

Abstract
Sarcomas are mesenchymal cancers consisting of tumors with various clinical and pathological features. Some of them compel affected individuals to lose important musculoskeletal functions, and some of them are highly malignant and life‐threatening. A great amount of genetic information for sarcomas has accumulated during the past two decades, contributing diagnoses and treatments. From the standpoint of molecular genetics, sarcomas are classified into two groups: those with defined genetic alterations and those with various genetic alterations. The genetic alterations in the first group include reciprocal translocations resulting in fusion oncoproteins and oncogenic mutations of defined genes such as those of the c‐kit gene in gastrointestinal stromal tumors. The function of fusion proteins includes transcription regulator, signal transducer, chromatic remodeling factor, and growth factor, some of which are suitable targets for the molecular therapy. In tumors belonging to the second group, the number of which is far larger than those of the first group, considerable genetic heterogeneity was found even among tumors with same pathological diagnosis. The disruption of the RB and p53 pathways was frequently found, resulting in the dysregulation of cell cycle and the genomic instability. The application of molecular target therapy for tumors in this group requires novel strategies to overcome cross talk between different signal pathways. Recent evidence from in vitro and in vivo experiments has indicated that the cells of origin of sarcomas are tissue stem cells such as mesenchymal stem cells, and the application of stem cell biology holds the promise of novel treatment options. (Cancer Sci 2009; 100: 1573–1580)
Sarcomas are defined as tumors derived from non‐epithelial tissues except hematopoietic tissues. In the latest edition of the World Health Organization classification, approximately 40 different types of sarcomas are listed.( 1 ) Due to a scarcity of tumors, less attention has been paid to the molecular genetics of sarcomas than that of carcinomas, the incidence of which is approximately 200‐fold higher, and until 20 years ago, we had little knowledge about the molecular genetics of sarcomas. Now a number of the genetic events that take place in sarcomas have been disclosed, some of which are specific enough to use in diagnosis, and others may provide a new avenue for therapy. Also with the help of recent advances in tissue stem cell biology and genetic engineering of mice, we are now able to analyze how sarcomatogenesis occur in particular types of cells. From the standpoint of molecular genetics, sarcomas can be divided into two groups: those with defined genetic alterations and simple karyotypes, and those with variable genetic alterations and complex unbalanced karyotypes. In this review, current information about tumors in each group has been complied, and issues that remain to be solved are discussed.
Sarcomas with defined genetic alterations
Sarcomas with defined genetic alterations are further divided into those with a reciprocal chromosomal translocation resulting in oncogenic fusion transcripts and those with an oncogenic mutation in defined genes.
Sarcomas with fusion genes. Although classical cytogenetic analyses had revealed tumor‐specific translocations in several types of sarcomas, the molecular mechanisms underlying them were not demonstrated until the discovery of the EWSR1–FLI1 fusion gene in Ewing's sarcoma.( 2 ) Since then 15 sarcomas have been demonstrated to have tumor type‐specific fusion genes (Table 1). Sarcomas with fusion genes are further divided by the structural and functional features of each fusion gene.
Table 1.
Sarcomas with fusion genes
| Tumor type | Translocation | N‐terminal partner | C‐terminal partner |
|---|---|---|---|
| Fusion genes involving TET genes | |||
| Ewing sarcoma/Primitive peripheral neurectodermal tumor (PNET) | t(11;22)(q24;q12) | EWSR1 | FLI1 |
| t(21;22)(q22;q12) | EWSR1 | ERG | |
| t(7;22)(p22;q12) | EWSR1 | ETV1 | |
| t(2;22)(q33;q12) | EWSR1 | FEV | |
| t(17;22)(q12;q12) | EWSR1 | ETV4 | |
| t(16;21)(p11;q22) | FUS | ERG | |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1 | WT1 |
| t(21;22)(q22;q12) | EWSR1 | ERG | |
| Myxoid liposarcoma | t(12;16)(q13;q11) | FUS | DDIT3 |
| t(12;22)(q13;q12) | EWSR1 | DDIT3 | |
| Clear cell sarcoma (CCS) | t(12;22)(q13;q12) | EWSR1 | ATF1 |
| Angiomatoid fibrous histiocytoma (AFH) | t(12;16)(q13;p11) | FUS | ATF1 |
| t(12;22)(q13;p12) | EWSR1 | ATF1 | |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1 | NR4A3 |
| t(9;17)(q22;q11) | TAFII68 | NR4A3 | |
| t(9;15)(q22;q21) | TCFI2 | NR4A3 | |
| t(9;22)(q22;q15) | TFG | NR4A3 | |
| Low grade fibromyxoid sarcoma | t(7;16)(q32;p11) | FUS | CREB3L2 |
| t(11;16)(p11;p11) | FUS | CREB3L1 | |
| Fusion genes involving RTK genes | |||
| Congenital fibrosarcoma | t(12;15)(p13;q25) | ETV6 | NTRK3 |
| Inflammatory myofibroblastic tumor | t(1;2)(q25;p23) | TMP3 | ALK |
| t(2;19)(p23;p13) | TMP4 | ALK | |
| t(2;17)(p23;q23) | CLTC | ALK | |
| Fusion genes involving chromatin remodeling genes | |||
| Synovial sarcoma | t(X;18)(p11;q11) | SS18 | SSX1 |
| t(X;18)(p11;q11) | SS18 | SSX2 | |
| t(X;18)(p11;q11) | SS18 | SSX4 | |
| t(X;20)(p11;q13) | SS18L1 | SSX1 | |
| Endometrial stromal sarcoma | t(7;17)(p15;q21) | JAZF1 | SUX12 |
| t(6;7)(p21;p15) | JAZF1 | PHF1 | |
| t(6;10)(p21;p11) | EPC1 | PHF1 | |
| Fusion gene involving growth factor gene | |||
| Dermatofibrosarcoma protuberance and giant‐cell fibroblastoma | t(17;22)(q22;q13) | COL1A1 | PDGFB |
| Giant cell fibroblastoma | t(17;22)(q22;q13) | COL1A1 | PDGFB |
| Other types of fusion genes | |||
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) | PAX3 | FOXO1 |
| t(1;13)(p36;q14) | PAX7 | FOXO1 | |
| t(2;X)(p35;q13) | PAX3 | MLLT7 | |
| t(2;2)(q35;p23) | PAX3 | NCOAI | |
| Alveolar soft part sarcoma | t(X;17)(p11;q25) | ASPL | TEF3 |
Fusion genes involving the TET gene family. Nearly half of the fusion proteins contain a portion of TET gene family products, including TAF15 (TATA‐binding protein‐associated factor 15), EWS (Ewing's sarcoma), and TLS (translocation in liposarcoma). TET gene products consist of an N‐terminal SYGQ‐rich region, an RNA recognition motif (RRM), a C2/C2 zinc finger motif, and at least one RNA‐binding domain with an Arg‐Gly‐Gly‐rich motif (RGG).( 3 ) The fusion protein retains the N‐terminal transactivation domain of the TET gene product, but the RNA‐binding domain in its C‐terminal region is replaced with the DNA‐binding domain (DBD) of the corresponding fusion partners (Fig. 1a). Among tumors with a fusion gene involving the TET gene family (Table 1), Ewing's sarcoma family tumors (ESFTs), clear cell sarcoma (CCS), and angiomatoid fibrous histiocytoma (AFH) were described in detail due to their unique features.
Figure 1.
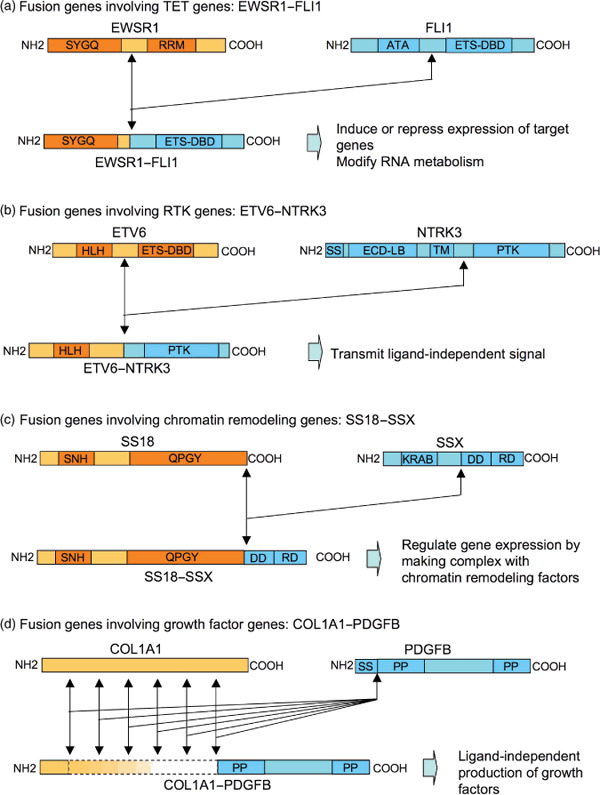
Structure and putative function of fusion proteins found in sarcomas. Representative cases are shown for each of four different types of fusion proteins. (a) Fusion genes involving TET genes: EWSR1–FLI1. The N‐terminus of EWSR1 containing a Ser‐Tyr‐Gly‐Gln rich region (SYGQ) fused with the C‐terminus of FLI1 containing Ets DNA‐binding domain (ETS–DBD). RRM, RNA recognition motif; ATA, amino‐terminal transactivation domain. (b) Fusion genes involving RTK genes: ETV6–NTRK3. The N‐terminus of ETV6 containing a basic helix‐loop‐helix domain (HLH) fused with the C‐terminus of NTRK3 containing a protein tyrosine kinase domain (PTK). SS, signal peptide; ECD‐LB, extracellular ligand‐binding domain; TM, transmembrane domain. (c) Fusion genes involving chromatin remodeling genes: SS18–SSX. The N‐terminus of SS18 containing an SS18 N‐terminal homology domain (SNH) and a Gln‐Pro‐Gly‐Tyr‐rich domain (QPGY) fused with the C‐terminus of SSX containing a dubbed divergent domain (DD) and an SSX repression domain (SSXRD). The former is highly divergent among the SSX protein family, while the latter has the highest similarity within the family. KRAB, Krüppel‐associated box‐like domain. (d) Fusion genes involving growth factor genes: COL1A1–PDGFB. The length of the N‐terminus of COL1A1 in the fusion protein is highly divergent, while the structure of PDGFB in the fusion protein is constant, containing an N‐terminal and C‐terminal propeptide (PP). SS, signal peptide.
TET–ETS in Ewing/primitive peripheral neurectodermal tumor (PNET). Ewing's sarcomas are the second most common bone sarcomas, arising in children and young adults, and have a characteristic phenotype consisting of sheets of small round cells. Along with PNETs, Ewing's sarcomas are referred to as ESFTs. ESFTs are the prototype of tumors with fusion genes involving the TET gene family. Except for FUS–ERG, all fusion transcripts identified so far in ESFTs consist of EWSR1 and an Ets family transcription factor, including the FLI1, ERG, ETV1, ETV4, and FEV genes.( 4 ) Approximately 85–90% of EFSTs are associated with the EWSR1–FLI1 fusion gene, 9–14% with EWR1–ERG, and the remaining 1–5% with other rare variants.( 4 )
TET–AFH in clear cell sarcoma (CCS) and angiomatoid fibrous histiocytoma (AFH). These two distinct tumors share a common fusion gene, EWSR1–ATF1.( 5 , 6 ) CCS or malignant melanoma of soft tissue is a rare and aggressive tumor that develops in young adults. AFH is also a rare low grade malignancy in soft tissues. ATF1 is a member of the CREB transcription factor family with bZip, via which ATF‐1 binds to CRE following activation by cAMP‐dependent protein kinase A (PKA). Upon fusion with EWSR1, the PKA site of ATF‐1 was replaced by the N‐terminus of EWSR1, resulting in a cAMP‐independent and constitutively active transactivator that is driven by the EWSR1 promoter.( 7 ) EWSR1–ATF1 binds to the promoter region and induced the expression of the MITF gene, a master regulator of melanocyte differentiation.( 8 ) Upregulation of MITF expression was found in CCS, endowing tumor cells with melanocytic features,( 9 ) but not in AFH. This suggests that cell context is an important factor determining the downstream repertoire of fusion gene products.
Fusion genes involving receptor tyrosine kinase (RTK). The second type of fusion protein consists of a catalytic domain of RTK and the strong transactivating domain of tissue‐specific genes, resulting in a ligand‐independent and constitutionally active RTK (Fig. 1b). This type of fusion gene is not restricted to one type of malignant tumor, but is found in tumors of various origins, even including hematological malignancies. This suggests that the fusion gene promotes oncogenic transformation without interfering with the differentiation of cell lineages.
ETV6–NTRK3 in congenital fibrosarcoma. Congenital fibrosarcoma is a pediatric soft tissue sarcoma (STS) with low grade malignancy. ETV6 is a member of the Ets family of transcription factors containing a basic helix‐loop‐helix (bHLH) dimerization domain, which was originally found at the breakpoint in translocations in leukemia.( 10 ) NTRK3 is the cell surface receptor for neurotropin 3 expressed primarily in the central nervous system.( 11 ) The ETV6–NTRK3 fusion protein forms a homodimer or heterodimer with wild‐type NTRK3, which displays RTK activity and undergoes autophosphorylation at tyrosine residues.( 12 )
Fusion genes involving chromatin remodeling factors. The third type of fusion gene involves chromatin remodeling factors, although the functions of fusion products are still equivocal (Fig. 1c).
SS18–SSX in synovial sarcoma (SS). SS is a soft tissue malignancy that develops from childhood up to middle age. Tumors arise in extremities in most cases, but also in other sites including visceral organs. The SS18 (also known as SYT) gene is expressed ubiquitously, and encodes a protein with an N‐terminal SNH domain, which is highly conserved among SS18 homolgues, and a C‐terminal QPGY domain which has regulatory roles in transcription.( 13 ) Through this SNH domain, SS18 protein interacts with BRM protein, becoming a component of the SWI/SNF complex involved in the remodeling of chromatin structures and functions as a transcriptional activator.( 14 ) SSX genes constitute a family of at least five (SSX1 to SSX5), and are expressed only in the testis and thyroid.( 15 ) SSX proteins have two putative transcriptional‐repressor domains; one is a Krüppel‐associated box‐like domain at the N‐terminal end and the other is an SSXRD domain in the C‐terminal region.( 15 ) The latter is preserved in the SS18–SSX fusion protein, which therefore contains both activator and repressor elements for transcription, the net result being that it seems to function as a transcriptional repressor of certain genes.( 16 )
Fusion genes involving growth factors. The fourth type of fusion transcript involves the growth factor gene (Fig. 1d).
COL1A1–PDGFB in dermatofibrosarcoma protuberance (DFSP). DFSP is a rare skin tumor of low grade malignancy which shows local recurrence. Either the reciprocal chromosomal translocation t(17;22) (q11;q13.1) or a supernumerary ring chromosome derived from t(17;22) is found in this type of tumor, which results in the fusion of the COL1A1 gene on chromosome 17 with the PDGFB gene on chromosome 22.( 17 ) Because the point of fusion is highly specific for PDGFB but spread over almost the entire locus for COL1A1, the role of the COL1A1 gene may be simply to up‐regulate the expression of PDGFB, which acts as an auto‐ or paracrine growth factor. The same fusion gene was found in giant‐cell fibroblastoma,( 17 ) an infantile tumor with intermediate malignancy showing local recurrence.
Sarcomas with oncogenic mutations
KIT and PDGFRA in gastrointestinal stromal tumors (GIST). GIST is the most common mesenchymal tumor in the gastrointestinal tract. Activating mutations of the KIT gene, which encodes a receptor for stem cell factor (SCF), were found in 75–80% of cases, and those of PDGFRA were found among the rest (5–10%).( 18 ) In all cases, mutated receptors transmit growth signals in a ligand‐independent manner, inducing dysregulated cell proliferation.
INI1 in malignant rhabdoid tumors. Malignant rhabdoid tumors are highly aggressive pediatric tumors that develop mainly in the kidney but also at virtually any extrarenal anatomic site. SMARCB1/hSN5/INI1 is a component of the SWI/SNF complex and represses the expression of cyclin D1, which promotes cell cycle progression.( 19 ) Thus INI1 is a tumor suppressor. Loss of function mutations of the INI1 gene accompanied by a loss of the wild‐type allele are frequently found in this type of tumor.( 20 ) Recently, frequent loss of INI1 protein expression was reported in epithelioid sarcomas,( 21 ) which are highly malignant adult sarcomas of unknown origin, although the frequency of genetic deletions was low.( 22 )
Therapeutic strategy for sarcomas with fusion genes
Targeting fusion products. Because proteins produced by fusion genes are absent in normal tissues, they are ideal targets for tumor‐specific therapy. Most fusion products, however, are a transcriptional factor located in the nucleus, which makes it difficult to apply new therapeutic tools such as antibodies. One approach currently being undertaken is vaccination immunotherapy against SS. Processed peptides derived from the SS18–SSX fusion protein were exhibited on the cell surface along with human leukocyte antigen (HLA) molecules, and vaccination with synthesized peptides primed SS18‐SSX‐specific cytotoxic T‐cells in patients.( 23 ) Although the result of a phase I study was not remarkable,( 24 ) this approach should be investigated further.
Targeting downstream molecules or signals of fusion products. A large number of studies have attempted to identify the genes regulated by fusion proteins in order to find targets for therapy. Most were based on gene expression profiling after the induction of expression in various normal cells by expression vectors, or the inhibition of expression in tumor cells with antisense or siRNA technology.
EWSR1–FLI1 in ESFT. Forced expression of the EWSR1–FLI1 gene in various cells resulted in the identification of up‐ or down‐regulated genes by EWSR1–FLI1 protein. A number of cell cycle–related genes were identified including p21, p27, p57kip, and cyclin E, and Tanaka and coworkers systemically investigated the mechanism of regulation and demonstrated possible therapeutic applications.( 25 , 26 , 27 ) In addition to these growth‐related genes, several candidates for transformation‐associated genes were identified, among which ID2 ( 28 ) and NKX2.2( 29 ) were proposed to be key factors controlling downstream genes.
SS18–SSX in SS. The downstream targets of SS18–SSX were still not clearly defined partially because the cell of origin was not known. Gene expression profiling of SS identified several possible targets such as ERBB2, IGF, FGF, and the WNT pathway.( 30 ) Frizzled homologue 10 (FZD10) is a member of the FZD family for the receptor of WNT signal, and is exclusively expressed in SS.( 31 ) A specific monoclonal antibody against FZD10 showed tumor‐specific accumulation in vivo and effectively killed cells when they were conjugated with a radioisotope.( 32 )
Sarcomas with variable genetic alterations
More than two‐thirds of sarcomas lack defined genetic alterations and are characterized by complex numerical and structural chromosomal aberrations. Most of the adult spindle and pleomorphic sarcomas belong to this group. Histopathological findings showed significant diversity even among tumors with the same diagnosis. Although not common to all tumors, disruption of the retinoblastoma (RB) and/or p53 regulatory pathways is frequently found. In contrast to carcinomas, oncogenic activating mutations in the ras gene family are extremely rare.
Osteosarcoma (OS). OS is the most frequent primary bone sarcoma, comprising approximately 20% of all bone tumors. Histologically, OS is defined as a tumor forming an immature bone matrix called an osteoid. They tend to develop in children and young adolescents, but also are found in the elderly. The most common locations are areas with rapid bone growth such as the metaphyseal region of the distant femur, but almost all bones can be involved. Therefore, OSs diagnosed by the current criteria are likely to be a mixture of tumors with heterogeneous cellular and genetic backgrounds.
Hereditary predisposition to OS. The genetic alterations in human OS were not disclosed until the discovery of the RB gene as the predisposing gene for hereditary RB.( 33 ) Subsequent analyses of somatic mutations revealed a loss of functional RB protein in approximately 60% of sporadic OSs.( 34 ) The second gene found to be involved in OS was also linked to a hereditary predisposition. Li‐Fraumeni syndrome is characterized by a high risk for various cancers including OS.( 35 ) The genetic defect lies in the p53 gene,( 36 ) the protein product of which is responsible for monitoring the integrity of the genome, and the control of cell cycle checkpoints after DNA damage. Somatic mutations of the p53 gene in sporadic OS were also found in 60% of tumors.( 37 ) Recent reports using conditional knockout mice clearly demonstrated that loss of function mutations of these two tumor suppressor genes were critical to the development of OS.( 38 )
Another hint to understand the molecular mechanism leading to OS was obtained again from cancer‐prone syndromes. DNA helicase disorders include Bloom syndrome (RECQL2 helicase), Werner syndrome (WRN or RECQL3), and Rothmund‐Thomosen syndrome (RTS or RECQL4 gene), and individuals with genetic defects in these genes manifest a number of disorders and are predisposed to cancers including OS.( 39 ) Mutations of the RECQL4 gene seem to be most closely linked with the development of OS,( 40 ) although somatic mutations of this gene in OS are extremely rare.( 41 )
Genetic alterations identified in OS. Studies of the molecular mechanisms of growth and progression in OS have identified more than 20 genetic alterations (Fig. 2). Identified molecules have been classified into two groups. The first group consists of molecules directly involved in regulating the cell cycle. Mutations of RB‐associated genes were also found such as amplification of the CDK4 ( 42 ) and cyclin D ( 43 ) genes and functional loss of the p16 gene.( 43 ) Mutations of p53‐associated genes were also found such as amplification of the MDM2 gene.( 44 ) When mutations of these genes were included, OSs were classified into four groups based on the status of the RB and p53 pathways (Fig. 3). Approximately half of OSs may have mutations in both the RB and p53 pathways, whereas 15% of tumors may have intact RB and p53, indicating alternative genetic mechanisms to dysregulate the cell cycle in OS.
Figure 2.
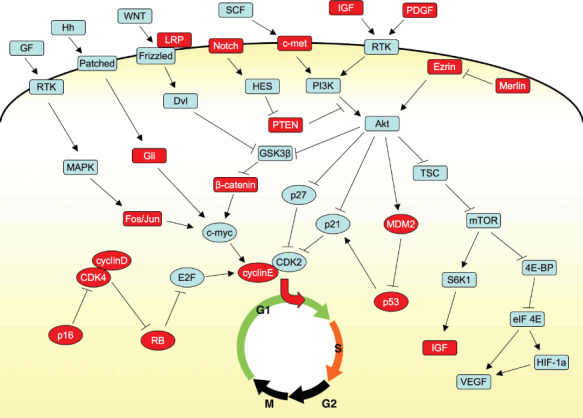
Molecules involved in the development and progression of osteosarcomas. Molecules involving the cell cycle and signal transduction are enclosed by circles and rectangles, respectively. Molecules in which abnormalities at the genetic or protein level have been shown in human or animal OSs are highlighted with red.
Figure 3.
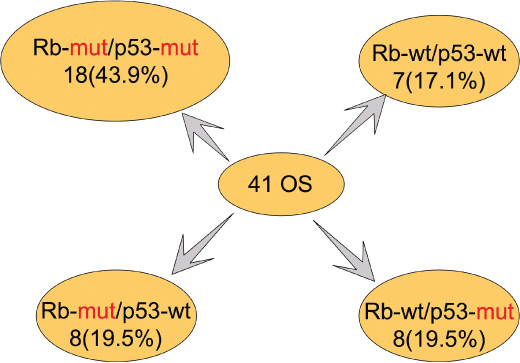
Classification of osteosarcomas by the status of genes in RB and p53 pathways. Forty‐one OSs were classified by the status of genes in RB (RB, p16, CDK4, and cyclin D) and p53 (p53 and MDM2) pathways. The classification was based on published data in references 34, 37, and unpublished data.
The second group consisted of growth signal transduction molecules. As well as mutation analyses, a number of studies have been performed to investigate the role of particular signaling pathways in OS such as WNT/β‐catenin,( 45 ) Hedgehog,( 46 ) and Notch,( 47 ) and identified several important molecules for the growth and survival of OS cells. Recent advances in this field, however, have revealed considerable cross talk between different signaling pathways, and so it is difficult and even inappropriate to define a particular signaling pathway as the main one in OS. Nevertheless, Akt signaling seemed to be a major pathway involved in OS, and a number of molecules related to this pathway were shown to be mutated in human and/or animal OS (Fig. 2). Akt is activated by growth factors such as IGF and PDGF via the activation of PI3K. This process is inhibited by phosphatase and tensin homolog deleted from chromosome 10 (PTEN), which is frequently deleted in canine OS.( 48 ) PTEN also inhibited the signal through c‐met, of which the receptor was strongly expressed in OS.( 49 ) The function of PTEN is inhibited by HES, whose expression is induced by activated Notch.( 50 ) Overexpression of ezrin, a cytoskeleton‐associated molecule, was shown to be necessary for the metastasis of OS through the activation of Akt.( 51 ) Interestingly, the conditional knockout of merlin, which inhibits the function of ezrin in mice, led to osteosarcomas.( 52 ) Activated Akt then activates mTOR via inhibition of TSC. The activated mTOR then activates S6K and eIF4E, resulting in the activation of invasion‐related protein such as vascular endothelial growth factor (VEGF). Activated Akt also inhibits the function of GSK‐3, resulting in the nuclear accumulation of β‐catenin,( 53 ) which then drives target such as c‐myc. GLI is a transcription factor located downstream of the hedgehog signaling pathway, the amplification of which was reported in OS.( 42 )
Based on these findings, inhibition of mTOR function seemed to be a promising molecular approach to the treatment of OS. The results of a clinical trial using novel mTOR inhibitors, however, showed minimum responses possibly due to feedback activation of Akt.( 53 ) This may illustrate the difficulty of targeting one signal mediator, and that the multi‐directional approach using an antibody against IGF‐1R may be necessary.( 54 )
Soft tissue sarcoma (STS). No definite tumor type‐specific genetic alterations have been found in most adult STSs, particularly those consisting predominantly of malignant spindle cells. Due to the lack of phenotypes indicating the particular differentiation, immunohistochemical studies have been the only way to obtain more information for diagnoses, which are notoriously inconsistent. Therefore, it is reasonable to try to understand these tumors based on gene expression signatures, and a number of studies have been performed to identify diagnosis‐specific signatures of these tumors (Table 2).
Table 2.
Representative expression profiling studies in soft tissue sarcomas (STSs)
| Author (ref. no.) | No. of samples | Sample content | Main messages | |
|---|---|---|---|---|
| Nagayama | 31 | 47 | MFH (14), SS (13), LMS (10), MPNST (4), PLS (3), DLS (3) | Synovial sarcoma shared gene expression profiles with MPNST |
| Nielsen | 57 | 41 | LMS (11), MFH (8), SS (8), GISTS (8), PLS (2), DLS (1), MLS (1), benign (2) | SS, GIST, and a subset of LMS, but not MFH, were clustered as a distinct group |
| Lee | 55 | 27 | MFH (9), SS (9), LMS (9) | 202 genes defined a cluster consisting of LMS and MFH |
| Segal | 65 | 51 | MFH (11), FBS (8), LMS (6), round‐cell LS (4), PLS (3), DLS (5), CCS (4), SS (5), GIST (5) | A subset of MFH was identified as a distinct group |
| Baird | 66 | 181 | MFH (38), LPS (33), EWS (19), LMS (17), SS (16), FBS (7), MPNST (6), OS (5), DFSP (5), malignant HPC (6), RMS (6), Schwannoma (3), mixed mullerian tumor (2), ASPS (1), CCS (1), CHS (1), NOS (10) | 2766 genes can be used to distinguish 16 different types of tumors |
| Nakayama | 56 | 105 | MFH (21), MLS (19), SS (16), MFS (15), DLS (15), LMS (6), FS (4), WDLS (3), MPNST (3), lipoma (3) | 18 of 21 MFH failed to be clustered with MFS, SS, DLS, LMS, FS, and MPNST |
Malignant fibrous histiocytoma (MFH). The pathological entity of MFH was established around 1970 by Enzinger, and since then MFH has been the most common STS in adults. Serious concern about the entity, however, has been raised by some pathologists, and in the latest edition of the World Health Organization classification, the term MFH was used with quotation marks accompanied by an equivocal description such as undifferentiated high grade pleomorphic sarcomas. In accordance with this pathological enigma, no clear molecular signatures of ‘MFH’ have been identified by gene expression profiling so far. Lee et al. found a subset of leiomyosarcomas to have similar profiles to MFH, making a cluster defined by 202 genes.( 55 ) Nakayama et al. compared the gene expression signature of ‘MFH’ with that of each of five group of spindle sarcomas which were relatively clearly defined by histological findings: dedifferentiated liposarcoma, leiomyosarcoma, myxofibrosarcoma, fibrosarcoma, and malignant peripheral nerve sheath tumor (MPNST).( 56 ) Three of the 21 MFH samples showed marked similarities to one of the five sarcoma types, which were supported by histological findings. Although the remaining 18 MFH samples showed little or no resemblance to any of the five sarcoma types by histology, 12 of them showed moderate similarities in terms of gene expression, suggesting the value of a reclassification using gene expression profiles.
Leiomyosarcoma (LMS). LMSs account for 5–10% of STSs, and are very heterogenous in terms of clinical (age or site) and pathological (well to poorly differentiated) findings. Nielsen et al. classified LMSs into two groups based on the expression of 24 genes. These genes were implicated in muscle structure and function, such as the genes for actin, myosin leiomodin, myosin phosphatase subunit, and calponin.( 57 ) Lee et al. defined a set of 355 genes for which the expression profiles indicated metastases of LMS.( 58 )
Insights into cells of origin for sarcomas
In addition to diagnostic and therapeutic information, molecular analyses, particularly recent reports using genetically engineered mice, have shed a light on the cells of origin for sarcomas.
From studies in vitro. In vitro transformation assays indicated the importance of cell‐context. Most of the TET gene‐containing fusion constructs could transform an established cell line, NIH3T3, but the results differed considerably when they were introduced into primary cells. Introduction of the expression vector for EWSR1 transformed mesenchymal stem cells (MSC), but not murine embryonic fibroblasts or fibroblasts,( 59 ) and transformed MSC lost their multi‐directional differentiation potential.( 59 ) Consistent with this result, EWS cells gained multidirectional differentiation potential when the EWSR1–FLI1 gene was silenced by siRNA, indicating MSC to be the cell of origin for ESFT.( 60 ) Similar results were reported in the case of FUS‐DDIT3,( 61 ) whereas EWSR1‐ATF1 failed to transform MSC,( 59 ) indicating that stemness is not the sole requirement for transformation.
From studies in vivo. The first engineered mouse with a fusion gene was the FUS–DDIT3 transgenic mouse.( 62 ) Surprisingly, even though the gene was driven by a ubiquitous promoter (EF1), the transgenic mice developed normally until tumors resembling human MLS formed.( 62 ) This result indicates that FUS–DDIT3 has few effects on cells except for the precursor cells of MLS. Most other fusion products, however, seemed toxic to ES cells, or to interfere with embryonic development, and no simple transgenic mice are available as models. To overcome this issue, the conditional knock‐in strategy was employed to create animal models using mice with a Cre gene driven by stage‐specific genes. When the expression of the PAX3–FOXOA1 gene was induced in Myf6‐expressing terminally differentiated muscle cells, tumors mimicking human alveolar rhabdomyosarcoma (ARMS) developed, although at a very low frequency (1/228).( 63 ) If the expression was induced in PAX7‐expressinng cells, severe developmental defects were observed without tumors. Similar results were reported in the case of SS. Tumors with features of human SS developed when the expression of the SS18–SSX gene was induced in Myf5‐expressing cells, which include muscle stem cells and satellite cells. Mice died in the embryonic stage when the SS18–SSX gene was expressed by the promoter of PAX3, or PAX7, and developed myopathy when the expression was induced in Myf6‐expressing cells.( 64 ) These results suggest that the window of permissive cells is relatively narrow for each type of fusion gene.
But one should be careful when interpreting these results. In these conditional mice, the expression of the fusion gene was induced in all cells expressing Cre‐driver genes, which may result in a complete loss of important functions in each individual. In the case of human malignancies, however, only one cell may suffer the effect of the fusion gene, and others with the same function may contribute to normal development. Therefore the window of cells of origin for human tumors with fusion genes might be much broader.
Why two types? The difference between two types of sarcomas may be due to two factors: type of genetic alteration and cell of origin (Fig. 4). These factors might not be independent of each other. In the case of tumors with defined genetic alterations, the window of cells of origin may be narrow, possibly tissue stem cells (MSC or neural crest stem cells). Genetic alterations may be toxic or have no effect in cells at different stages. In addition to driving transformation, such genetic alterations may restrict differentiation in a particular lineage, and therefore the phenotype of final tumors may be relatively homogeneous with the remnant of differentiation. On the other hand, genetic instability due to loss of RB and p53 function or DNA helicases may induce sarcomatogenesis in tumors without defined genetic alterations. Such mechanisms may not require a particular cell context, and therefore differentiated cells may be permissive for this type of transformation. Also, further differentiation of tumor cells may be allowed, although the process may be random and/or dysregulated. This causes the extensively heterogeneous phenotype of tumors in this group such as osteosarcomas and so‐called MFH. These hypotheses may have relevance with the prevailing concept of cancer stem cells. If the cell of origin in tumors with fusion genes is tissue stem cells such as MSC, the stemness is not an exclusive feature of rare sarcoma stem cells in each tumor, and the ability to form tumor tissues might be defined by factors unrelated to stemness.
Figure 4.
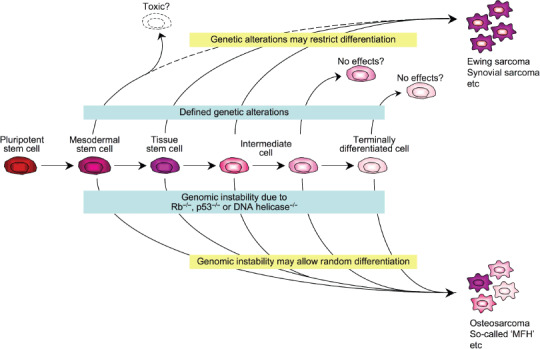
Hypothetical model of sarcomatogenesis.
Conclusions
Whenever a new fusion gene is discovered in one particular type of sarcoma, we cannot help admiring the pathologists who discern its entity and establish a concept. Without their careful and accurate observations, no molecular diagnoses would be possible. The number of patients with STS, particularly undifferentiated pleomorphic sarcomas, seems to be increasing, and no effective therapy has been invented. The cross talk between different growth and survival signaling pathways is a life‐guarding system under physiological conditions, but also it provides malignant cells with a way to escape from molecular targeting therapies. Future innovations in analytical methods such as whole genomic and protein sequencing analyses may provide a way to overcome this hurdle and open a new era for patients with sarcomas.
Acknowledgments
We thank Dr Yusuke Nakamura for continuous support with the studies in our group. These studies were supported in part by a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Fletcher CD, Van Den Berg E, Molenaar WM. Pleomorphic malignant fibrous histiocytoma/undifferentiated high grade pleomorphic sarcoma. In: Fletcher CD, Unni KK, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Washington, DC: IARC Press, 2002; 120–2. [Google Scholar]
- 2. Delattre O, Zucman J, Plougastel B et al . Gene fusion with an ETS DNA‐binding domain caused by chromosome translocation in human tumours. Nature 1992; 359: 162–5. [DOI] [PubMed] [Google Scholar]
- 3. Law WJ, Cann KL, Hicks GG. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic 2006; 5: 8–14. [DOI] [PubMed] [Google Scholar]
- 4. De Alava E, Gerald WL. Molecular biology of the Ewing's sarcoma/primitive neuroectodermal tumor family. J Clin Oncol 2000; 18: 204–13. [DOI] [PubMed] [Google Scholar]
- 5. Zucman J, Delattre O, Desmaze C et al . EWS and ATF‐1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 1993; 4: 341–5. [DOI] [PubMed] [Google Scholar]
- 6. Waters BL, Panagopoulos I, Allen EF. Genetic characterization of angiomatoid fibrous histiocytoma identifies fusion of the FUS and ATF‐1 genes induced by a chromosomal translocation involving bands 12q13 and 16p11. Cancer Genet Cytogenet 2000; 121: 109–16. [DOI] [PubMed] [Google Scholar]
- 7. Fujimura Y, Ohno T, Siddique H, Lee L, Rao VN, Reddy ES. The EWS‐ATF‐1 gene involved in malignant melanoma of soft parts with t(12;22) chromosome translocation, encodes a constitutive transcriptional activator. Oncogene 1996; 12: 159–67. [PubMed] [Google Scholar]
- 8. Tachibana M, Takeda K, Nobukuni Y et al . Ectopic expression of MITF, a gene for Waardenburg syndrome type 2, converts fibroblasts to cells with melanocyte characteristics. Nat Genet 1996; 14: 50–4. [DOI] [PubMed] [Google Scholar]
- 9. Davis IJ, Kim JJ, Ozsolak F et al . Oncogenic MITF dysregulation in clear cell sarcoma: defining the MiT family of human cancers. Cancer Cell 2006; 9: 473–84. [DOI] [PubMed] [Google Scholar]
- 10. Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets‐like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 1994; 77: 307–16. [DOI] [PubMed] [Google Scholar]
- 11. Lamballe F, Klein R, Barbacid M. trkC, a new member of the trk family of tyrosine protein kinase, is a receptor for neurotrophin‐3. Cell 1991; 66: 967–79. [DOI] [PubMed] [Google Scholar]
- 12. Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PHB. A novel ETV6‐NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998; 18: 184–7. [DOI] [PubMed] [Google Scholar]
- 13. Thaete C, Brett D, Monaghan P et al . Functional domains of the SYT and SYT‐SSX synovial sarcoma translocation proteins and co‐localization with the SNF protein BRM in the nucleus. Hum Mol Genet 1999; 8: 585–91. [DOI] [PubMed] [Google Scholar]
- 14. Perani M, Ingram CJ, Cooper CS, Garrett MD, Goodwin GH. Conserved SNH domain of the proto‐oncoprotein SYT interacts with components of the human chromatin remodeling complexes, while the QPGY repeat domain forms homo‐oligomers. Oncogene 2003; 22: 8156–67. [DOI] [PubMed] [Google Scholar]
- 15. Gure AO, Türeci O, Sahin U et al . SSX: a multigene family with several members transcribed in normal testis and human cancer. Int J Cancer 1997; 72: 965–71. [DOI] [PubMed] [Google Scholar]
- 16. De Bruijin DRH, Nap J‐P, Van Kessel AG. The (Epi) genetics of human synovial sarcoma. Genes Chromosomes Cancer 2007; 46: 107–17. [DOI] [PubMed] [Google Scholar]
- 17. Simon MP, Pedeutour F, Sirvent N et al . Deregulation of the platelet‐derived growth factor B‐chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant‐cell fibroblastoma. Nat Genet 1997; 15: 95–8. [DOI] [PubMed] [Google Scholar]
- 18. Subramanian S, West RB, Corless CL et al . Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene 2004; 23: 7780–90. [DOI] [PubMed] [Google Scholar]
- 19. Kingston RE, Narlikar GJ. ATP‐dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev 1999; 13: 2339–52. [DOI] [PubMed] [Google Scholar]
- 20. Versteege I, Sévenet N, Lange J et al . Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998; 394: 203–6. [DOI] [PubMed] [Google Scholar]
- 21. Modena P, Lualdi E, Facchinetti F et al . SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005; 65: 4012–9. [DOI] [PubMed] [Google Scholar]
- 22. Kohashi K, Izumi T, Oda Y et al . Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol 2009; 40: 349–55. [DOI] [PubMed] [Google Scholar]
- 23. Sato Y, Nabeta Y, Tsukahara T et al . Detection and induction of CTLs specific for SYT‐SSX‐derived peptides in HLA‐A24 (+) patients with synovial sarcoma. J Immunol 2002; 169: 1611–8. [DOI] [PubMed] [Google Scholar]
- 24. Kawaguchi S, Wada T, Ida K et al . Phase I vaccination trial of SYT‐SSX junction peptide in patients with disseminated synovial sarcoma. J Transl Med 2005; 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakatani F, Tanaka K, Sakimura R et al . Identification of p21WAF1/CIP1 as a direct target of EWS‐Fli1 oncogenic fusion protein. J Biol Chem 2003; 278: 15105–15. [DOI] [PubMed] [Google Scholar]
- 26. Matsunobu T, Tanaka K, Matsumoto Y et al . The prognostic and therapeutic relevance of p27kip1 in Ewing's family tumors. Clin Cancer Res 2004; 10: 1003–12. [DOI] [PubMed] [Google Scholar]
- 27. Li X, Tanaka K, Nakatani F et al . Transactivation of cyclin E gene by EWS‐Fli1 and antitumor effects of cyclin dependent kinase inhibitor on Ewing's family tumor cells. Int J Cancer 2005; 116: 385–94. [DOI] [PubMed] [Google Scholar]
- 28. Nishimori H, Sasaki Y, Yoshida K et al . The Id2 gene is a novel target of transcriptional activation by EWS–ETS fusion proteins in Ewing family tumors. Oncogene 2002; 21: 8302–9. [DOI] [PubMed] [Google Scholar]
- 29. Owen LA, Kowalewski AA, Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing's sarcoma. PLoS ONE 2008; 3: e1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Allander SV, Illei PB, Chen Y et al . Expression profiling of synovial sarcoma by cDNA microarrays: association of ERBB2, IGFBP2, and ELF3 with epithelial differentiation. Am J Pathol 2002; 161: 1587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagayama S, Katagiri T, Tsunoda T et al . Genome‐wide analysis of gene expression in synovial sarcomas using a cDNA microarray. Cancer Res 2002; 62: 5859–66. [PubMed] [Google Scholar]
- 32. Fukukawa C, Hanaoka H, Nagayama S et al . Radioimmunotherapy of human synovial sarcoma using a monoclonal antibody against FZD10. Cancer Sci 2008; 99: 432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friend SH, Bernards R, Rogelj S. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986; 323: 643–6. [DOI] [PubMed] [Google Scholar]
- 34. Wadayama B, Toguchida J, Shimizu T et al . Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res 1994; 54: 3042–8. [PubMed] [Google Scholar]
- 35. Li FP, Fraumeni JF Jr. Prospective study of a family cancer syndrome. JAMA 1982; 247: 2692–4. [PubMed] [Google Scholar]
- 36. Malkin D, Li FP, Strong LC, Fraumeni JF Jr. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–8. [DOI] [PubMed] [Google Scholar]
- 37. Toguchida J, Yamaguchi T, Ritchie B et al . Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res 1992; 52: 6194–9. [PubMed] [Google Scholar]
- 38. Walkley CR, Qudsi R, Sankaran VG et al . Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Gene Dev 2008; 22: 1662–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hanada K, Hickson D. Molecular genetics of RecQ helicase disorders. Cell Mol Life Sci 2007; 64: 2306–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang LL, Gannavarapu A, Kozinetz CA et al . Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothumund‐Thomson syndrome. J Natl Cancer Inst 2003; 95: 669–74. [DOI] [PubMed] [Google Scholar]
- 41. Nishijo K, Nakayama T, Aoyama T et al . Mutation analysis of the RECQL4 gene in sporadic osteosarcomas. Int J Cancer 2004; 111: 367–72. [DOI] [PubMed] [Google Scholar]
- 42. Wei G, Lonardo F, Ueda T et al . CDK4 gene amplification in osteosarcoma: reciprocal relationship with INK4A gene alterations and mapping of 12q13 amplicons. Int J Cancer 1999; 80: 199–204. [DOI] [PubMed] [Google Scholar]
- 43. Maelandsmo GM, Berner JM, Florences VA et al . Homozygous deletion frequency and expression level of the CDKN2 gene in human sarcomas – relationship to amplification and mRNA levels of CDK4 and CCND1. Br J Cancer 1995; 72: 393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lonardo F, Ueda T, Huvos AG, Healey J, Ladanyi M. p53 and MDM2 alterations in osteosarcomas: correlation with clinicopathologic features and proliferative rate. Cancer 1997; 79: 1541–7. [PubMed] [Google Scholar]
- 45. Haydon RC, Deyrup A, Ishikawa A et al . Cytoplasmic and/or nuclear accumulation of the beta‐catenin protein is a frequent event in human osteosarcoma. Int J Cancer 2002; 102: 338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang P, Yang Y, Zweidler‐McKay PA, Hughes DPM. Critical role of Notch signaling in osteosarcoma invasion and metastasis. Clin Cancer Res 2008; 14: 2962–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47. Warzecha J, Göttig S, Chow KU et al . Inhibition of osteosarcoma cell proliferation by the Hedgehog‐inhibitor cyclopamine. J Chemother 2007; 19: 554–61. [DOI] [PubMed] [Google Scholar]
- 48. Levine RA, Forest T, Smith C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol 2002; 39: 372–8. [DOI] [PubMed] [Google Scholar]
- 49. Coltella N, Manara MC, Cerisano UV. Role of the MET/HGF receptor in proliferation and invasive behavior of osteosarcoma. FASEB J 2003; 17: 1162–4. [DOI] [PubMed] [Google Scholar]
- 50. Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT pathway in NOTCH1‐induced leukemia. Cell Cycle 2008; 7: 965–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin‐mediated metastatic behavior in a murine model of osteosarcoma. Cancer Res 2005; 65: 2406–11. [DOI] [PubMed] [Google Scholar]
- 52. McClatchey AI, Saotome I, Mercer K et al . Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev 1998; 12: 1121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang W, Chang HY, Fei T, Wu H, Chen Y‐G. GSKβ mediates suppression of cyclin D2 expression by tumor suppressor PTEN. Oncogene 2007; 26: 2471–82. [DOI] [PubMed] [Google Scholar]
- 54. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF‐1R‐dependent mechanism. Oncogene 2007; 26: 1932–40. [DOI] [PubMed] [Google Scholar]
- 55. Lee YF, John M, Edwards S et al . Molecular classification of synovial sarcomas, leiomyosarcomas and malignant fibrous histiocytomas by gene expression profiling. Br J Cancer 2003; 88: 510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakayama R, Nemoto T, Takahashi H et al . Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous histiocytoma. Mod Pathol 2007; 20: 749–59. [DOI] [PubMed] [Google Scholar]
- 57. Nielsen TO, West RB, Linn SC et al . Molecular characterization of soft tissue tumours: a gene expression study. Lancet 2002; 359: 1301–7. [DOI] [PubMed] [Google Scholar]
- 58. Lee YF, John M, Falconer A et al . A gene expression signature associated with metastatic outcome in human leiomyosarcomas. Cancer Res 2004; 64: 7201–4. [DOI] [PubMed] [Google Scholar]
- 59. Riggi N, Suvà ML, Suvà D et al . EWS‐FLI‐1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res 2008; 68: 2176–85. [DOI] [PubMed] [Google Scholar]
- 60. Tirode F, Laud‐Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007; 11: 421–9. [DOI] [PubMed] [Google Scholar]
- 61. Riggi N, Cironi L, Provero P et al . Expression of the FUS‐CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res 2006; 66: 7016–23. [DOI] [PubMed] [Google Scholar]
- 62. Perez‐Losada J, Pintado B, Gutierrez‐Adan A et al . The chimeric FUS/TLS‐CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene 2000; 19: 2413–22. [DOI] [PubMed] [Google Scholar]
- 63. Keller C, Arenkiel BR, Coffin CM, El‐Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3: Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 2004; 18: 2614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell 2007; 11: 375–88. [DOI] [PubMed] [Google Scholar]
- 65. Segal NH, Pavlidis P, Antonescu CR et al . Classification and subtype prediction of adult soft tissues by functional genomics. Am J Pathol 2003; 163: 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Baird K, Davis S, Antonescu CR et al . Gene expression profiling of human sarcomas: Insights into sarcoma biology. Cancer Res 2005; 65: 9226–35. [DOI] [PubMed] [Google Scholar]