E7070 was given in 1000 mL of normal saline over 2 h at 700, 800, and 900 mg/m2. Patients were hospitalized for the first course of E7070 and remained hospitalized for close observation for 21 days thereafter. Subsequent courses could be administered on an outpatient basis with weekly patient evaluations by the investigator.
Patients were enrolled in cohorts of three patients per dose level and observed for 21 days; the observation period was extended to 42 days if a longer recovery period was needed. If one of the three patients experienced DLT, then three additional patients were treated at the same dose. If two or more of three or six patients experienced DLT, that dose level was regarded as the MTD. If none of the first three patients demonstrated DLT, then the next three patients were treated at the next (higher) dose level. Individual patients who did not demonstrate DLT and showed no evidence of disease progression could receive E7070 at the originally assigned dose.
After the MTD had been determined, a dose below the MTD was evaluated in a total of six patients for identification of the proposed recommended dose for phase II study. The recommended dose was the highest dose at which less than 33.3% of treated patients experienced DLT.
Definition of dose‐limiting toxicity
The DLT was defined as the occurrence of any of the following events during cycle 1 that were attributable to E7070: National Cancer Institute Common Toxicity Criteria (NCI CTC) (version 2.0) grade 3 or 4 non‐hematological toxicity (except nausea, vomiting effectively managed with symptomatic treatment, or alopecia), grade 4 leukopenia, grade 4 neutropenia accompanied by fever of ≥ 38.5°C, or that persisted ≥ 5 days, and platelet count < 25 000/µL. Prophylactic use of colony‐stimulating factors was not permitted during the first cycle; however, patients who had neutropenia that had met the criteria for DLT were permitted to receive concomitant treatment with G‐CSF.
Evaluation of patients
Safety was evaluated on the basis of physical findings, vital signs, adverse events, and laboratory parameters obtained at baseline and periodically throughout the study. During the first cycle, hematology studies were performed at least twice a week, while vital signs, physical examinations (including evaluation of performance status) and serum biochemistry were measured on days 1, 8 and 15. Toxicity evaluations of subjective and objective findings were performed according to the NCI CTC (version 2.0) on days 1, 8 and 15. For the second and subsequent cycles, vital signs, laboratory tests and toxicity evaluations based on subjective and objective findings were performed on days 1, 8, and 15 of each cycle. Blood glucose was monitored before the dose of E7070 and after the end of infusion. Blood coagulation studies were carried out before each dose for all cycles and also at day 8 of the first cycle. Efficacy was assessed by the physician on the basis of antitumor effect according to the Response Evaluation Criteria in Solid Tumors (RECIST).( 11 ) If an antitumor effect was observed, the disease site was re‐evaluated at least 4 weeks later to confirm the response.
Pharmacokinetics
Pharmacokinetic studies were performed during the first cycle of treatment. On day 1, blood samples (4 mL each) were drawn from an indwelling intravenous cannula in the arm contralateral to that bearing the infusion line. Samples were collected in heparinized tubes, preinfusion, at 30 min after the start of the infusion, at the end of the 1‐ or 2‐h infusion, and at 10, 30, and 60 min and 2, 4, 6, 10, 24, 48, 72, 96, 168, and 240 h after the infusion. The samples were centrifuged at 1500× g for 10 min at 5°C, and the resulting plasma samples were stored at −20°C until analysis. Urine samples were collected before the start of E7070 infusion and over three 24‐h intervals for 72 h after the start of the infusion. The concentrations of E7070 in plasma and in urine were analyzed at Eisai Co. by means of validated high‐performance liquid chromatography methods with UV detection (HPLC‐UV). The lower limit of quantification was 20 ng/mL. N‐(3‐Chloro‐7‐indolyl)‐4‐(N‐methylsulfamoyl)benzenesulfonamide (ER‐67771)( 12 ) was used as an internal standard.
Plasma, the internal standard and 0.1 mol/L phosphate buffer (pH 6.8) were vortexed. After adding ethyl acetate, the mixture was shaken and centrifuged. The organic layer was collected and transferred into a glass tube, then evaporated under nitrogen flow in a drying block. The residue was dissolved in CH3CN‐6.7 mmol/L phosphate buffer (pH 6.6), and the solution was injected into an HPLC apparatus.
Chromatographic separation was achieved by using a column switching method with Asahipak C8P‐50 (Showa Denko, Tokyo, Japan) as a separation column and YMC‐pack ODS‐AM‐312 (YMC) as an analytical column. Mobile phases were CH3CN: 6.7 mmol/L phosphate buffer (pH 6.6; 360:640 [v:v]) for separation and CH3CN: 6.7 mmol/L phosphate buffer (pH 7.4; 360:640 [v:v]) for analysis. E7070 was monitored by UV detection at 280 nm.
Pharmacokinetic parameters of E7070 after a single dose administration during the first cycle were determined by non‐compartmental analysis using WinNONLIN (Pharsight Corporation, CA, USA). The apparent elimination rate constant at the terminal phase (λz) was estimated by linear regression analysis from the terminal log‐linear declining phase to the last quantifiable concentration. The elimination half‐life (t1/2) was calculated as t1/2 = ln(2)/λz. The area under the plasma concentration–time curve from zero to the last quantifiable sampling time, AUC(0–tn), was obtained by the log trapezoidal rule. The AUC from zero to infinity was calculated as AUC(0–tn)+ Cn/λz, where Cn was the last quantifiable concentration. The clearance was calculated as dose/AUC. The mean residence time (MRT) was estimated from AUMC/AUC, where AUMC is the first moment curve. The volume of distribution was calculated as MRT × clearance.
Genotyping procedures for CYP2C9 and CYP2C19
Genotyping was conducted using the Invader assay (BML, Tokyo, Japan). Genomic DNA was isolated from whole blood with the QIAamp DNA Blood Kit (Qiagen, CA, USA). The primary probes (wild type and mutant probes) were used to detect C430T (*2) and A1075C (*3) mutations of CYP2C9, and G681A (*2) and G636A (*3) of CYP2C19, respectively. The invader assay fluorescent resonance energy transfer (FRET)‐detection 96‐well plates (Third Wave Technologies, WI, USA) contained Cleavase enzyme, FRET probes, MOPS buffer and polyethylene glycol. Eight microliters of mixtures consisting of an appropriate primary probe, Invader oligonucleotide and MgCl2 was added to the wells, followed by addition of 7 µL of the heat‐denatured genomic DNA, and this was overlaid with 15 µL of mineral oil. For only CYP2C9*2 (C430T) detection, a dilution of the CYP2C9‐specific polymerase chain reaction (PCR) product was used instead of genomic DNA, because the CYP2C9*2 (C430T) detection point has the same sequence on CYP2C19. The plates were incubated at 63°C for 4 h for genomic DNA or 1 h for the PCR product. The fluorescence intensities were measured on a Cytofluor 4000 fluorescence plate reader (Applied Biosystems, CA, USA) with excitation at 485/20 nm (wavelength/bandwidth) and emission at 530/25 nm for FAM dye detection, and excitation at 560/20 nm and emission at 620/40 nm for RED dye detection.
Subjects having either the *2 or *3 allele (*1/*2 or *1/*3) were defined as hetero extensive metabolizers (hetero EM), those with two mutated alleles (*2/*2, *2/*3 or *3/*3) as poor metabolizers (PM), and those with no mutated alleles (*1/*1) as homo EM.
Results
Patients’ characteristics
Twenty‐one patients (15 male and six female) were enrolled into the study (Table 1). All patients had a good performance status and had received previous chemotherapy regimens. The colon/rectum was the most commonly noted primary disease site. All patients were evaluable for toxicity during the first cycle, and for efficacy. Twenty‐one patients received 42 cycles of treatment. The median number of cycles administered per patient was one (range, 1–8).
Table 1.
Patients’ characteristics
| No. entered | 21 |
| Age (years) | |
| Median | 57 |
| Range | 35–70 |
| Male:female (no. patients) | 15:6 |
| WHO performance status (no. patients) | |
| 0 | 7 |
| 1 | 14 |
| Tumor type (no. of patients) | |
| Colorectal | 15 |
| SCLC | 2 |
| Gastric | 1 |
| NSCLC | 1 |
| Liposarcoma | 1 |
| Mesothelioma | 1 |
| Prior treatment | |
| Chemotherapy | |
| No. prior regimens (no. patients) | |
| 0 | 0 |
| 1 | 1 |
| 2 | 5 |
| >3 | 15 |
| Surgery (no. patients) | 18 |
| Radiation therapy (no. patients) | 7 |
SCLC, small cell lung cancer; NSCLC, non‐small cell lung cancer; WHO, World Health Organization.
Dose escalation and identification of DLT, MTD, and the recommended phase II dose
The DLT in this study were leukopenia, neutropenia, thrombocytopenia, elevation of AST, colitis, and ileus. None of the patients treated at any dose of less than 800 mg/m2 experienced DLT. At a dose of 900 mg/m2, two of three patients experienced dose‐limiting leukopenia, neutropenia, and thrombocytopenia, identifying this dose level as the MTD. At the same dose, grade 3 colitis and grade 3 AST elevation were observed in one patient. Therefore, three additional patients were enrolled at 800 mg/m2. One of the additional three patients evaluated for safety at 800 mg/m2 experienced DLT of grade 3 ileus. This patient had previously undergone intestinal surgery for colon cancer. On the basis of these findings, a total of six patients were treated at the dose of 800 mg/m2 and one of the six patients experienced DLT. Thus, based on protocol‐defined criteria, the MTD was estimated to be 900 mg/m2. Therefore, a dose of 800 mg/m2 is the recommended dose for single‐agent phase II studies.
Hematological toxicity
Neutropenia, leukopenia, and thrombocytopenia were the hematological toxicities observed most commonly during the first cycle (Table 2). Neutropenia was the principal hematological toxicity in this study and was dose‐limiting at 900 mg/m2. Eight patients treated at 600, 800 and 900 mg/m2 experienced grade 3 or more neutropenia. In these patients the median times to nadir neutrophil counts were 12.5 (8–25) days in the first cycle and 15.5 (8–25) days in all cycles, and the median times to recovery from nadir to grade 1 were 5.0 (3–15) days in the first cycle and 6.0 (3–15) days in all cycles. Neutrophil counts recovered to grade 1 within 21 days after E7070 infusion in all patients treated with 400 mg/m2, but had not recovered by day 22 in two, one and two patients at 600, 700 and 800 mg/m2, respectively. Neutrophil counts recovered by day 29 after E7070 infusion in all patients. G‐CSF support was provided during cycle 1 in two of three patients treated at 900 mg/m2. One patient treated at 800 mg/m2 and three patients treated at 900 mg/m2 experienced grade 3 thrombocytopenia. In patients treated at 800 and 900 mg/m2, the median time to nadir platelet counts was 10.0 (7–12) days, and the median time to recovery from nadir to grade 1 was 5.0 (2–9) days in the first cycle. Anemia, reported in 13 (62%) patients, did not exceed grade 1–2 severity except in two patients at 800 and 900 mg/m2. The numbers of patients with blood cell count toxicity did not tend to increase with increasing number of courses of treatment, suggesting that the hematological toxicity of E7070 is not cumulative.
Table 2.
Hematological toxicities during the first cycle of treatment with E7070
| Toxicity | Grade | Dose (mg/m2) | ||||
|---|---|---|---|---|---|---|
| 400 (n = 3) | 600 (n = 3) | 700 (n = 6) | 800 (n = 6) | 900 (n = 3) | ||
| Neutropenia | 1/2 | 0 | 0 | 3 | 1 | 0 |
| 3/4 | 0 | 2 | 0 | 3 | 3 | |
| Leukopenia | 1/2 | 1 | 2 | 3 | 3 | 1 |
| 3/4 | 0 | 0 | 0 | 2 | 2 | |
| Thrombocytopenia | 1/2 | 0 | 2 | 1 | 2 | 0 |
| 3/4 | 0 | 0 | 0 | 1 | 3 | |
| Anemia | 1/2 | 1 | 2 | 4 | 2 | 2 |
| 3/4 | 0 | 0 | 0 | 1 | 1 | |
Non‐hematological toxicity
The non‐hematological toxicities reported commonly during the first cycle were rash, fatigue, stomatitis, alopecia, injection site reaction, diarrhea and constipation (Table 3). These toxicities were generally mild. Grade 3 and grade 4 toxicities were reported in patients treated with 800 or 900 mg/m2. Grade 3 ileus and grade 4 constipation associated with the ileus developed in one patient at 800 mg/m2. Grade 3 AST elevation, grade 3 colitis and grade 3 diarrhea accompanying the colitis were observed in one patient at 900 mg/m2. The toxicities reported most commonly in subsequent cycles were similar in terms of number of patients affected and severity to those reported during the first cycle of treatment.
Table 3.
Non‐hematological toxicities during the first cycle of treatment with E7070
| Toxicity | Grade | Dose (mg/m2) | ||||
|---|---|---|---|---|---|---|
| 400 (n = 3) | 600 (n = 3) | 700 (n = 6) | 800 (n = 6) | 900 (n = 3) | ||
| Diarrhea | 1/2 | 0 | 0 | 0 | 5 | 2 |
| 3 | 0 | 0 | 0 | 0 | 1 | |
| Constipation | 1/2 | 0 | 1 | 2 | 3 | 1 |
| 3/4 | 0 | 0 | 0 | 1 | 0 | |
| Nausea | 1/2 | 0 | 1 | 3 | 1 | 1 |
| 3 | 0 | 0 | 0 | 1 | 0 | |
| Vomiting | 1/2 | 0 | 1 | 0 | 0 | 0 |
| 3 | 0 | 0 | 0 | 1 | 0 | |
| Anorexia | 1/2 | 0 | 1 | 2 | 1 | 1 |
| 3 | 0 | 0 | 0 | 1 | 0 | |
| Stomatitis | 1/2 | 1 | 1 | 2 | 3 | 3 |
| 3 | 0 | 0 | 0 | 0 | 0 | |
| Injection site reaction | 1/2 | 1 | 3 | 3 | 2 | 0* |
| 3 | 0 | 0 | 0 | 0 | 0* | |
| Rash | 1/2 | 1 | 1 | 5 | 2 | 3 |
| 3 | 0 | 0 | 0 | 0 | 0 | |
| Fatigue | 1/2 | 1 | 2 | 2 | 3 | 3 |
| 3 | 0 | 0 | 0 | 0 | 0 | |
| Headache | 1/2 | 1 | 1 | 4 | 2 | 0 |
| 3 | 0 | 0 | 0 | 0 | 0 | |
| Alopecia | 1/2 | 0 | 1 | 2 | 3 | 3 |
E7070 was administered through a central vein at a dose of 900 mg/m2.
Gastrointestinal toxicity, usually mild, was the most common non‐hematological toxicity associated with E7070. Diarrhea (grades 1–3) was noted in eight (38%) patients during the first cycle, and the incidence was greater at the 800 mg/m2 (5/6) and 900 mg/m2 (3/3) doses than at the 400–600 mg/m2 doses (none). Severe diarrhea (grade 3) was observed in only one patient, who received 900 mg/m2 and had previously undergone surgery for primary colorectal cancer. In almost all cases, nausea and vomiting responded well to antiemetic therapies and patients were able to maintain good oral intake. Mild constipation (grades 1–2) was noted at 600–900 mg/m2, except for one patient with grade 4 constipation associated with grade 3 ileus at 800 mg/m2. Alopecia was observed in nine (43%) patients. Grades 1–2 injection site reaction, including irritation, pain, or phlebitis, developed in one patient at 400 mg/m2 and three patients at 600 mg/m2. Therefore, E7070 in 1000 mL of normal saline was given over 2 h at 700, 800, and 900 mg/m2. However, three patients at 700 mg/m2 and two patients at 800 mg/m2 showed injection site reaction, and thus E7070 was given to patients at 900 mg/m2 through a central vein. There were no deaths within 28 days of E7070 administration, and none of the deaths that occurred after the study was considered to have been treatment‐related.
Pharmacokinetics
Complete pharmacokinetic data sets were obtained in 21 patients. The mean (+SD) plasma concentration‐time curves of E7070 are shown in Figure 1. The mean (± SD) pharmacokinetic parameters derived from the plasma concentration are listed in Table 4. After the end of the infusion, plasma concentration of E7070 decreased rapidly for several hours, followed by a slower elimination phase (Fig. 1). During the elimination phase, the E7070 plasma concentration‐time profile was convex, which is characteristic of non‐linear pharmacokinetics. Maximum plasma concentrations (Cmax) of E7070 at the 700–900 mg/m2 doses were lower than that at 600 mg/m2 (Table 4). This is probably related to the change of the infusion time of E7070 from 1 h to 2 h at doses over 600 mg/m2 because of injection site reaction. The AUC increased more than expected with increasing dose. The clearance decreased between 400 and 900 mg/m2, with mean values of 6.3 mL/min per m2 to 2.5 mL/min per m2. The mean plasma half‐life (t1/2) was between 20 and 32 h at the examined doses. Mean 72‐h urinary excretion was 0.82% to 2.47% of the administered dose of E7070 in the five cohorts.
Figure 1.
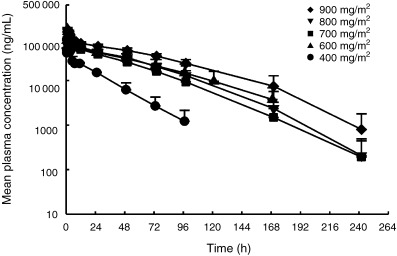
Mean plasma concentrations of E7070 after single intravenous infusion at each dose level. Circles, 400 mg/m2; triangles, 600 mg/m2; squares, 700 mg/m2; inverted triangles, 800 mg/m2; diamonds, 900 mg/m2. Each point represents the mean with standard deviation.
Table 4.
Pharmacokinetic parameters of E7070
| Dose (mg/m2) | No. patients | Cmax (µg/mL) | AUC (µg·h/mL) | CL (mL/min per m2) | MRT (h) | t1/2 (h) | Vss (L/m2) | Urinary excretion (%) |
|---|---|---|---|---|---|---|---|---|
| 400 | 3 | 82.2 ± 15.4 | 1066 ± 140 | 6.3 ± 0.9 | 26 ± 8 | 20 ± 5 | 9.8 ± 1.6 | 0.82 ± 0.22 |
| 600 | 3 | 142.8 ± 12.3 | 4204 ± 1353 | 2.6 ± 1.0 | 53 ± 17 | 32 ± 11 | 7.6 ± 0.0 | 1.67 ± 0.13 |
| 700 | 6 | 116.1 ± 11.3 | 3300 ± 1058 | 3.9 ± 1.3 | 41 ± 12 | 21 ± 7 | 8.7 ± 0.9 | 1.57 ± 0.39 |
| 800 | 6 | 117.7 ± 8.6 | 3943 ± 1243 | 3.6 ± 1.0 | 45 ± 11 | 22 ± 4 | 9.2 ± 0.8 | 1.77 ± 0.80 |
| 900 | 3 | 133.8 ± 0.7 | 6095 ± 1009 | 2.5 ± 0.4 | 59 ± 10 | 27 ± 8 | 8.7 ± 0.7 | 2.47 ± 1.33 |
Cmax, maximum plasma concentration; AUC, area under the plasma concentration–time curve; CL, clearance; MRT, mean residence time; t1/2, terminal elimination half‐life; Vss, distribution volume at steady state; urinary excretion, cumulative excreted amount of E7070 in urine.
Pharmacodynamics
The pharmacodynamic analysis was performed by focusing on leukopenia, neutropenia and thrombocytopenia, because these were the DLT of E7070. Figure 2 shows that the nadirs of white blood cells (WBC), neutrophils, and platelets were related to the AUC of E7070. The percentage decrease rate from the value before dosing to the nadir of WBC, neutrophil or platelet count also showed a good correlation with the AUC of E7070.
Figure 2.
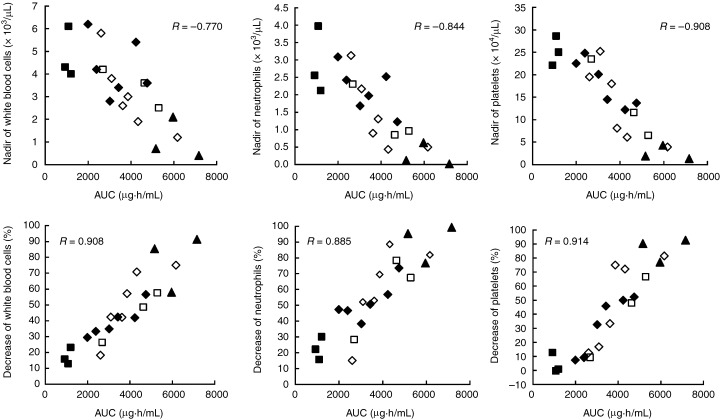
Relationship between the area under the plasma concentration–time curve (AUC) of E7070 and white blood cells, neutrophil and platelet counts. Closed squares, 400 mg/m2; open squares, 600 mg/m2; closed diamonds, 700 mg/m2; open diamonds, 800 mg/m2; closed triangles, 900 mg/m2. R, Pearson's correlation coefficient.
Genotyping of CYP2C9 and CYP2C19
The CYP2C9 and CYP2C19 genotypes were studied in 21 patients. Two (10%) were hetero EM for CYP2C9 (*1/*3), and 19 (90%) were homo EM for CYP2C9 (*1/*1). Five (24%) were PM for CYP2C19 (*2/*2 or *2/*3), eight (38%) were hetero EM for CYP2C19 (*1/*2 or *1/*3) and eight (38%) were homo EM for CYP2C19 (*1/*1). Figure 3 shows the relationship between dose and AUC of E7070 with respect to CYP2C9 and CYP2C19 genotypes. At a dose level of 600 mg/m2 or more, the AUC of patients with mutant allele(s) (PM and hetero EM) of CYP2C9 or CYP2C19 were higher than those of the patients without mutant alleles (homo EM). DLT was observed in one CYP2C19 PM patient at 800 mg/m2 and two CYP2C19 hetero EM or PM patients at 900 mg/m2.
Figure 3.
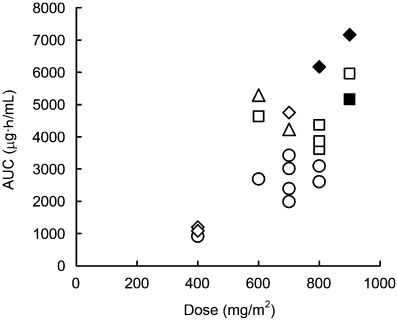
Relationship between dose and area under the plasma concentration–time curve (AUC) of E7070 in relation to CYP2C9 and CYP2C19 genotypes. Circles, homo extensive metabolizers (EM) for both CYP2C9 and CYP2C19; squares, homo EM for CYP2C9 and hetero EM for CYP2C19; triangles, hetero EM for both CYP2C9 and CYP2C19; diamonds, homo EM for CYP2C9 and poor metabolizers (PM) for CYP2C19. Patients with dose‐limiting toxicity are indicated with closed symbols.
Antitumor activity
No objective clinical responses were observed, but liver metastasis was reduced by 22.5% at the 8th cycle of 700 mg/m2 in one colorectal cancer patient, who had previously received 5‐fluorouracil.
Discussion
This phase I study was conducted to determine the MTD of E7070 administered by intravenous infusion over 1–2 h every 21 days, to determine the recommended single‐agent dose for phase II studies, and to characterize the safety, pharmacokinetic and pharmacodynamic profiles of E7070. The MTD in this study was estimated to be 900 mg/m2, and the recommended dose for phase II studies is 800 mg/m2. DLT observed at 900 mg/m2 included leukopenia, neutropenia, thrombocytopenia, elevation of AST, and colitis.
The hematological abnormalities most commonly reported during the first cycle of treatment were neutropenia, leukopenia, and thrombocytopenia. Neutropenia, which tended to be dose‐dependent, but not course‐dependent, was the principal hematological toxicity in this study and was dose‐limiting at 900 mg/m2. At the recommended dose of 800 mg/m2, the mean recovery time of neutrophils from day 1 to grade 1 neutropenia was 24.0 ± 6.1 days. Therefore, bone marrow recovery should be confirmed before the start of successive treatment cycles. Hematological toxicities were also dose‐limiting in four previous phase I trials of E7070.( 5 , 6 , 7 , 8 )
The non‐hematological toxicities most commonly reported during the first cycle of treatment were rash, fatigue, stomatitis, alopecia, injection site reaction, diarrhea and constipation. The type and incidence of the frequently noted events were generally consistent across dosages and cycles of treatment. Gastrointestinal toxicity, the most common non‐hematological type of toxicity associated with E7070, was usually mild and well‐controlled with medication. The frequency of diarrhea increased with dose, and grade 3 severe diarrhea and colitis were observed only in one patient at 900 mg/m2; this patient had previously undergone intestinal surgery for colon cancer. Diarrhea was a dose‐limiting toxicity in two previous phase I trials of E7070.( 6 , 7 ) Because of the relatively high frequency and dose‐dependency of diarrhea in this study, patients receiving E7070 should be carefully monitored for diarrhea. Grade 3 ileus followed by grade 4 constipation was reported in one patient treated with 800 mg/m2 of E7070. Although this event appeared to be related to peritoneal dissemination, its onset after 7 days of E7070 infusion suggested that it might have been induced by E7070. None of the patients treated at less than 800 mg/m2 had grade 2 or higher nausea, vomiting, or anorexia.
Grades 1–2 rash, commonly localized to the face, anterior chest, and upper back, with mild itching, was observed in 12 patients given 400–900 mg/m2 of E7070. Its frequency and severity were not dose‐dependent. Rashes recovered within a week after the administration of E7070, and skin toxicity did not interrupt the therapy in any patient. Injection site reaction (grades 1–2) was reported in nine (43%) patients. The frequency of this event did not seem to be dose‐dependent, suggesting that it was related to infusion irritation by E7070, rather than hemolysis or thrombosis. E7070 shows similarities to chloroquinoxaline sulfonamide, which is known to cause hypoglycemia and cardiac tachycardia.( 13 ) However, no hypoglycemia or cardiac arrhythmia was observed in this phase I trial of E7070.
The results of pharmacokinetic analysis suggested that the AUC of E7070 was non‐linearly related to dose within the dose range of 400–900 mg/m2. The clearance seemed to decrease, with a disproportionate increase in AUC. These results were in agreement with those obtained in other phase I trials with Caucasian patients.( 5 , 6 , 7 , 8 ) This non‐linearity was prominent at higher dose levels and is likely to be a complex consequence of saturation of metabolism, protein binding and distribution of E7070.( 14 ) The absolute values of nadirs and the decrease ratios of WBC, neutrophil and platelet counts were apparently correlated with the AUC of E7070. In vitro experiments have shown that E7070 has the potential to inhibit CYP2C9 and CYP2C19, suggesting that these CYP may be involved in the metabolism of E7070.( 10 ) In fact, other in vitro experiments have shown that CYP2C9 and CYP2C19 are responsible for the metabolism of E7070 (unpublished data). Since these CYP show genetic polymorphism, there is a possibility that subjects with one or more mutant alleles of these CYP have decreased clearance for any compound that is mainly metabolized by these polymorphic CYP. Therefore, we were prompted to investigate the relationship of the pharmacokinetics of E7070 with CYP2C9 and CYP2C19 genotype in this trial. At a dose level of 600 mg/m2 or more, the AUC of patients with mutant allele(s) (PM and hetero EM) of CYP2C19 were higher than those of the patients without mutant alleles (homo EM). These results imply that the presence of mutant allele(s) of CYP2C19 may result in a decrease in the clearance of E7070 (Fig. 3), and support the involvement of CYP2C19 in the metabolism of E7070, as suggested from in vitro studies. The difference of AUC between CYP2C19 homo EM and PM was not clear at a dose of 400 mg/m2. This was probably because metabolic capacity was less saturated at the low dose of 400 mg/m2 compared with the higher doses, and so intergenotypic differences did not appear. The influence of the CYP2C9 genotype on the AUC of E7070 was not clarified because only two subjects had a mutant allele of this gene. The incidence of CYP2C9 PM is known to be less in Asian (< 1%) than in Caucasian (< 10%) people,( 15 , 16 ) whereas CYP2C19 PM is more frequent in Asian (20%) than in Caucasian (< 1%) people.( 17 ) Due to the low frequency of mutation of CYP2C9 in Asian populations, investigation of the effect of CYP2C9 on the pharmacokinetics of E7070 might be difficult in Japanese subjects. Research on subjects with various racial origins would be necessary for evaluation of the clinical impact of the CYP2C9 genotype. In any case, because of the small number of subjects in the present study, further studies should be taken into consideration to assess whether either the CYP2C9 or CYP2C19 genotype is of any clinical significance from the viewpoints of safety and efficacy of E7070. Urinary excretion of unchanged E7070, up to 72 h after the start of administration, was only 0.82–2.47% of the administered dose, indicating that renal clearance plays only a minor role in the elimination of E7070.
Although clinical efficacy (in terms of confirmed partial or complete responses) of E7070 was not demonstrated in this study, one patient with liver metastasis from colon cancer had a reduction in tumor size of ≤ 22.5% and demonstrated stable disease lasting 5 months. A phase II trial of E7070 as a single agent in 5‐fluorouracil‐resistant or refractory colorectal cancer showed limited activity with a 4% response rate,( 18 ) and thus further clinical studies of combination therapy with irinotecan (CPT‐11) are ongoing for the treatment of this tumor type.
In conclusion, the MTD of E7070 administered intravenously in a 1–2 h infusion every 3 weeks was estimated to be 900 mg/m2 and the recommended dose for a phase II study is 800 mg/m2. At 800 mg/m2, hematological toxicities were manageable. Gastrointestinal toxicity was the most common non‐hematological toxicity associated with E7070, but was generally well controlled with premedication. However, this recommended dose might be influenced by the CYP2C19 genotype and possibly by the CYP2C9 genotype as well. E7070 seems to be an interesting agent with novel cell‐cycle‐inhibitory effects. Additional phase I and II studies are currently ongoing in various tumor types to explore further the antitumor activity of this drug as a single agent and in combination with other chemotherapeutic agents.
Acknowledgments
We thank Mr Nozomu Koyanagi, Mr Makoto Shiba, Ms. Mayumi Suzuki, Mr Masaki Tanaka, Mr Masakiyo Kato, Mr Yuichi Inai and Dr Tatsuo Watanabe of Eisai Co. for data management and clinical research coordination.
References
- 1. Fukuoka K, Usuda J, Iwamoto Y et al. Mechanism of action of the novel sulfonamide anticancer agent E7070 on cell cycle progression in human non‐small cell lung cancer cells. Invest New Drugs 2001; 19: 219–27. [DOI] [PubMed] [Google Scholar]
- 2. Sugi N, Ozawa Y, Watanabe T et al. A novel agent ER‐35744, targeting G1 phase. II: Antitumor activities in vitro and in vivo . Proc Am Assoc Cancer Res 1996; 37: A2668. [Google Scholar]
- 3. Yokoi A, Kuromitsu J, Kawai T et al. Profiling novel sulfonamide antitumor agents with cell‐based phenotypic screens and array‐based gene expression analysis. Mol Cancer Ther 2002; 1: 275–86. [PubMed] [Google Scholar]
- 4. Ozawa Y, Sugi NH, Nagasu T et al. E7070, a novel sulphonamide agent with potent antitumour activity in vitro and in vivo . Eur J Cancer 2001; 37: 2275–82. [DOI] [PubMed] [Google Scholar]
- 5. Raymond E, Ten Bokkel Huinink WW, Taieb J et al. Phase I and pharmacokinetic study of E7070, a novel chloroindolyl sulfonamide cell‐cycle inhibitor, administered as a one‐hour infusion every three weeks in patients with advanced cancer. J Clin Oncol 2002; 20: 3508–21. [DOI] [PubMed] [Google Scholar]
- 6. Punt CJ, Fumoleau P, van de Walle B, Faber MN, Ravic M, Campone M. Phase I and pharmacokinetic study of E7070, a novel sulfonamide, given at a daily times five schedule in patients with solid tumors. A study by the EORTC‐early clinical studies group (ECSG). Ann Oncol 2001; 12: 1289–93. [DOI] [PubMed] [Google Scholar]
- 7. Dittrich C, Dumez H, Calvert H et al. Phase I and pharmacokinetic study of E7070, a chloroindolyl‐sulfonamide anticancer agent, administered on a weekly schedule to patients with solid tumors. Clin Cancer Res 2003; 9: 5195–204. [PubMed] [Google Scholar]
- 8. Terret C, Zanetta S, Roche H et al. Phase I clinical and pharmacokinetic study of E7070, a novel sulfonamide given as a 5‐day continuous infusion repeated every 3 weeks in patients with solid tumours. A study by the EORTC Early Clinical Study Group (ECSG). Eur J Cancer 2003; 39: 1097–104. [DOI] [PubMed] [Google Scholar]
- 9. Thijssen HH, Flinois JP, Beaune PH. Cytochrome P4502C9 is the principal catalyst of racemic acenocoumarol hydroxylation reactions in human liver microsomes. Drug Metab Dispos 2000; 28: 1284–90. [PubMed] [Google Scholar]
- 10. Van Den Bongard HJGD, Sparidans RW, Critchley DJP, Beijnen JH, Schellens JHM. Pharmacokinetic drug‐drug interaction of the novel anticancer agent E7070 and acenocoumarol. Invest New Drugs 2004; 22: 151–8. [DOI] [PubMed] [Google Scholar]
- 11. Therasse P, Arbuck SG, Eisenhauer EA et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 12. Owa T, Yoshino H, Okauchi T et al. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J Med Chem 1999; 42: 3789–99. [DOI] [PubMed] [Google Scholar]
- 13. Rigas JR, Tong WP, Kris MG, Orazem JP, Young CW, Warrell RP Jr. Phase I clinical and pharmacological study of chloroquinoxaline sulfonamide. Cancer Res 1992; 52: 6619–23. [PubMed] [Google Scholar]
- 14. Van Den Bongard HJGD, Pluim D, van Waardenburg RC, Ravic M, Beijnen JH, Schellens JHM. In vitro pharmacokinetic study of the novel anticancer agent E7070: red blood cell and plasma protein binding in human blood. Anticancer Drugs 2003; 14: 405–10. [DOI] [PubMed] [Google Scholar]
- 15. Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999; 353: 717–9. [DOI] [PubMed] [Google Scholar]
- 16. Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40: 587–603. [DOI] [PubMed] [Google Scholar]
- 17. De Morais SM, Wilkinson GR, Blaisdell J, Meyer UA, Nakamura K, Goldstein JA. Identification of a new genetic defect responsible for the polymorphism of (S)‐mephenytoin metabolism in Japanese. Mol Pharmacol 1994; 46: 594–8. [PubMed] [Google Scholar]
- 18. Mainwarning PN, van Cutsem E, van Laethem JL et al. A multicentre randomised phase II study of E7070 in patients with colorectal cancer who have failed 5‐fluorouracil based chemotherapy. Proc Am Soc Clin Oncol 2002; 21: 611. [Google Scholar]