

Abstract
(Cancer Sci 2010; 101: 700–706)
Wnt signaling plays key roles in development, cell growth, differentiation, polarity formation, neural development, and carcinogenesis. DIX Domain Containing 1 (DIXDC 1), a novel component of the Wnt pathway, was recently cloned. DIXDC1 is the human homolog of Ccd1, a positive regulator of the Wnt signaling pathway during zebrafish neural patterning. Little has been known about DIXDC1 gene expression regulation. In the present study, we showed that the DIXDC1 protein was induced upon Wnt‐3a stimulation, whereas the DIXDC1 mRNA level was not significantly increased after Wnt‐3a treatment. Positive DIXDC1 staining was detected in colon cancer cells and was colocalized with β‐catenin staining. However, the DIXDC1 mRNA expression decreased in human colon cancer cells compared to the matched normal colon epithelial cells. Our further investigation showed that the DIXDC1 protein was degraded through the proteasome pathway, and the activation of canonical Wnt signaling decreased the ubiquitin‐dependent degradation of both the ectopic and endogenous DIXDC1 protein. In order to explore the possible mechanism of the ubiquitination of DIXDC1, we found that the phosphorylation of DIXDC1 was inhibited by Wnt‐3a. Collectively, these results indicate that canonical Wnt/β‐catenin pathway activation might upregulate DIXDC1 through a post‐translational mechanism by inhibiting the ubiquitin‐mediated degradation of the DIXDC1 protein.
The Wnt pathway plays pivotal roles in development, cell proliferation, differentiation, and carcinogenesis.( 1 , 2 , 3 ) Wnt signaling includes two separate pathways: one is a canonical pathway that is mediated by a key signaling transducer, β‐catenin, and the other is a non‐canonical pathway that is conducted via Ca2+signaling or small G‐protein Rho/Rac.( 4 , 5 )
In the canonical Wnt pathway, the degradation of β‐catenin is regulated by Dishevelled and Axin. The Dishevelled–Axin (DIX) domain, an amino sequence motif conserved in these two proteins, is indispensable for mediating the Wnt signaling transduction.( 6 ) Frame‐shift mutations of the Axin2 gene, which caused the complete deletion of the C‐terminal DIX domain of the Axin2 protein, were detected in more than 25% of colorectal cancers with microsatellite instability.( 7 ) DIXDC1, the human homolog of Ccd1 (coiled‐coil‐DIX1), was recently identified as an Axin2 C‐terminus‐binding protein.( 6 ) DIXDC1 is a third type of DIX domain‐possessing protein and a positive regulator in the Wnt signaling pathway.( 8 , 9 , 10 ) Recently, we showed that DIXDC1 was upregulated in human colorectal adenocarcinoma cells, and activated DIXDC1 could promote colon cancer cell proliferation both in vivo and in vitro through PI3K pathway activation.( 11 ) However, little has been known about DIXDC1 expression regulation until now. In the current study, we report the identification of DIXDC1, a recently recognized new component in Wnt signaling, as a target of the ubiquitin–proteasome pathway. The activation of the canonical Wnt pathway stabilizes the DIXDC1 protein via inhibiting the ubiquitin‐dependent degradation of DIXDC1.
Materials and Methods
Reagents. The rabbit anti‐DIXDC1 antibody was a kind gift from Dr. Wanguo Liu (Louisiana State University Health Science Center, New Orleans, LA, USA).( 6 ) The mouse anti‐β‐catenin antibody was purchased from BD Biotechnology (Franklin Lakes, NJ, USA). The mouse anti‐myc, anti‐Hemagglutinin (HA), anti‐β‐actin, antiphosphoserine, antiphosphothreonine, and antiphosphotyrosine antibodies were purchased from Sigma–Aldrich (St Louis, MO, USA). The mouse anti‐ubiquitin antibody was purchased from Santa Cruz (Santa Cruz, CA, USA). Supervision antirabbit and antimouse detection reagents (horseradish peroxidase [HRP]) were purchased from Dako (Glostrup, Denmark). Carbobenzoxy‐l‐leucyl‐l‐leucyl‐l‐leucinal (MG132), a proteasome inhibitor, was purchased from Calbiochem (Darmstadt, Germany). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA, USA).The RNAeasy micro kit was purchased from Qiagen (Duesseldorf, Germany). The enhanced chemiluminescence (ECL) western blotting substrate system was purchased from Pierce (Rockford, IL, USA).
Cell culture and transient transfection. HeLa cells were maintained in Minimum Essential Media (MEM) medium (Gibco, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco, USA) at 37°C under 5% CO2. Transient transfection was performed using Lipofectamine 2000 according to the manufacturer’s instructions.
Specimens. The study was approved by the institutional review board at Shanghai Hua Shan Hospital (Shanghai, China) with the patients’ consent. All cases were from the files collected from the Department of Pathology, Hua Shan Hospital, affiliated to Fudan University (Shanghai, China). The specimens included 20 cases of human colon carcinoma with both tumoral and matched normal tissues. Patients suffering from familial cancer syndrome or those previously treated with chemotherapy were not included.
Immunoblotting analysis. Cells were lysed in lysis buffer (50 mM Tris–HCl [pH 8.0], 150 mM NaCl, 15 mM MgCl2, 5 mM ethylenediaminetetraacetic acid, and 1% Nonidet P‐40 [NP‐40]) with a freshly added protease inhibitor cocktail (Roche, Mennheim, Switzerland). Proteins were quantified according to the instructions of a bicinchoninic acid protein assay kit (Pierce, USA).The cell lysates were separated by electrophoresis on 10% sodium dodecylsulfate (SDS)–polyacrylamide gel and transferred onto a polyvinylidene fluoride membrane. The blots were blocked with 1% bovine serum albumin followed by incubation in a primary antibody solution (rabbit anti‐DIXDC1 antibody; 1:200, mouse anti‐β‐catenin antibody, 1:1000) at 4°C overnight. After incubation in the peroxidase‐conjugated secondary antibody, signals were detected using the ECL western blotting substrate system. The protein band intensities were measured using a FluroChem8800 imaging system (Alpha Innotech, San Leandro, CA, USA).
Immunohistochemistry. Serial sections of formalin‐fixed and paraffin‐embedded tissues were subjected to antigen retrieval by microwaving in 0.1 M citrate solution (pH 6.0) for 10 min, then incubated with either the mouse anti‐β‐catenin antibody (1:200; BD Biotechnology, USA) or rabbit antihuman DIXDC1 antibody( 6 ) (1:100) at 4°C overnight. DIXDC1 and β‐catenin immunocomplexes were visualized using the supervision antirabbit or antimouse detection reagent (HRP). Stained slides were examined under a light microscope by two pathologists. The staining of DIXDC1 was coded as positive (tumor cells displaying cytoplasmic immunoreactivity for DIXDC1) or negative (absence of cytoplasmic staining). The staining patterns of β‐catenin were divided into three types: (i) membranous pattern; (ii) cytoplasmic pattern; and (iii) nuclear pattern.
Laser‐capture microdissection and RNA extraction. Five 8‐μm‐thick sections were cut from frozen colon cancer tissues, mounted on slides, and stained with hematoxylin. The tumor cells and matched normal epithelial cells were accurately captured by the Lcc1704 Veritas laser‐capture microdissection (LCM) and laser cutting system (Arcturus, Sunnyvale, CA, USA). Total RNA was isolated using the RNAeasy micro kit according to the manufacturer’s instructions.
DIXDC1 mRNA expression analysis. HeLa cells at 70–80% confluence were incubated in serum‐free MEM medium overnight, treated with 80 ng/mL Wnt‐3a (R&D Systems, Minneapolis, MN, USA) for the indicated intervals, and then subjected to real‐time reverse transcription–polymerase chain reaction analysis (RT–PCR). Total RNA isolated from the HeLa cells or colon cancer tissues were reverse transcribed with the oligo(dT) primer. The logarithm of the ratio of tumor/normal tissue mRNA levels of DIXDC1 was estimated by quantitative real‐time PCR analysis and normalized to the GAPDH level. The following primers were used: DIXDC1 forward, 5′‐GGT GAT CCT CAT TCC AGT TTC CA‐3′, DIXDC1 reverse, 5′‐AAT GCC ACC AGG CGA CAA TAC TA‐3′ and GAPDH forward, 5′‐AAC GGA TTT GGT CGT ATT G‐3′, GAPDH reverse, 5′‐GGA AGA TGG TGA TGG GAT T‐3′. The reaction was performed in a Rotor‐Gene RG‐3000 (Corbett Research, Sydney, NSW, Australia) using the SYBR premix ex TaqTM kit (TaKaRa Biotechnology, Dalian, China). Each reaction was carried out at a final volume of 25 μL containing 2 μL cDNA product sample; 0.2 μM each primer; 2× reaction mix, including DNA polymerase, reaction buffer, dNTP, and SYBR Green. The thermal cycles for both DIXDC1 and GAPDH were initiated with a denaturation step of 95°C for 30 s, and consisted of 40 cycles (denaturation at 95°C for 5 s, annealing at 60°C for 30 s, and elongation at 72°C for 30 s). At the end of the PCR cycles, melting curve analyses and electrophoresis of the products on non‐denaturing 8% polyacrylamide gels were performed to ensure the generation of the expected PCR product. The ratio of the DIXDC1 mRNA expression in tumoral versus normal tissues was calculated as log T/N. Log T/N > 0 was counted as the increased DIXDC1 mRNA expression in colon cancer cells.
Cycloheximide chase assay. HeLa cells at 70–80% confluence were transiently transfected with pcDNA3.0–myc–DIXDC1 using Lipofectamine 2000 according to the manufacturer’s instructions. The cells were treated with phosphate‐buffered saline (PBS) or Wnt‐3a (80 ng/mL) in serum‐free medium for 16 h. The cells were then treated with cycloheximide (CHX) at a final concentration of 100 μg/mL (Calbiochem, Germany) and were harvested at 0, 2, 4, and 8 h after treatment. The ectopically expressed or endogenous DIXDC1 protein levels were analyzed by immunoblotting using the anti‐myc antibody or anti‐DIXDC1 antibody.
Co‐immunoprecipitation and ubiquitination assay. The myc‐tagged DIXDC1 plasmid (kindly provided by Dr. Wanguo Liu, Louisiana State University Health Science Center, USA) was transiently cotransfected into the HeLa cells with the HA‐tagged ubiquitin plasmid (kindly provided by Professor Jianxin Gu, Fudan University, China). After being treated with Wnt‐3a (80 ng/mL) in serum‐free medium for 16 h, the cells were resuspended in modified Radio Immunoprecipitation (RIPA) buffer (50 mM Tris‐HCl [pH 7.5], 150 mM NaCl, 0.5% NP‐40, 0.1% (w/v) deoxycholate, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 10 mg/mL aprotinin, and 5 mg/mL leupeptin) with 1%SDS to disrupt the protein–protein interaction, boiled for 10 min, then diluted in 10 volumes of RIPA buffer. The lysates were sonicated at 4°C and subsequently precleared with protein G agarose (Roche, Switzerland) for 2 h at 4°C. Identical amounts (2 mg protein) of precleared cell lysates were immunoprecipitated using the anti‐myc antibody and analyzed by immunoblotting using anti‐HA and anti‐myc antibodies. The ubiquitination and phosphorylation statuses of endogenous DIXDC1 were detected by immunoprecipitation using the anti‐DIXDC1 antibody and analyzed using anti‐ubiquitin, anti‐DIXDC1, antiphosphoserine, antiphosphothreonine, and antiphosphotyrosine antibodies.
Results
Tissue localization of DIXDC1 and β‐catenin in colon cancer tissues. We previously showed that the DIXDC1 protein was upregulated in colon cancer tissues to promote cancer cell proliferation through PI3K pathway activation.( 11 , 12 ) To explore the mechanism of DIXDC1 activation, we first tried to examine both the expression of the DIXDC1 and β‐catenin proteins in human colon cancer tissues, in which Wnt signaling is activated because of Adenomatous Polyposis Coli (APC) or β‐catenin gene mutations. The expressions of the DIXDC1 and β‐catenin proteins were examined by immunohistochemistry in tumoral and matched normal tissues from 20 patients who had undergone surgery for colon carcinoma. Serial sections of these 20 cases were stained with the antirabbit polyclonal DIXDC1 antibody( 6 ) and mouse monoclonal anti‐β‐catenin antibody to assess the degree of colocalization of these two antigens. Positive DIXDC1 staining was detected in 16 of 20 colon cancer cases (80%), presenting as focal or diffuse cytoplasmic staining in colon cancer cells. None of the normal colonic epithelial cells displayed positive DIXDC1 staining. In the normal colon tissues, β‐catenin was detected at the cell–cell interface. On the contrary, in 15 cases (75%), cancer cells displayed diffuse and intense cytoplasmic and/or nuclear β‐catenin staining, suggesting an increase of the β‐catenin protein level and activation of the canonical Wnt pathway( 13 ) (Fig. 1). All 15 cases with intense β‐catenin staining also displayed enhanced anti‐DIXDC1 antibody reactivity. Neither β‐catenin nor DIXDC1 were detected at high levels in the surrounding normal tissue. All of the specimens displayed identical DIXDC1 and β‐catenin distributions in the tumor area. These results indicate the upregulation of the DIXDC1 protein in the tumoral colon specimens with the activation of canonical Wnt signaling.
Figure 1.
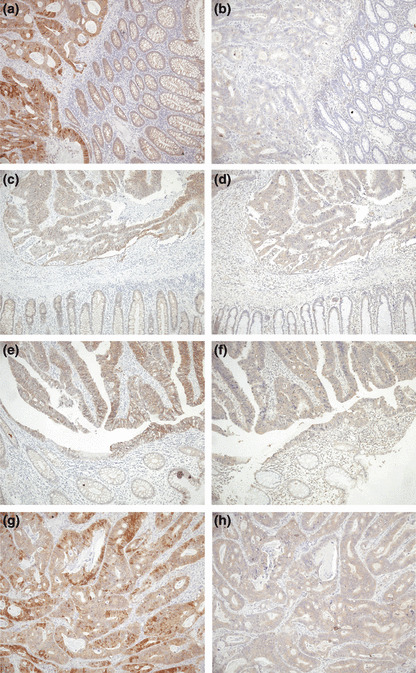
Tissue localizations of the DIX Domain Containing 1 (DIXDC1) and β‐catenin proteins in human colorectal adenocarcinoma tissues. Serial sections of formalin‐fixed and paraffin‐embedded human colon cancer tissue were immunostained with antibodies against β‐catenin (a,c,e) (×100) and DIXDC1 (b,d,f) (×100). Normal colon epithelial cells displayed weak membrane staining of β‐catenin, while colon cancer cells displayed strong cytoplasm/nuclear staining, suggesting the activation of the canonical Wnt pathway. Positive DIXDC1 protein expression was exhibited as diffuse or focal cytoplasmic staining. A significant consistence of DIXDC1 expression and β‐catenin overexpression was observed in colon cancer cells. (g) High power view of (a). (h) High power view of (b).
Downregulation of DIXDC1 mRNA in human colorectal adenocarcinoma tissues. We then evaluated DIXDC1 mRNA expression in 20 pairs of human colon tumoral and normal tissues using real‐time RT–PCR analysis. To successfully tackle the problem of tissue heterogeneity in the DIXDC1 mRNA analysis, LCM was used to precisely separate tumor cells from the corresponding normal colon epithelial cells to make the gene expression assay more reliable( 14 , 15 , 16 ) (Fig. 2a). As shown in Figure 2(b), in 14 cases (14/20, 70%), the DIXDC1 mRNA level was downregulated in tumors compared to the normal tissues.
Figure 2.
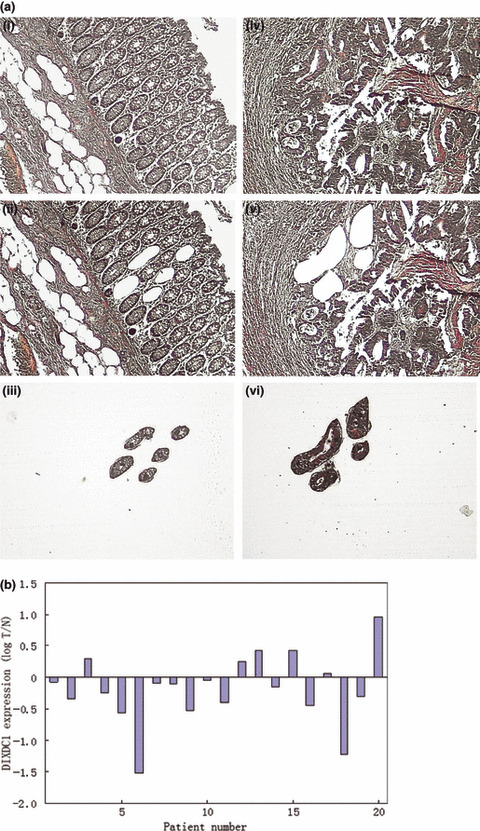
DIX Domain Containing 1 (DIXDC1) mRNA level was downregulated in colorectal adenocarcinoma cancer cells compared to the normal colon epithelium. (a) Normal colon mucosa and colon cancer cells were precisely isolated by laser‐capture microdissection (LCM). Normal colon mucosa: (i) before LCM (×100); (ii) after LCM (×100); (iii) normal colon mucosa captured by LCM (×100). Colon cancer: (iv) before LCM (×100); (v) after LCM (×100); (vi) colon cancer cells captured by LCM (×100). (b) Total mRNA extracted from LCM tissues was reverse transcribed and amplified. Logarithm of the ratio of tumor/normal tissue (log T/N) RNA level of DIXDC1 was determined by quantitative real‐time polymerase chain reaction. Log T/N > 0 indicates an elevated DIXDC1 mRNA expression in tumor cells compared to the normal colon mucosa.
The upregulation of the DIXDC1 protein level and downregulation of the DIXDC1 mRNA level detected in human colon cancer tissues excluded our initial assumption that the activation of DIXDC1 upon Wnt activation was through a β‐catenin‐targeted gene transcriptional activation mechanism.
DIXDC1 protein level, but not the mRNA level, is upregulated by Wn‐t3a in vitro. We then performed an in vitro experiment to detect if canonical Wnt signaling activation affected the DIXDC1 expression. The HeLa cells were treated with Wnt‐3a, a canonical Wnt activator, for 0, 4, 8, 12, and 24 h,( 17 ) then collected and subjected to immunoblotting and real‐time RT–PCR analysis. As shown in Figure 3, the DIXDC1 protein was endogenously expressed in HeLa cells, but the expression amount is relatively low. We found that both the β‐catenin and DIXDC1 protein levels increased significantly within 8 h after Wnt‐3a treatment (Fig. 3a). However, only weak DIXDC1 mRNA induction was observed at 4 and 8 h after Wnt‐3a treatment. The DIXDC1 mRNA expression decreased at 12 and 24 h after Wnt‐3a treatment (Fig. 3b).
Figure 3.

Wnt‐3a increased the DIX Domain Containing 1 (DIXDC1) protein level, but not the DIXDC1 mRNA expression in vitro. (a) HeLa cells were cultured in Minimum Essential Media with or without Wnt‐3a (80 ng/mL) for the indicated time periods. β‐Catenin and DIXDC1 proteins were examined by western blot analysis. Protein levels were normalized to β‐actin. (b) DIXDC1 mRNA levels in Wnt‐3a‐treated HeLa cells at different time intervals were evaluated by real‐time reverse transcription–polymerase chain reaction assay. Only weak mRNA expression stimulation was observed at 4 and 8 h after Wnt‐3a treatment. DIXDC1 mRNA expression decreased after 12‐h treatment with Wnt‐3a. Consistent results were obtained in at least three independent experiments. Data represent the mean ± SD of three independent experiments. Least significant difference t‐test was used to identify significant differences in multiple comparisons. *P < 0.01 compared with 0 h; **P < 0.01 compared with 4 h, ***P < 0.01 compared with 8 h, and ****P < 0.05 compared with 12 h.
Wnt‐3a increases DIXDC1 protein stability. Since the DIXDC1 mRNA level was not induced upon Wnt activation, we wondered if the elevation of the DIXDC1 protein by canonical Wnt activation was acquired through a post‐translational mechanism, in which Wnt activation can stabilize the DIXDC1 protein. We measured the decay rate of the DIXDC1 protein in the presence or absence of Wnt‐3a using the CHX chase assay.( 18 , 19 ) The HeLa cells were transiently transfected with pcDNA3.0–myc–DIXDC1. At 24 h post‐transfection, the cells were treated with PBS or Wnt‐3a in serum‐free medium for 16 h, followed by CHX treatment for a subsequent 8‐h period, over which myc–DIXDC1 protein decay was monitored. As shown in Figure 4(a), the ectopically‐expressed DIXDC1 protein in the HeLa cells could not be detected by immunoblotting at 8 h after CHX treatment in the absence of Wnt‐3a pretreatment. However, the pretreatment of cells with Wnt‐3a obviously slowed down the decay rate of the myc–DIXDC1 protein in the HeLa cells. Similar results were obtained upon assaying the decay rate of the endogenous DIXDC1 protein in HeLa cells; Wnt‐3a was found to delay the degradation of the endogenous DIXDC1 protein (Fig. 4b). These data suggested that the degradation of the DIXDC1 protein was delayed by Wnt activation.
Figure 4.
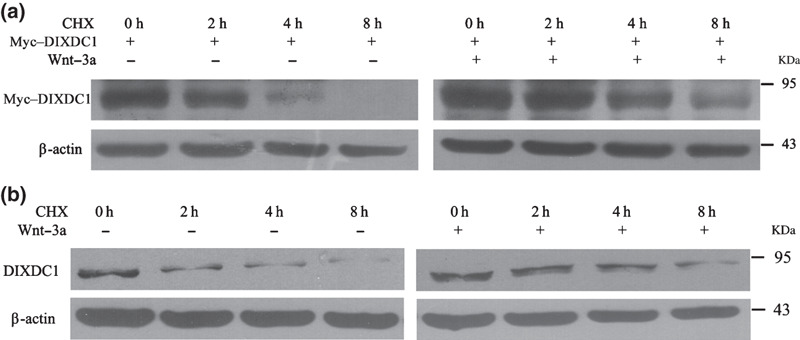
Wnt‐3a increased the stability of the DIX Domain Containing 1 (DIXDC1) protein. HeLa cells were treated with phosphate‐buffered saline or Wnt‐3a (80 ng/mL) in serum‐free medium for 16 h. Cells were then treated with cycloheximide (CHX) (100 μg/mL) and harvested at the indicated time intervals. Cell lysates were subjected to western blotting to analyze the decay rate of the DIXDC1 protein. Stabilities of the ectopic (a) and endogenous DIXDC1 protein (b) were increased after Wnt‐3a treatment.
Wnt‐3a stabilizes the DIXDC1 protein via inhibiting its ubiquitin‐mediated degradation. The discovery that the DIXDC1 protein could be stabilized upon Wnt‐3a treatment led us to hypothesize that Wnt‐3a causes the accumulation of the DIXDC1 protein by preventing its proteasome‐mediated degradation. To test this, the pcDNA3.0–myc–DIXDC1 plasmid was transiently transfected into the HeLa cells, and the steady‐state level of the myc–DIXDC1 protein was analyzed using western blot assay. As shown in Figure 5(a), the steady‐state level of the DIXDC1 protein was increased by Wnt‐3a treatment; the DIXDC1 protein amount was also increased after MG132 treatment (10 μM), indicating that Wnt‐3a might regulate the DIXDC1 protein level via proteasome‐dependent degradation.
Figure 5.
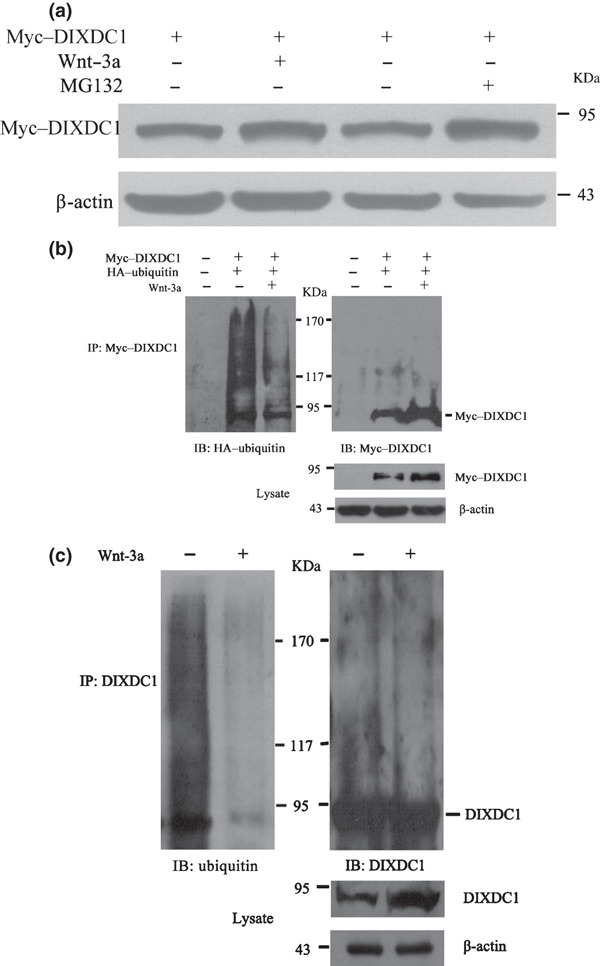
Wnt‐3a increased the DIX Domain Containing 1 (DIXDC1) protein level via inhibiting its ubiquitin‐mediated degradation. (a) steady‐state level of the myc‐tagged DIXDC1 protein was increased after carbobenzoxy‐l‐leucyl‐l‐leucyl‐l‐leucinal (MG132) (10μM) or Wnt‐3a (80 ng/mL) treatment. HeLa cells transiently transfected with the myc‐tagged DIXDC1 plasmid were treated with Wnt‐3a or MG132 and then subjected to immunoblotting analysis of DIXDC1 protein expression. Protein levels were normalized to β‐actin. (b) Wnt‐3a decreased the ubiquitination of the ectopically expressed DIXDC1 protein. HeLa cells were cotransfected with myc‐tagged DIXDC1 and hemagglutinin‐tagged ubiquitin constructs. Cells were treated with Wnt‐3a or phosphate‐buffered saline at 24 h after transfection. Cell extracts were subjected to immunoprecipitation using the anti‐myc antibody. Ubiquitinated myc–DIXDC1 was analyzed by immunoblotting using the anti‐HA antibody. Same blot was stripped and reprobed with the anti‐myc antibody. Steady‐state level of the myc–DIXDC1 protein was examined by western blotting using the anti‐myc antibody and normalized to β‐actin. (c) Wnt‐3a decreased the ubiquitination of endogenous DIXDC1. HeLa cell lysates were subjected to immunoprecipitation using the anti‐DIXDC1 rabbit polyclonal antibody. Ubiquitinated DIXDC1 was detected by immunoblotting using the anti‐ubiquitin antibody. After stripping, the same blot was stained with the anti‐DIXDC1 antibody. Endogenous DIXDC1 protein level was detected by western blot using the anti‐DIXDC1 antibody and normalized to β‐actin. Similar results were obtained in three independent experiments.
The efficient degradation of proteins by the proteasome generally requires the conjugation of multiple molecules of ubiquitin to the substrate.( 20 ) To determine whether the DIXDC1 protein was targeted for ubiquitination, co‐immunoprecipitation was performed to detect the association of myc–DIXDC1 and HA–ubiquitin, which were ectopically expressed in the HeLa cells. The cell lysates were immunoprecipitated using the anti‐myc antibody and analyzed by immunoblotting using the anti‐HA antibody. A high molecular weight of HA‐marked molecules was detected, suggesting that myc–DIXDC1 was polyubiquitinated. However, the presence of high‐molecular weight HA–ubiquitin conjugates in the myc–DIXDC1 immunoprecipitates decreased markedly with Wnt‐3a treatment. The western blot analysis revealed that Wnt‐3a increased the steady‐state level of myc–DIXDC1 (Fig. 5b). To assess the contribution of Wnt‐3a in regulating the ubiquitin‐dependent degradation of endogenous DIXDC1, HeLa cell extracts were immunoprecipitated with the anti‐DIXDC1 antibody and analyzed by immunoblotting using the anti‐ubiquitin antibody. Compared to the PBS treatment, the presence of ubiquitin conjugates in the endogenous DIXDC1 co‐immunoprecipitates decreased obviously upon Wnt‐3a treatment (Fig. 5c). Furthermore, we obtained similar results using anti‐myc or anti‐DIXDC1 antibodies to detect the same blot after stripping (Fig. 5b,c). These results indicate that Wnt‐3a increased the DIXDC1 protein level through inhibiting the ubiquitin‐dependent degradation of the DIXDC1 protein.
Wnt‐3a affects DIXDC1phosphorylation. There is a molecular link between the Wnt signaling pathway and the ubiquitin–proteasome degradation pathway.( 21 ) The phosphorylation of β‐catenin by glycogen synthase kinase‐3β (GSK‐3β) in the absence of Wnt signaling would represent the initial event for the ubiquitination and degradation of β‐catenin. Therefore, we further explored the phosphorylated state of DIXDC1 when Wnt signaling was activated. However, to date, there is no antibody against phosphorylated DIXDC1. The antiphosphoserine, antiphosphothreonine, and antiphosphotyrosine antibodies were used to detect the phosphorylation of endogenous DIXDC1 after immunoprecipitation by the anti‐DIXDC1 antibody. As shown in Figure 6, Wnt‐3a inhibited the phosphorylation of DIXDC1 at the serine and tyrosine residues, but not the threonine residues in HeLa cells because the DIXDC1 levels were high, while the phosphorylation levels increased only slightly. Therefore, we cannot exclude the possibility that Wnt‐3a affected the phosphorylation of DIXDC1, resulting in the accumulation of DIXDC1. However, the positions of the serine or tyrosine residues in DIXDC1, which might mediate a phosphorylation‐dependent ubiquitination, should be explored.
Figure 6.

Wnt‐3a inhibited the phosphorylation of DIX Domain Containing 1 (DIXDC1). Phosphorylated state of endogenous DIXDC1 was detected by immunoprecipitation using the anti‐DIXDC1 antibody and probed with antiphosphoserine and antiphosphotyrosine antibodies. After Wnt‐3a treatment, the DIXDC1 protein level increased significantly, while the phosphorylation levels increased slightly. Results shown (a) are representative of three independent experiments. (b) Ratio of phosphorylated DIXDC1: total DIXDC1 decreased upon Wnt‐3a treatment. *P < 0.01 compared with the control groups without Wnt‐3a treatment. IB, immunoblotting; IP, immunoprecipitation. ( ), Wnt‐3a–; (
), Wnt‐3a–; ( ), Wnt‐3a+.
), Wnt‐3a+.
Discussion
DIXDC1, the human homolog of Ccd1, was recently identified as a novel DIX domain‐containing protein and a positive regulator of the Wnt pathway, functioning downstream of Wnt and upstream of Axin.( 6 , 8 , 9 , 10 ) The current study aimed to illustrate how canonical Wnt pathway activation affected the expression of the DIXDC1 gene. Genetic studies have identified the mutations in the key regulators of this pathway in human colon tumors that cause the augmentation of Wnt/β‐catenin signaling.( 22 , 23 ) Biochemical analyses have shown that the activation of Wnt/β‐catenin signaling impairs the degradation process for β‐catenin and leads to its accumulation in the cytoplasm, and subsequently, the transactivation of the T cell factor/lymphoid enhancer factor (TCF)/β‐catenin target gene.( 24 , 25 ) In searching for the mechanism of DIXDC1 activation in colon cancer cells, we first hypothesized it as a downstream target in Wnt signaling because there were four likely TCF‐binding sites on its upstream promoter area. However, the discovery that the DIXDC1 protein, but not DIXDC1 mRNA, was upregulated upon Wnt activation also excluded our original hypothesis.
These data also raised the possibility that DIXDC1 might be activated by canonical Wnt signaling through a post‐translational mechanism. Our results indicated that a proteasome‐dependent mechanism might participate in DIXDC1 protein degradation. Protein ubiquitination has been well characterized as a mechanism for targeting the protein to the proteasome for degradation.( 26 ) Selective proteolytic degradation by the ubiquitin–proteasome pathway effectively lowers the concentration of a target protein in a timely fashion, thereby regulating numerous important biological processes, such as cell cycle progression, signaling transduction, cell transformation, and cell apoptosis.( 20 , 27 , 28 ) In the present study, we also provide the first evidence that the activation of canonical Wnt signaling increases the DIXDC1 protein level through the inhibition of the ubiquitin‐dependent degradation of the DIXDC1 protein. To date, there has been very little research on the human DIXDC1 gene, since it was first identified as an actin‐binding protein in 2006.( 6 ) We recently reported that the overexpression of the DIXDC1 protein could promote colon cancer cell proliferation through PI3K pathway activation by targeting p21 and cyclin D1.( 11 ) These findings, together with our own in the current study, further indicate that the stabilization of DIXDC1 is crucial for its effects on tumor cell growth and migration in colorectal cancers.
There is cross‐talk between the Wnt signaling pathway and the ubiquitin–proteasome degradation pathway. Wnt signaling inhibits the GSK‐3β‐dependent phosphorylation of β‐catenin,( 29 ) thereby leading to the inhibition of ubiquitination and the accumulation of β‐catenin. Another important finding in this study is that Wnt signaling can also inhibit the phosphorylated state of DIXDC1. We cannot exclude the possibility that Wnt affected the phosphorylation of DIXDC1, resulting in the accumulation of DIXDC1. The possible kinases that phosphorylate DIXDC1 and the amino acid sequences at which there are phosphorylation sites for ubiquitination and degradation will be the focus of a future study. Although the direct links between the phosphorylation machinery and the ubiquitination of DIXDC1 is largely unknown, our study sheds some new light on the mechanism of Wnt pathway gene activation in human colon carcinogenesis.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30700363) and research funding from the Shanghai Health Bureau Municipal in China (No. 2007087) and the Science and Technology Commission of Shanghai Municipality (No. 08PJ1407400) to Dr Li Zheng.
References
- 1. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell 2000; 103: 311–20. [DOI] [PubMed] [Google Scholar]
- 2. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 1997; 11: 3286–305. [DOI] [PubMed] [Google Scholar]
- 3. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–50. [DOI] [PubMed] [Google Scholar]
- 4. Giles RH, Van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta 2003; 1653: 1–24. [DOI] [PubMed] [Google Scholar]
- 5. Miller JR. The Wnts. Genome Biol 2002; 3: REVIEWS3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X, Zheng L, Zeng Z et al. DIXDC1 isoform, l‐DIXDC1, is a novel filamentous actin‐binding protein. Biochem Biophys Res Commun 2006; 347: 22–30. [DOI] [PubMed] [Google Scholar]
- 7. Liu W, Dong X, Mai M et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta‐catenin/TCF signalling. Nat Genet 2000; 26: 146–7. [DOI] [PubMed] [Google Scholar]
- 8. Shiomi K, Kanemoto M, Keino‐Masu K, Yoshida S, Soma K, Masu M. Identification and differential expression of multiple isoforms of mouse Coiled‐coil‐DIX1 (Ccd1), a positive regulator of Wnt signaling. Brain Res Mol Brain Res 2005; 135: 169–80. [DOI] [PubMed] [Google Scholar]
- 9. Shiomi K, Uchida H, Keino‐Masu K, Masu M. Ccd1, a novel protein with a DIX domain, is a positive regulator in the Wnt signaling during zebrafish neural patterning. Curr Biol 2003; 13: 73–7. [DOI] [PubMed] [Google Scholar]
- 10. Soma K, Shiomi K, Keino‐Masu K, Masu M. Expression of mouse Coiled‐coil‐DIX1 (Ccd1), a positive regulator of Wnt signaling, during embryonic development. Gene Expr Patterns 2006; 6: 325–30. [DOI] [PubMed] [Google Scholar]
- 11. Wang L, Cao XX, Chen Q, Zhu TF, Zhu HG, Zheng L. DIXDC1 targets p21 and cyclin D1 via PI3K pathway activation to promote colon cancer cell proliferation. Cancer Sci 2009; 100: 1801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu T, Wang L, Chen Q, Zheng L, Zhu H. A novel monoclonal antibody specific to DIXDC1 protein. Hybridoma (Larchmt) 2009; 28: 183–8. [DOI] [PubMed] [Google Scholar]
- 13. Ghiselli G, Coffee N, Munnery CE, Koratkar R, Siracusa LD. The cohesin SMC3 is a target the for beta‐catenin/TCF4 transactivation pathway. J Biol Chem 2003; 278: 20259–67. [DOI] [PubMed] [Google Scholar]
- 14. Domazet B, Maclennan GT, Lopez‐Beltran A, Montironi R, Cheng L. Laser capture microdissection in the genomic and proteomic era: targeting the genetic basis of cancer. Int J Clin Exp Pathol 2008; 1: 475–88. [PMC free article] [PubMed] [Google Scholar]
- 15. Heath PR, Tomkins J, Ince PG, Shaw PJ. Quantitative assessment of AMPA receptor mRNA in human spinal motor neurons isolated by laser capture microdissection. Neuroreport 2002; 13: 1753–7. [DOI] [PubMed] [Google Scholar]
- 16. Lindeman N, Waltregny D, Signoretti S, Loda M. Gene transcript quantitation by real‐time RT‐PCR in cells selected by immunohistochemistry‐laser capture microdissection. Diagn Mol Pathol 2002; 11: 187–92. [DOI] [PubMed] [Google Scholar]
- 17. Niida A, Hiroko T, Kasai M et al. DKK1, a negative regulator of Wnt signaling, is a target of the beta‐catenin/TCF pathway. Oncogene 2004; 23: 8520–6. [DOI] [PubMed] [Google Scholar]
- 18. Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. Axin and GSK3‐ control Smad3 protein stability and modulate TGF‐ signaling. Genes Dev 2008; 22: 106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li M, Chen D, Shiloh A et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002; 416: 648–53. [DOI] [PubMed] [Google Scholar]
- 20. Hoff H, Zhang H, Sell C. Protein degradation via the proteosome. Methods Mol Biol 2004; 285: 79–92. [DOI] [PubMed] [Google Scholar]
- 21. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J 1997; 16: 3797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis – a look outside the nucleus. Science 2000; 287: 1606–9. [DOI] [PubMed] [Google Scholar]
- 23. Van Noort M, Clevers H. TCF transcription factors, mediators of Wnt‐signaling in development and cancer. Dev Biol 2002; 244: 1–8. [DOI] [PubMed] [Google Scholar]
- 24. Krieghoff E, Behrens J, Mayr B. Nucleo‐cytoplasmic distribution of beta‐catenin is regulated by retention. J Cell Sci 2006; 119: 1453–63. [DOI] [PubMed] [Google Scholar]
- 25. Staal FJ, Noort Mv M, Strous GJ, Clevers HC. Wnt signals are transmitted through N‐terminally dephosphorylated beta‐catenin. EMBO Rep 2002; 3: 63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta 2004; 1695: 55–72. [DOI] [PubMed] [Google Scholar]
- 27. Ben‐Neriah Y. Regulatory functions of ubiquitination in the immune system. Nat Immunol 2002; 3: 20–6. [DOI] [PubMed] [Google Scholar]
- 28. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67: 425–79. [DOI] [PubMed] [Google Scholar]
- 29. Liu C, Li Y, Semenov M et al. Control of beta‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 2002; 108: 837–47. [DOI] [PubMed] [Google Scholar]