

Abstract
Aprepitant is a new neurokinin‐1 (NK1) receptor antagonist developed as a treatment for chemotherapy‐induced nausea and vomiting (CINV). To evaluate the efficacy and safety of aprepitant used in combination with standard therapy (granisetron and dexamethasone), we conducted a multicenter, phase II, placebo‐controlled, double‐blind, randomized study in Japanese cancer patients who received cancer chemotherapy including cisplatin (≥70 mg/m2). Aprepitant was administered for 5 days. A total of 453 patients were enrolled. In the three study groups, (i) standard therapy, (ii) aprepitant 40/25 mg (40 mg on day 1 and 25 mg on days 2–5) and (iii) aprepitant 125/80 mg (125 mg on day 1 and 80 mg on days 2–5), the percentage of patients with complete response (no emesis and no rescue therapy) was 50.3% (75/149 subjects), 66.4% (95/143 subjects) and 70.5% (103/146 subjects), respectively. This shows that efficacy was significantly higher in the aprepitant 40/25 mg and 125/80 mg groups than in the standard therapy group (χ2 test [closed testing procedure]: P = 0.0053 and P = 0.0004, respectively) and highest in the aprepitant 125/80 mg group. The delayed phase efficacy (days 2–5) was similar to the overall phase efficacy (days 1–5), indicating that aprepitant is effective in the delayed phase when standard therapy is not very effective. In terms of safety, aprepitant was generally well tolerated in Japanese cancer patients. (ClinicalTrials.gov number, NCT00212602.) (Cancer Sci 2010; 101: 2455–2461)
Chemotherapy‐induced nausea and vomiting (CINV) is a common adverse event observed in more than 90% of patients treated with highly emetogenic antitumor agents, especially cisplatin.( 1 , 2 )
In general, CINV persists for approximately 5 days.( 3 ) The CINV that occurs within 24 h after administration of antitumor agents is defined as acute phase CINV, and delayed phase CINV occurs 2–5 days after administration of antitumor agents. It has been reported that the incidence of nausea/vomiting induced by cisplatin, the most highly emetogenic antitumor agent, is 98% in the acute phase and 77% in the delayed phase after administration of 50 mg/m2 or higher doses without preventive treatment.( 4 )
As of October 2009 in Japan, the standard antiemetic therapy for CINV is a 5‐HT3 receptor antagonist plus dexamethasone. In the presence of this therapy, CINV is known to occur in approximately 25 and 50% of patients treated with highly emetogenic antitumor agents in the acute and delayed phases, respectively.( 5 ) In addition, the percentage of patients who developed CINV under standard antiemetic therapy increased from approximately 50% in the first course of cancer chemotherapy to approximately 75% in the sixth course.( 6 , 7 ) In several clinical studies of a 5‐HT3 receptor antagonist with dexamethasone, no efficacy was demonstrated for CINV in the delayed phase.( 3 , 8 )
Aprepitant is a neurokinin‐1 (NK1) receptor antagonist developed as a treatment for CINV. It acts by inhibiting the binding of substance P to the NK1 receptor in the vomiting center, and when used with standard antiemetic therapy (5‐HT3 receptor antagonist and dexamethasone) it has been shown to be effective for CINV (especially for delayed CINV) induced by highly and moderately emetogenic cancer chemotherapy.( 9 , 10 , 11 , 12 ) Overseas guidelines recommend the use of aprepitant in combination with a 5‐HT3 receptor antagonist and dexamethasone to prevent nausea/vomiting induced by highly and moderately emetogenic cancer chemotherapy.( 13 , 14 , 15 ) While the efficacy and safety of aprepitant has been established in other countries, no study has been conducted in Japanese patients.
Therefore, we conducted a multicenter, placebo‐controlled, double‐blind, randomized, parallel comparative study to evaluate the efficacy and safety of aprepitant plus standard therapy (granisetron and dexamethasone) to prevent CINV in Japanese cancer patients undergoing treatment with chemotherapy including a highly emetogenic cisplatin‐based regimen (≥70 mg/m2).
Materials and Methods
Patient selection. Japanese cancer patients aged 20 years and older who received cancer chemotherapy including cisplatin at a dose of ≥70 mg/m2 were included in the present study. If at least moderately (Hesketh level ≥3) emetogenic antitumor agent other than cisplatin was concomitantly used, it had to be administered on the same day with cisplatin (day 1). With a Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0–2 and an estimated life expectancy of at least 3 months, patients had to meet the following laboratory criteria: white blood cell count ≥3000/mm3; neutrophil count ≥1500/mm3; platelet count ≥100 000/mm3; aspartate aminotransferase (AST) (glutamic oxaloacetic transaminase (GOT)) and alanine aminotransferase (ALT) (glutamic pyruvic transaminase (GPT)) ≤2.5 × upper limit of the normal range at the facility; total bilirubin ≤1.5 × upper limit of the normal range at the facility; and creatinine ≤1.5 × upper limit of the normal range at the facility. The following patients were excluded from the study: patients with a risk of vomiting for other reasons (symptomatic brain metastasis, meningeal infiltration, epilepsy, active peptic ulcer, gastrointestinal obstruction, concomitant abdominal, pelvic radiotherapy, etc.); and pregnant, nursing or possibly pregnant women. After the protocol and informed consent form were approved by the Institutional Review Board (IRB) at each facility, patients who gave written informed consent were enrolled.
Study design. This was a multicenter, placebo‐controlled, double‐blind, randomized, parallel comparative study and conducted in a total of 127 institutions in Japan. Patients who met all of the inclusion criteria and none of the exclusion criteria were allocated to the aprepitant 125/80 mg group (oral administration at a dose of 125 mg on day 1 and a dose of 80 mg on days 2–5), aprepitant 40/25 mg group (oral administration at a dose of 40 mg on day 1 and a dose of 25 mg on days 2–5) or the standard therapy group (oral administration of placebo on days 1–5). Treatment assignment (dynamic allocation) was performed using a minimization method for balancing four factors (sex, presence or absence of at least one emetogenic antitumor agent used in combination with cisplatin, presence or absence of previous treatment with cisplatin, and institution) between the treatment and control groups. All patients received standard therapy consisting of intravenous granisetron (40 μg/kg on day 1) and dexamethasone. The dose of each drug in each group is shown in Table 1. Because it is a substrate and inhibitor of CYP3A4, aprepitant is known to increase the plasma dexamethasone concentration.( 9 ) Therefore, to achieve comparable plasma levels of dexamethasone in the presence and absence of aprepitant in this study, the dose of dexamethasone was 6 mg on day 1 and 4 mg on days 2 and 3 in the 125/80 mg group (50% of the dose in the absence of aprepitant), and 8 mg on day 1 and 6 mg on days 2 and 3 in the 40/25 mg group (75% of the dose in the absence of aprepitant).
Table 1.
Dose of each drug in each group
| Treatment group | Drug | Day 1 | Days 2–3 | Days 4–5 |
|---|---|---|---|---|
| Aprepitant 125/80 mg regimen | Aprepitant (po) | 125 mg | 80 mg | 80 mg |
| Dexamethasone (i.v.) | 6 mg | 4 mg | – | |
| Granisetron (i.v.) | 40 μg/kg | – | – | |
| Aprepitant 40/25 mg regimen | Aprepitant (po) | 40 mg | 25 mg | 25 mg |
| Dexamethasone (i.v.) | 8 mg | 6 mg | – | |
| Granisetron (i.v.) | 4l0 μg/kg | – | – | |
| Standard therapy | Aprepitant (po) | Placebo | Placebo | Placebo |
| Dexamethasone (i.v.) | 12 mg | 8 mg | – | |
| Granisetron (i.v.) | 40 μg/kg | – | – |
i.v., intravenous; po, per os.
On day 1, administration of the first at least moderately (Hesketh level ≥3) emetogenic antitumor agent (including cisplatin) was started 1.5 h after oral administration of aprepitant or placebo and 30 min after intravenous administration of granisetron and dexamethasone (over 30 min or less). On day 2 and thereafter, aprepitant or placebo was orally administered in the morning, followed by intravenous administration of dexamethasone 1 h later.
Concomitant use of other antiemetics was prohibited from 48 h before day 1 to the morning of day 6, except for rescue therapy for CINV.
Assessments. Patients recorded the onset of vomiting and nausea in a symptom diary from day 1 to the morning of day 6. Vomiting was defined as at least one episode of emesis or gagging and was distinguished from other episodes if emesis was not observed for at least 1 min. For nausea, patients recorded the most severe intensity during the previous 24‐h period based on a 4‐point scale (0, none; 1, mild; 2, moderate; 3, severe). After rescue therapy was administered (defined as a drug prescribed by a physician to reduce nausea/vomiting), the date/time, name of the drug, dose and reason for use were recorded. Efficacy was evaluated from the start of administration of the first at least moderately emetogenic antitumor agent (including cisplatin) on day 1 (also defined as 0 h) to the morning on day 6 (120 h).
Safety was evaluated on the basis of physical examination findings (which included vital signs, bodyweight, general laboratory tests and electrocardiogram) and adverse events (clinical findings and laboratory values recorded until day 15). Toxicity grades were assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI‐CTCAE) v3.0.
Statistical analysis. Based on the results of combined analysis from overseas phase III studies (studies 052 and 054)( 16 ) that the percentage of patients with complete response in the overall phase was 67.7% in the 125/80 mg group and 47.8% in the placebo group, a sample size of 115 subjects per group was estimated to be required to provide a power of approximately 80%. On the assumption that approximately 15–20% of subjects would be withdrawn or drop out, a target sample size of 130–140 subjects per group (390–420 subjects in total) was selected. The analysis for efficacy was performed on the full analysis set (FAS) data. The FAS population was the set of all randomized subjects after minimal and justified elimination, who were treated with granisetron hydrochloride and dexamethasone phosphate (at least one dose), who kept a symptom diary, and who received at least one dose of the study drug. The primary efficacy end‐point was the percentage of patients with complete response (defined as no emetic episode and no rescue therapy). The secondary efficacy end‐points were the percentage of patients with: (i) no emesis; (ii) no rescue therapy; (iii) complete protection (no emesis, no rescue therapy and no significant nausea [nausea score: 0 and 1]); (iv) total control (no emesis, no rescue therapy and no nausea [nausea score: 0]); (v) no significant nausea (nausea score: 0 and 1); and (vi) no nausea (nausea score: 0). Both the primary and secondary end‐points were assessed in the overall phase (days 1–5), acute phase (day 1) and delayed phase (days 2–5). The χ2 test was performed at a two‐tailed significance level of 0.05 to compare the efficacy between standard therapy and the 125/80 mg groups, and between standard therapy and the 40/25 mg groups. For a complete response in the overall phase, a closed testing procedure was used to control the overall Type I error at 0.05 beginning with the 125/80 mg group and then the 40/25 mg group.
The population used for analysis of the safety data included subjects with the target disease who received at least one dose of the study drug. The incidence of adverse events and adverse drug reactions (adverse events for which a causal relationship could not be ruled out) was calculated in each group and compared between groups using the χ2 test at a two‐tailed significance level of 0.05.
Results
Patients. A total of 453 patients were enrolled in the present study and allocated to one of three groups (151 patients per group) (Fig. 1). Of these, 449 patients were included in the safety analysis set, 439 subjects were included in the FAS. Table 2 shows their demographic characteristics. All baseline factors were similar across the groups, including age, sex, height, bodyweight and cisplatin dose, as well as known risk factors for CINV (female, motion sickness, history of CINV, etc.).
Figure 1.
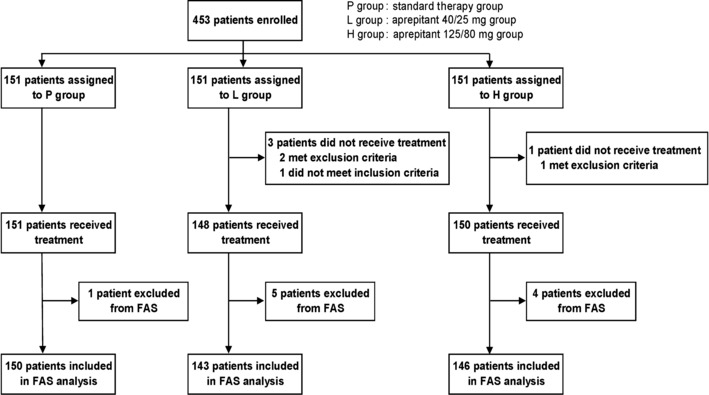
Study flow chart. FAS, full analysis set.
Table 2.
Characteristics of patients
| Characteristics | Aprepitant 125/80 mg + standard therapy (n = 146) | Aprepitant 40/25 mg + standard therapy (n = 143) | Standard therapy (n = 150) |
|---|---|---|---|
| Sex (%) | |||
| Female | 24.0 | 25.2 | 25.3 |
| Male | 76.0 | 74.8 | 74.7 |
| Age (%) | |||
| ≥65 years | 37.0 | 51.7 | 42.0 |
| <65 years | 63.0 | 48.3 | 58.0 |
| Mean (SD) | 60.5 (9.7) | 63.3 (9.4) | 62.2 (9.8) |
| Use of concurrent emetogenic chemotherapy† (% of patients) | 17.8 | 15.4 | 20.0 |
| Cisplatin dose (% of patients) | |||
| <70 | 0.0 | 0.0 | 0.0 |
| ≥70, <80 | 41.8 | 42.0 | 46.7 |
| ≥80, <90 | 56.2 | 57.3 | 52.7 |
| ≥90, <100 | 0.0 | 0.0 | 0.0 |
| ≥100 | 2.1 | 0.7 | 0.7 |
| Mean dose (mg/m2) | 76.9 | 76.9 | 76.2 |
| Alcoholic drinks/week (at the time of informed consent) (% of patients) | |||
| None | 57.5 | 61.5 | 58.0 |
| Several times per month | 10.3 | 6.3 | 10.7 |
| 3–4 times per week | 6.2 | 3.5 | 7.3 |
| Almost every day | 26.0 | 28.7 | 24.0 |
| History of morning sickness (% of patients) | 43.3 | 38.2 | 44.1 |
| History of motion sickness (% of patients) | 9.6 | 4.2 | 11.3 |
| History of cisplatin chemotherapy (% of patients) | 17.8 | 15.4 | 17.3 |
| History of chemotherapy except cisplatin (% of patients) | 19.9 | 24.5 | 18.7 |
| History of CINV except cisplatin chemotherapy (% of patients) | 41.4 | 37.1 | 42.9 |
| Primary cancer diagnosis (% of patients)‡ | (n = 150) | (n = 148) | (n = 151) |
| Respiratory | 73.3 | 73.0 | 70.2 |
| Urogenital | 16.7 | 13.5 | 14.6 |
| Digestive | 4.0 | 5.4 | 4.6 |
| Eyes/ears/nose/throat | 3.3 | 4.7 | 7.3 |
| Other | 3.3 | 3.4 | 3.3 |
†Hesketh level ≥3; analysis population: full analysis set. ‡Analysis population: safety analysis set. SD, standard deviation.
Efficacy. The primary end‐point was the percentage of patients with complete response (no emesis and no rescue therapy) over the entire treatment course, and the results for each treatment are shown in Figure 2. Efficacy of aprepitant was significantly higher than efficacy of standard therapy (125/80 mg group, 70.5% [103/146]; 40/25 mg group, 66.4% [95/143]; standard therapy group, 50.3% [75/149]; 125/80 mg group versus standard therapy group, P < 0.001; 40/25 mg group versus standard therapy group, P < 0.01). The acute‐ and delayed‐phase efficacies are shown in Figure 3. While the delayed phase efficacy (125/80 mg group, 72.6% [106/146]; 40/25 mg group, 69.9% [100/143]; standard therapy group, 51.7% [77/149]; 125/80 mg group versus standard therapy group, P < 0.001; 40/25 mg group versus standard therapy group, P < 0.01) was similar to the overall phase efficacy, the percentage of patients with a complete response was higher (but not significantly higher) in both aprepitant groups than in the standard therapy group in the acute phase (125/80 mg group, 87.0% [127/146]; 40/25 mg group, 90.2% [129/143]; standard therapy group, 83.3% [125/150]). In addition, subgroup analysis of patients with a complete response in the overall phase performed after stratification for sex, age and previous treatment with cisplatin showed that the overall phase efficacy of aprepitant was consistently higher than that of standard therapy, irrespective of these factors (Table 3).
Figure 2.
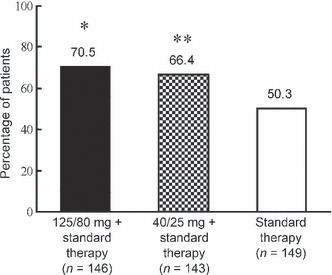
Percentage of patients with a complete response (no emesis and no rescue therapy) in the overall phase (days–5) of aprepitant treatment. *P < 0.001 versus standard therapy group. **P < 0.01 versus standard therapy group.
Figure 3.
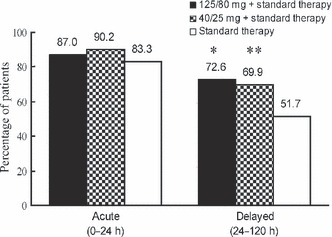
Percentage of patients with a complete response (no emesis and no rescue therapy) in the acute phase (day 1) and the delayed phase (days 2–5). *P < 0.001 versus standard therapy group. **P < 0.01 versus standard therapy group.
Table 3.
Subgroup analysis of the percentage of patients with complete response over the course of treatment
| Patients with complete response (%) | |||
|---|---|---|---|
| Aprepitant 125/80 mg + standard therapy (n = 146) | Aprepitant 40/25 mg + standard therapy (n = 143) | Standard therapy (n = 149) | |
| Sex | |||
| Female | 68.6 | 50.0 | 36.8 |
| Male | 71.2 | 72.0 | 55.0 |
| Age (years) | |||
| ≥65 years | 72.2 | 71.6 | 51.6 |
| <65 years | 69.6 | 60.9 | 49.4 |
| History of cisplatin chemotherapy | |||
| Yes | 65.4 | 54.5 | 19.2 |
| No | 71.7 | 68.6 | 56.9 |
For each secondary end‐point and each treatment, the overall phase, acute phase and delayed phase efficacies are shown in Table 4. In the overall phase, the percentage of patients with “no emesis” was significantly higher in the 125/80 mg and 40/25 mg groups than in the standard therapy group (P < 0.001 for both). The percentage of patients with “complete protection” and “no significant nausea” was significantly higher in the 125/80 mg group than in the standard therapy group (P < 0.01 and P < 0.05, respectively), but was not significantly different between the 40/25 mg and standard therapy groups. The percentage of patients with “total control,”“no rescue therapy” or “no nausea” was numerically higher in the 125/80 mg and 40/25 mg groups, but not significantly different from the standard therapy group. In the acute phase, secondary end‐points were not significantly different between the aprepitant groups and the standard therapy group. In the delayed phase, on the other hand, the percentage of patients with “no emesis” was significantly higher in the 125/80 mg and 40/25 mg groups than in the standard therapy group (P < 0.0001 for both), whereas the percentage of patients with “complete protection” and “no significant nausea” was significantly higher in the 125/80 mg group than in the standard therapy group (P < 0.01 for both), but was not significantly different between the 40/25 mg and standard therapy groups.
Table 4.
Percentage of patients reaching efficacy end‐points, by study phase and treatment group, using data obtained after dose adjustment
| Treatment group | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Overall phase (0–120 h) | Acute phase (0–24 h) | Delayed phase (24–120 h) | |||||||
| End‐point | A 125/80 | A 40/25 | ST | A 125/80 | A 40/25 | ST | A 125/80 | A 40/25 | ST |
| Total no. | 146 | 143 | 149 | 146 | 143 | 150 | 146 | 143 | 149 |
| No emesis (%) | 76.7* | 74.1* | 51.0 | 89.7 | 90.2 | 83.3 | 78.8* | 77.6* | 53.0 |
| No rescue (%) | 80.8 | 80.4 | 79.2 | 95.2 | 98.6 | 96.0 | 82.2 | 81.1 | 79.9 |
| No nausea (%) | 34.2 | 28.0 | 24.2 | 67.1 | 63.6 | 66.0 | 34.9 | 30.1 | 26.2 |
| No significant nausea (%) | 69.2 | 60.8 | 55.7 | 90.4 | 84.6 | 88.0 | 72.6** | 60.8 | 56.4 |
| Complete protection (%) | 61.6** | 53.1 | 43.0 | 83.6 | 80.4 | 82.0 | 65.1** | 55.2 | 44.3 |
| Total control (%) | 33.6 | 28.0 | 24.2 | 66.4 | 63.6 | 64.7 | 34.2 | 30.1 | 26.2 |
*P < 0.001. **P < 0.01. A 125/80: standard therapy plus aprepitant 125 mg on day 1 and aprepitant 80 mg on days 2–5; A 40/25: standard therapy plus aprepitant 40 mg on day 1 and 25 mg on days 2–5; No nausea: nausea score 0; No significant nausea: nausea score 0 and 1; Complete protection: no emesis, no rescue therapy and no significant nausea (nausea score 0 and 1); Total control: no emesis, no rescue therapy and no nausea (nausea score 0). ST, standard therapy.
Tolerability. All 453 enrolled subjects were included in the safety analysis. Adverse events that occurred within 15 days after the start of treatment with the study drug are summarized in Table 5. In all groups, the incidence of adverse events was high and not different across the groups. The incidence of drug‐related adverse events was also not significantly different between each of the aprepitant groups and the standard therapy group. In addition, the distribution of toxicity grades (NCI‐CTCAE grades indicating severity of adverse events or drug‐related adverse events) was not markedly different across the groups. In terms of clinical findings, the most common adverse event was anorexia. Other adverse events (clinical findings) with an incidence of ≥10% in any group were constipation, hiccups, malaise, diarrhea, nausea, vomiting, pyrexia and insomnia. In terms of laboratory values, the incidence of common adverse events (including decreased white blood cell count, neutrophil count, platelet count, lymphocyte count and decreased hemoglobin) were similar across the groups. The incidence of the most common drug‐related adverse events (hiccups) was similar across the groups (125/80 mg group, 10.0%; 40/25 mg group, 6.1%; standard therapy group, 9.3%). The incidence of febrile neutropenia as well as that of other infection‐related adverse events was not different across the groups. Since interactions between aprepitant (which has an inhibitory effect on CYP3A4) and antitumor agents metabolized by CYP3A4 are possible, the correlation of the incidence of adverse events and drug‐related adverse events with the concomitant use of antitumor agents metabolized by CYP3A4 (cyclophosphamide, etoposide, vincristine sulfate, vinblastine sulfate, vindesine sulfate, irinotecan hydrochloride, docetaxel hydrate, vinorelbine ditartrate, ifosfamide and gefitinib) was examined. Antitumor agents metabolized by CYP3A4 were used in 103 (68.7%) of 150 patients in the 125/80 mg group, 93 (62.8%) of 148 patients in the 40/25 mg group and 93 (61.6%) of 151 patients in the standard therapy group. No apparent correlation was observed between the incidence of adverse events or adverse drug reactions and concomitant use of antitumor agents metabolized by CYP3A4.
Table 5.
Summary of adverse events
| Percentage of patients | Treatment group | ||
|---|---|---|---|
| Aprepitant 125/80 mg + standard therapy (n = 150) | Aprepitant 40/25 mg + standard therapy (n = 148) | Standard therapy (n = 151) | |
| With ≥1 adverse event | 99.3 | 99.3 | 99.3 |
| With drug‐related adverse events† | 23.3 | 18.9 | 19.9 |
| With serious adverse events | 6.0 | 6.8 | 2.6 |
| Discontinued due to adverse events | 0.7 | 1.4 | 0.0 |
| With most common adverse events‡ | |||
| Anorexia | 48.0 | 59.5 | 53.6 |
| Constipation | 38.7 | 42.6 | 45.7 |
| Hiccups | 43.3 | 33.1 | 37.1 |
| Malaise | 25.3 | 31.8 | 17.9 |
| Diarrhea | 21.3 | 26.4 | 26.5 |
| Nausea | 36.7 | 41.9 | 35.1 |
| Vomiting | 14.7 | 14.9 | 19.2 |
| Pyrexia | 9.3 | 12.8 | 13.9 |
| Insomnia | 4.7 | 7.4 | 10.6 |
| With febrile neutropenia | 4.0 | 4.1 | 6.6 |
†Determined by the investigator as possibly drug related, probably drug related or definitely drug related. ‡Incidence ≥10% in at least one group. There were no statistically significant (P > 0.1) differences in the risk of adverse events between the treatment groups. Statistical testing was not performed for individual common adverse events. Nausea and vomiting were considered adverse events if they occurred after day 5 of the study, or at any time if they were determined by the investigator to be serious or drug related, or if they resulted in discontinuation.
The incidence of serious adverse events was not significantly different across the groups. No serious adverse event was considered by the investigator to be related to aprepitant. Serious adverse events led to the death of one patient in the standard therapy group and one in the 125/80 mg group. The former died of febrile neutropenia, acute respiratory distress syndrome (ARDS) and septic shock, and the latter died of cardiac failure. Neither case was considered to be related to aprepitant.
In addition, no clinically significant abnormality was observed in the vital signs, 12‐lead electrocardiogram or bodyweight in the aprepitant groups.
Discussion
As of October 2009 in Japan, 5‐HT3 receptor antagonist plus dexamethasone is the only standard antiemetic therapy for CINV. Approximately 25 and 50% of patients treated with highly emetogenic antitumor agents fail to respond to such therapy in the acute and delayed phases, respectively.( 5 ) This study was conducted in Japanese cancer patients who received cancer chemotherapy including cisplatin at a dose of ≥70 mg/m2 to evaluate the efficacy and safety of adding aprepitant to standard antiemetic therapy (5‐HT3 receptor antagonist and dexamethasone). It was shown that the percentage of patients with a complete response (the primary efficacy end‐point) in the overall phase including both the acute (day 1) and delayed (days 2–5) phases was significantly higher in the aprepitant groups than in the standard therapy group, irrespective of sex, age or previous treatment with cisplatin. In the acute phase, the percentage of patients with a complete response was not significantly different between the aprepitant and the standard therapy groups. In the delayed phase as well as the overall phase, on the other hand, the percentage of patients with a complete response was significantly higher in the aprepitant groups. These results demonstrated the efficacy of aprepitant for CINV in the delayed phase, when 5‐HT3 receptor antagonist plus dexamethasone, the current standard antiemetic therapy in Japan, is not very effective. Although the percentage of patients with a complete response in the overall phase, the primary efficacy end‐point, was significantly higher in both aprepitant groups (40/25 and 125/80 mg) than in the standard therapy group, the percentages of patients with “complete protection” and “no significant nausea” in the overall phase and delayed phase, which were secondary end‐points, were statistically significantly higher only in the 125/80 mg group. In addition, the incidence or severity of adverse events was not markedly different between each aprepitant and standard therapy groups. Based on these results, the recommended dose of aprepitant is considered to be 125/80 mg (oral administration at a dose of 125 mg on day 1 and a dose of 80 mg on days 2–5) in Japanese cancer patients.
In the present study, unlike the overseas studies,( 9 , 10 ) efficacy estimated using either the primary measure (the percentage of patients with complete response) or other secondary measures was not significantly greater in either aprepitant group in the acute phase. Nonetheless, the percentage of patients with a complete response (125/80 mg group, 87.0%; 40/25 mg group, 90.3%) in the acute phase in the present study was not inferior to that in overseas studies (89.2%,( 9 ) 82.8%( 10 )). In this study, the percentage of patients with a complete response in the standard therapy group in the acute phase was substantially higher (83.3%) than in the overseas studies (78.1%,( 9 ) 68.4%,( 10 )), indicating that the sample size was too small to detect any additional efficacy attributable to aprepitant for CINV in the acute phase.
In terms of safety, the incidence of adverse events was not different between the aprepitant and standard therapy groups, and the severity of adverse events was not markedly different across the groups. The incidence of serious adverse events was not significantly different across the groups, and no serious adverse event was considered by the investigator to be related to aprepitant. Since aprepitant has an inhibitory effect on CYP3A4, interactions between aprepitant and antitumor agents metabolized by CYP3A4 were a concern. Supporting overseas reports that failed to find notable interactions between aprepitant and docetaxel or vinorelbine,( 17 , 18 ) the present study showed that the incidence of adverse events was not affected by combining aprepitant with antitumor agents metabolized by CYP3A4. These results showed that the safety of aprepitant is maintained irrespective of which metabolic pathways are disrupted by the antitumor agents.
It is known that aprepitant increases the plasma concentration of dexamethasone administered in combination,( 19 ) and that this increase probably accounts for the higher incidence of serious infections such as febrile neutropenia associated with the concomitant use of aprepitant.( 11 ) Therefore, in this study the dose of dexamethasone was adjusted so that comparable dexamethasone levels could be achieved in all groups. Population pharmacokinetic analysis of the plasma dexamethasone concentration found that aprepitant at doses of 125/80 mg and 40/25 mg reduced the clearance of dexamethasone to approximately 50% and 75%, respectively, of that in the absence of aprepitant in Japanese patients,( 20 ) demonstrating the appropriateness of dose adjustment of dexamethasone in the present study. The appropriateness was also supported by data showing no increase in the incidence of serious infections such as febrile neutropenia in the aprepitant combination groups.
In conclusion, aprepitant used in combination with standard antiemetic therapy (5‐HT3 receptor antagonist and corticosteroid) was well tolerated and very effective in preventing CINV associated with highly emetogenic antitumor agents in Japanese cancer patients.
Trial members. Koichi Goto (protocol evaluation committee), Nobuyuki Yamamoto (protocol evaluation committee), Hiroshi Saito (protocol evaluation committee), Yoichi Nakanishi (protocol evaluation committee), Hiroyuki Iinuma (ECG committee), Chikuma Hamada (medical statistical adviser).
Principal investigators. Yuka Fujita (National Hospital Organization Dohoku National Hospital); Masahiro Yanase (Sunagawa City Medical Center); Koichi Yamazaki (Hokkaido University Hospital); Atsushi Taira (Sapporo Medical University Hospital); Satoshi Takahashi (Sapporo Medical University Hospital); Kenji Numahata (Tohoku University Hospital); Akira Inoue (Tohoku University Hospital); Masashi Tanaka (Miyagi Cancer Center); Tomonori Habuchi (Akita University Hospital); Yoshihiko Tomita (Yamagata University Hospital); Satoru Ishikawa (Hitachi General Hospital); Takefumi Saito (National Hospital Organization Ibaraki Higashi National Hospital); Hideyuki Akaza (Tsukuba University Hospital); Koji Kikuchi (Tsukuba Medical Center Hospital); Kiyoshi Mori (Tochigi Cancer Center); Yoshiki Ishii (Dokkyo Medical University Hospital); Ken‐ichiro Yoshida (Dokkyo Medical University Hospital); Kazuhiro Suzuki (Gunma University Hospital); Shinichi Ishihara (National Hospital Organization, Nishigunma National Hospital); Kyoko Tanaka (National Hospital Organization Saitama National Hospital) Katsuhiko Aoyama (National Hospital Organization Higashi Saitama Hospital); Shuichi Yoneda (Saitama Cancer Center); Eishin Hoshi (Saitama Cardiovascular and Respiratory Center): Hidemitsu Funabashi (National Hospital Organization Chiba Medical Center); Tsutomu Numata (National Hospital Organization Chiba Medical Center); Yuichi Takiguchi (Chiba University Hospital); Tetsuro Moriya (Matsudo City Hospital); Toshio Yoshida (Nihon University Itabashi Hospital); Shigeo Horie (Teikyo University Hospital); Masahiro Tsuboi (Tokyo Medical University Hospital); Yoshikazu Hirano (The Fraternity Memorial Hospital); Kazuhiro Ishizaka (Kanto Central Hospital of the Mutual Aid Association of Public School Teachers); Ikuo Sekine (National Cancer Center Hospital); Takeshi Horai (The Cancer Institute Hospital of JFCR [Japanese Foundation for Cancer Research]); Akihiko Gemma (Nippon Medical School Hospital); Masahiko Shibuya (Tokyo Metropolitan Komagome Hospital); Yasuyuki Yoshizawa (Tokyo Medical and Dental University Hospital Faculty of Medicine); Tadashi Arai (Sempo Tokyo Takanawa Hospital); Tetsuo Sato (Jikei University Hospital); Masato Fujii (National Hospital Organization Tokyo Medical Center); Makoto Miyahara (Takagi Hospital); Shinichi Odama (Ome Municipal General Hospital); Shigeyuki Aoki (Showa General Hospital); Tomoyuki Yoshida (Tokyo Medical University Hachioji Medical Center); Akira Fujita (Tokyo Metropolitan Fuchu Hospital); Tomoyuki Goya (Kyorin University Hospital); Atsuhisa Tamura (National Hospital Organization Tokyo National Hospital); Haruhiko Kaguraoka (Higashi Yamato Hospital); Shigeru Minowada (International Medical Center of Japan); Takashi Ogura (Kanagawa Cardiovascular and Respiratory Center); Hiroshi Kunikane (Yokohama Municipal Citizen’s Hospital); Junichi Ishitoya (Yokohama City University Medical Center); Kazumasa Noda (Kanagawa Cancer Center); Michiaki Narusima (Showa University Fujigaoka Hospital); Noriyuki Masuda (Kitasato University Hospital); Sumio Noguchi (Yokosuka Kyosai Hospital); Kenji Eguchi (Tokai University Hospital); Tetsuyuki Morikawa (Yokohama Rosai Hospital); Kunio Yamaguchi (Yokohama Rosai Hospital); Mamoru Tsukuda (Yokohama City University Hospital); Hirohisa Yoshizawa (Niigata University Medical and Dental Hospital); Akira Iwashima (Nagaoka Chuo General Hospital); Muneharu Maruyama (Toyama University Hospital); Kiyoshi Koshida (National Hospital Organization Kanazawa Medical Center); Kazuo Kasahara (Kanazawa University Hospital); Koichi Nishi (Ishikawa Prefectural Central Hospital); Masayoshi Shimamura (Ishikawa Prefectural Central Hospital); Kazuhiro Okafuji (Fukuiken Saiseikai Hospital); Keishi Kubo (Shinshu University Hospital); Sekiya Koyama (National Hospital Organization National Chushin‐Matsumoto Hospital); Toshiyuki Sawa (Gifu Municipal Hospital); Jo Shindo (Ogaki Municipal Hospital); Takashi Kato (Shizuoka Red Cross Hospital); Kingo Chida (Hamamatsu University School of Medicine, University Hospital); Masakazu Takagi (Shizuoka General Hospital); Toshiaki Takahashi (Shizuoka Cancer Center); Yoshinori Hasegawa (Nagoya University Hospital); Toyoaki Hida (Aichi Cancer Center Hospital); Hiroaki Iwase (National Hospital Organization Nagoya Medical Center); Masashi Yamamoto (Nagoya Ekisaikai Hospital); Atsushi Watanabe (Anjo Kosei Hospital); Hiroshi Saito (Aichi Cancer Center Aichi Hospital); Yoshimasa Tanikawa (Kamo Hospital); Takuya Ikeda (Yokkaichi Municipal Hospital); Hidenori Ibata (National Hospital Organization Mie Chuo Medical Center); Hiroshi Hara (Kyoto Second Red Cross Hospital); Taketoshi Shimada (Kyoto Prefectural University of Medicine Hospital); Yoshiyuki Sasaki (National Hospital Organization Kyoto Medical Center); Mitsuo Nonomura (Kyoto‐Katsura Hospital); Atsuo Sato (National Hospital Organization Minami‐Kyoto Hospital); Shinzo Kudo (Osaka City University Hospital); Takashi Yana (Federation of National Public Service Personnel Mutual Aid Associations Otemae Hospital); Fumio Imamura (Osaka Medical Center for Cancer and Cardiovascular Diseases); Tatsuya Ioka (Osaka Medical Center for Cancer and Cardiovascular Diseases); Michiyuki Usami (Osaka Medical Center for Cancer and Cardiovascular Diseases); Shinji Atagi (National Hospital Organization Kinki‐Chuo Chest Medical Center); Kaoru Matsui (Osaka Prefectural Medical Center for Respiratory and Allergic Diseases); Kazuhiko Nakagawa (Kinki University Hospital); Tatsuhiko Kashii (Osaka City General Hospital); Yoshihiro Nishimura (Kobe University Hospital); Takashi Nishimura (Kobe City General Hospital); Shunichi Negoro (Hyogo Medical Center for Adults); Shigemichi Iwae (Hyogo Medical Center for Adults); Nobuyuki Katakami (Institute of Biomedical Research and Innovation); Shuichi Yano (National Hospital Organization Matsue National Hospital); Takeshi Isobe (Shimane University Hospital); Kenichi Gemba (Okayama Rosai Hospital); Minoru Fukuda (Kawasaki Medical School Hospital); Takuo Shibayama (National Hospital Organization Minami‐Okayama Medical Center); Isao Murakami (National Hospital Organization Higashihiroshima Medical Center); Akihito Yokoyama (Hiroshima University Hospital); Yukio Kimura (National Hospital Organization Iwakuni Clinical Center); Jun Araki (Yamaguchi Grand Medical Center); Keisuke Aoe (National Hospital Organization Sanyo Hospital); Tetsu Shinkai (National Hospital Organization Shikoku Cancer Center); Fumitaka Ogushi (National Hospital Organization Kochi National Hospital); Sadamu Ando (Kitakyushu Municipal Medical Center); Hiroshi Matsuura (Saiseikai Fukuoka General Hospital); Kentaro Watanabe (Fukuoka University Hospital); Akito Yamaguchi (Harasanshin Hospital); Koichi Takayama (Kyushu University Hospital); Hiroshi Aso (National Hospital Organization Fukuoka National Hospital); Masayuki Kawasaki (National Hospital Organization Fukuoka‐Higashi Medical Center); Shinichiro Hayashi (Saga University Hospital); Hiroaki Kikukawa (National Hospital Organization Kumamoto Medical Center); Mitsuhiro Matsumoto (Kumamoto University Hospital); Eiji Moriyama (National Hospital Organization Kumamoto Saishunso National Hospital); Hideki Yokoyama (National Hospital Organization Beppu Medical Center); Mutsuo Kuba (National Hospital Organization Okinawa National Hospital).
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This study was designed and funded by Ono Pharmaceutical Co., Ltd, and Merck & Co., Inc., the manufacturer of aprepitant.
References
- 1. Grunberg SM, Hesketh PJ. Control of chemotherapy‐induced emesis. N Engl J Med 1993; 329: 1790–6. [DOI] [PubMed] [Google Scholar]
- 2. Bender CM, McDaniel RW, Murphy‐Ende K et al. Chemotherapy‐induced nausea and vomiting. Clin J Oncol Nurs 2002; 6: 94–102. [DOI] [PubMed] [Google Scholar]
- 3. Tavorath R, Hesketh PJ. Drug treatment of chemotherapy‐induced delayed emesis. Drugs 1996; 52: 639–48. [DOI] [PubMed] [Google Scholar]
- 4. Kris MG, Cubeddu LX, Gralla RJ et al. Are more antiemetic trials with a placebo necessary? Report of patient data from randomized trials of placebo antiemetics with cisplatin Cancer 1996; 78: 2193–8. [DOI] [PubMed] [Google Scholar]
- 5. Gralla RJ, Osoba D, Kris MG et al. Recommendations for the use of antiemetics: evidence‐based, clinical practice guidelines. J Clin Oncol 1999; 17: 2971–94. Erratum in: J Clin Oncol 1999; 17: 3860. J Clin Oncol 2000; 18: 3064. [DOI] [PubMed] [Google Scholar]
- 6. De Wit R, Schmitz PIM, Verweij J et al. Analysis of cumulative probabilities shows that the efficacy of 5HT3 antagonist prophylaxis is not maintained. J Clin Oncol 1996; 14: 644–51. [DOI] [PubMed] [Google Scholar]
- 7. De Wit R, Van Den Berg H, Burghouts J et al. Initial high anti‐emetic efficacy of granisetron with dexamethasone is not maintained over repeated cycles. Br J Cancer 1998; 77: 1487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kris MG, Gralla RJ, Tyson LB, Clark RA, Cirrincione C, Groshen S. Controlling delayed vomiting: double‐blind, randomized trial comparing placebo, dexamethasone alone, and metoclopramide plus dexamethasone in patients receiving cisplatin. J Clin Oncol 1989; 7: 108–14. [DOI] [PubMed] [Google Scholar]
- 9. Hesketh PJ, Grunberg SM, Gralla RJ et al. The oral neurokinin‐1 antagonist aprepitant for the prevention of chemotherapy‐induced nausea and vomiting: a multinational, randomized, double‐blind, placebo‐controlled trial in patients receiving high‐dose cisplatin‐‐the Aprepitant Protocol 052 Study Group. J Clin Oncol 2003; 21(22): 4112–9. Epub 2003 Oct 14. [DOI] [PubMed] [Google Scholar]
- 10. Poli‐Bigelli S, Rodrigues‐Pereira J, Carides AD et al. Addition of the neurokinin 1 receptor antagonist aprepitant to standard antiemetic therapy improves control of chemotherapy‐induced nausea and vomiting. Results from a randomized, double‐blind, placebo‐controlled trial in Latin America. Cancer 2003; 97(12): 3090–8. [DOI] [PubMed] [Google Scholar]
- 11. Chawla SP, Grunberg SM, Gralla RJ et al. Establishing the dose of the oral NK1 antagonist aprepitant for the prevention of chemotherapy‐induced nausea and vomiting. Cancer 2003; 97(9): 2290–300. [DOI] [PubMed] [Google Scholar]
- 12. Warr DG, Hesketh PJ, Gralla RJ et al. Efficacy and tolerability of aprepitant for the prevention of chemotherapy‐induced nausea and vomiting in patients with breast cancer after moderately emetogenic chemotherapy. J Clin Oncol 2005; 23: 2822–30. [DOI] [PubMed] [Google Scholar]
- 13. American society of clinical oncology , Kris MG, Hesketh PJ, Somerfield MR et al. American society of clinical oncology guideline for antiemetics in oncology: update 2006. J Clin Oncol 2006; 24: 2932–47. [DOI] [PubMed] [Google Scholar]
- 14. Roila F, Hesketh PJ, Herrstedt J. Antiemetic Subcommittee of the Multinational Association of Supportive Care in Cancer (MASCC) . . Prevention of chemotherapy‐ and radiotherapy‐induced emesis: results of the 2004 Perugia International Antiemetic Consensus Conference. Ann Oncol 2006; 17: 20–8. [DOI] [PubMed] [Google Scholar]
- 15. NCCN Clinical Practice Guidelines in OncologyTM . Antiemesis V.1. 2007.
- 16. Warr DG, Grunberg SM, Gralla RJ et al. The oral NK1 antagonist aprepitant for the prevention of acute and delayed chemotherapy‐induced nausea and vomiting: pooled data from 2 randomised, double‐blind, placebo controlled trials. Eur J Cancer 2005; 41(9): 1278–85. [DOI] [PubMed] [Google Scholar]
- 17. Nygren P, Hande K, Petty KJ et al. Lack of effect of aprepitant on the pharmacokinetics of docetaxel in cancer patients. Cancer Chemother Pharmacol 2005; 55(6): 609–16. [DOI] [PubMed] [Google Scholar]
- 18. Loos WJ, De Wit R, Freedman SJ et al. Aprepitant when added to a standard antiemetic regimen consisting of ondansetron and dexamethasone does not affect vinorelbine pharmacokinetics in cancer patients. Cancer Chemother Pharmacol 2007; 59(3): 407–12. [DOI] [PubMed] [Google Scholar]
- 19. McCrea JB, Majumdar AK, Goldberg MR et al. Effects of the neurokinin1 receptor antagonist aprepitant on the pharmacokinetics of dexamethasone and methylprednisolone. Clin Pharmacol Ther 2003; 74(1): 17–24. [DOI] [PubMed] [Google Scholar]
- 20. Nakade S, Ohno T, Kitagawa J et al. Population pharmacokinetics of aprepitant and dexamethasone in the prevention of chemotherapy‐induced nausea and vomiting. Cancer Chemother Pharmacol 2008; 63: 75–83. Epub 2008 Mar 4. [DOI] [PubMed] [Google Scholar]