

Abstract
Special AT‐rich sequence binding protein (SATB) 1 has been proposed to act as a determinant for the acquisition of metastatic activity by controlling expression of a specific set of genes that promote metastatic activity. Here we found that SATB1 expression is upregulated in multidrug‐resistant breast cancer cells that exhibit higher invasive potential than the parental cells. Apart from accelerating metastasis and inducing epithelial–mesenchymal transition, SATB1 was demonstrated to confer resistance to both P‐glycoprotein‐related and P‐glycoprotein‐non‐related drugs on MCF7 cells, which was accompanied by decreasing accumulation of adriamycin in SATB1‐overexpressing transfectants. SATB1 depletion could partially reverse the multidrug resistance (MDR) phenotype of MCF7/ADR in vitro and in vivo. The SATB1‐induced P‐glycoprotein‐mediated MDR could be reversed by treatment with anti‐P‐glycoprotein mAb. Moreover, SATB1 plays an important role in anti‐apoptotic activity in MCF7/ADR cells in response to adriamycin treatment, which suggests another mechanism contributing to SATB1‐related MDR of breast cancers. These data provide new insights into the mode by which breast tumors acquire the MDR phenotype and also imply a role for SATB1 in this process. (Cancer Sci 2009)
Special AT‐rich sequence binding protein (SATB) 1, the global chromatin organizer and transcription factor, has emerged as a key factor in regulating gene expression during the differentiation and activation of T cells, making it a key player in the immune system.( 1 ) Previous studies have unraveled the role of SATB1 in the organization of the chromatin “loopscape” and its dynamic nature in response to physiological stimuli. At the genome‐wide level, SATB1 seems to play a role in the organization of the transcriptionally poised chromatin.( 2 , 3 ) SATB1 organizes the MHC class I locus into distinct chromatin loops by tethering matrix association regions to the nuclear matrix at fixed distances.( 4 , 5 , 6 ) Silencing of SATB1 mimics the effects of interferon (IFN)‐γ treatment on the chromatin loop architecture of the MHC class I locus and alters the expression of genes within the locus.( 5 )
Besides the immune system, SATB1 is also expressed in many cancer cells, such as colorectal cancer cells,( 7 ) lymphoma cells,( 8 ) and breast cancer cells.( 9 ) SATB1 has now revealed a darker side. Rygiel et al. found that SATB1 expression in colorectal cancer seems to be associated with an aggressive phenotype.( 7 ) SATB1 correlates with high expression of tumor necrosis factor (TNF)‐α, which mediates the inflammatory processes in tumor invasion, angiogenesis, and metastasis. On the other hand, SATB1 influences the expression of anti‐inflammatory interleukin (IL)‐4 and IL‐10, which in turn are known to lead to escape from cancer immune surveillance. In addition, SATB1 is also an essential contributing factor in the most aggressive forms of breast cancer.( 9 ) By introducing SATB1 into otherwise non‐metastatic breast cancer cells, invasive tumors can be induced in mice; conversely, removing SATB1 from metastatic cells not only abolishes metastasis and tumor growth in mice but also returns cells to their normal appearance in vitro.
In our preliminary work, a modified subtractive hybridization method was used to identify upregulated or downregulated genes from an adriamycin (ADM)‐resistant human breast carcinoma cell line that was derived from the parental cells. SATB1 was found to be overexpressed in MCF7/ADR cells compared with MCF7 cells. The MCF7/ADR cell line displays cross‐resistance to various anticancer drugs, although it is selected with a single anticancer drug, ADM. The overexpression of SATB1 in MCF7/ADR cells indicated that SATB1 might be related to the occurrence and development of the multidrug resistance (MDR) phenotype in breast cancers.
Here, we addressed the role of SATB1 in breast cancers and particularly its biological actions within MDR of breast carcinoma. Our results suggest that SATB1 expression plays an important role in the regulation of P‐glycoprotein (P‐gp), the drug transporter that mediates resistance to chemotherapeutic drugs, and seems to correlate with epithelial–mesenchymal transition (EMT). On the other hand, SATB1 suppresses the apoptosis rate, which is known to allow cells to escape from chemotherapy. Future studies targeting the expression of SATB1 may lead to a novel approach to inhibit or reverse the development of MDR and EMT in breast carcinomas.
Materials and Methods
Cell culture. The human breast carcinoma cell lines MCF7, Hs578T, MX‐1, and their multidrug‐resistant counterparts MCF‐7/ADR, MCF7/mitoxantrone (MCF7/MX), Hs578T/doxorubicin (Hs578T/Dox), and MX‐1/Taxol (MX‐1/T) were from American Type Culture Collection (Manassas, VA, USA) and cultured under the conditions specified by the manufacturer. Multidrug‐resistant sublines of MCF7 were obtained by culturing the cells in gradually increasing doses of ADM as described previously.( 10 ) Cells that grew in 1, 3, 5, and 10 μM ADM were obtained after 2, 4, 7, and 11 months of culture with ADM, respectively. The stability of the resistant phenotype was determined by culturing continuously in medium with corresponding concentrations of ADM and assessing relative resistance after various periods of time up to 5 months.
Reverse transcription and quantitative RT‐PCR. Quantitative RT‐PCR was carried out using the basic procedure described previously.( 11 ) The primers used in the real‐time PCR reactions were designed based on information from the human genomic database. The following are the primers used for the specific amplification of GAPDH, SATB1, and MDR1: GAPDH forward primer 5′‐catcaagaaggtggtgaagc‐3′ and reverse primer 5′‐ggaaattgtgagggagatgc‐3′; SATB1 forward primer 5′‐aggtgtcttccgaaatcta‐3′ and reverse primer 5′‐cactttagcacgcttcat‐3′; MDR1 forward primer 5′‐cccatcattgcaatagcagg‐3′ and reverse primer 5′‐gttcaaacttctgctcctga‐3′.
Immunoblotting. Total protein was extracted from cells using RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Fifty micrograms of protein extract/lane was electrophoresed, transferred to PVDF membranes, and incubated overnight with antibodies against SATB1 (Santa Cruz Biotechnology), P‐gp (Chemicon International, Temecula, CA, USA), multidrug resistant protein (MRP) 1 (Santa Cruz Biotechnology), and and breast cancer resistant protein (BCRP) (Santa Cruz Biotechnology) respectively. Membranes were treated with the appropriate HRP‐conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA). Detection was carried out using the reagents provided in the ECL+Plus kit (GE Healthcare, Wauwatosa, WI, USA).
Plasmid construction and transfection of MCF7 or MCF7/ADR cells. The SATB1 cDNA was amplified from total cDNA of MCF7/ADR cells. Primers for SATB1 cDNA were: forward 5′‐aactggtaaccacctcatt‐3′ and reverse 5′‐ctgccacatcgacctcta‐3′. The resulting PCR products were then introduced into the SacI and EcoRI sites of the pEGFP vector. This recombinant plasmid (named pEGFP‐SATB1) was confirmed by restriction digestion and full‐length sequencing. shRNA was designed, based on the SATB1 sequence (NM_002971) identified with siRNA Target Finder (Ambion, Austin, TX, USA): shRNA 5′‐ggatttggaagagagtgtc‐3′. The oligoduplexes were cloned into the pSUPER vector (Oligoengine). Transfection of MCF7 with pEGFP‐SATB1 or transfection of MCF7/ADR with pSUPER‐shRNASATB1 was done using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Luciferase reporter assay. The 2946‐bp SATBl 5′ untranslated region was cloned by PCR amplification using the upstream primer 5′‐cggggtaccccgattgggggaacactaacattca‐3′ and the downstream primer 5′‐cccaagcttgggggagcgaggcgagga‐3′ (the KpnI and HindIII sites, respectively, are underlined). After digestion with the restriction endonucleases KpnI, HindIII, SacI, and XhoI, different fragments of the SATB1 5′ untranslated region were cloned into pGL3‐basic (Promega, Madison, WI, USA) (named pGL3‐2946‐luciferase, pGL3‐1718‐luc, and pGL3‐751‐luc respectively. Cells at 50% confluence in 24‐well plates were transfected using Lipofectamine2000. A firefly luciferase reporter gene construct (200 ng) and 1 ng of the pRL‐SV40 Renilla luciferase construct (for normalization) were cotransfected per well. Cell extracts were prepared 48 h after transfection, and the luciferase activity was measured using the Dual‐Luciferase Reporter Assay System (Promega).
Inhibition of SATB1 expression by RNAi. To generate SATB1‐negative MCF7/ADR cells, SATB1‐targeted RNAi experiments were done. Cells (2 × 105) were seeded in six‐well plates in triplicate and, after an overnight incubation, the cells were transfected with various concentrations of siRNA before ADM treatment in serum‐free Opti‐MEM medium using HiPerfect Reagent (Qiagen, Valencia, CA, USA) as suggested by the manufacturer’s instructions. Total protein was extracted and gene expression was determined by western blotting. Anti‐β‐actin was used as a protein loading control. The target sequence 5′‐tgggtacgcgatgaactgaaa‐3′ was synthesized by Qiagen.
Immunohistochemical assay. Briefly, slides were dehydrated in xylene and a graded alcohol series. Antigen retrieval was carried out with 0.01 M citrate buffer at pH 6.0 at 95°C for 10 min. Then slides were incubated with diluted primary antibody for 12 h, followed by incubations with biotinylated secondary antibody for 1 h, peroxidase‐labeled streptavidin for 15 min (LSAB‐2 System; DAKO, Glostrup, Denmark), and diaminobenzidine and hydrogen peroxide chromogen substrate plus diaminobenzidine enhancer (DAKO) for 10 min. Slides were again counterstained with Mayer’s hematoxylin. Known positive and negative tissue controls were processed at the same time and under the same conditions. To evaluate SATB1 levels, immunostained slides were scored on the following scale: score 0, negative nuclear staining for all tumor cells; score 1, weak nuclear staining representing all positive staining other than score 2; score 2, moderate nuclear staining 50% or strong nuclear staining in 5% of the tumor cells. The intensity of immunohistochemical reactions with P‐gp was appraised using the semiquantitative immunoreactive score scale ( 12 ).
In vitro drug sensitivity assay. The multidrug chemosensitivity of cells under investigation and their corresponding controls was assessed as described previously.( 13 ) The relative inhibitory rate of cell growth by different concentrations of ADM was calculated according to the following formula: R = (A 2 − A 1)/A 2, where R is the relative inhibitory rate of cell growth by ADM, A 1 is the absorbance value of cells in the presence of ADM for 48 h, and A 2 is the absorbance value of control cells without ADM treatment.
Laser cytometric analysis of ADM accumulation efflux. Cells were seeded in chambered borosilicate coverglass slides (1 × 106 cells/well) and incubated for 36 h to allow cells to adhere to chamber slides. Cells were then incubated with ADM (10 μM) for approximately 48 h, washed once with Dulbecco’s PBS, and used immediately for experiments with the laser cytometer (Meridian Ultima Workstation; Meridian Instruments, Okemos, MI, USA) to quantitate intracellular fluorescence intensity. In each experiment, 1 × 104 cells were analyzed at a flow rate of 200–400 cells/s. To measure the fluorescence intensity, drug‐exposed cells were analyzed with excitation at 488 nm (1 W power) with emission integrated above 530 nm, and were selected by gating the scattered light from the cell suspension. To relate the fluorescence intensities of ADM obtained by laser cytometry from different treatments to intracellular ADM concentration, excitation and detection parameters were kept constant, and a suspension calibration curve was generated with graded concentrations (0–2 mM) of ADM in suspension. Ten microscopic fields, each containing aggregates of 10–15 cells, were analyzed for each treatment. At least two experiments were carried out on different days.
In vivo drug sensitivity assay. The subrenal capsule assay was carried out to evaluate the sensitivity of different cell lines (MCF7‐con, MCF7‐SATB1, MCF7/ADR‐con, and MCF7/ADR‐shRNASATB1) to ADM as described previously.( 14 ) Briefly, exponentially growing cells (1 × 107) were harvested, resuspended in 250 μL serum‐free medium, and then mixed with 12.5 μL fibrinogen (40 mg/mL) and 12.5 μL thrombin (200 μg/mL) to prepare the cell‐fibrinogen clot. Nude mice, 8–12 weeks old, were anesthetized with sodium pentobarbital at a concentration of 70 mg/kg. The left kidney was exteriorized and the cell‐fibrinogen clot of approximately 1 mm3 was inserted under the renal capsule. ADM was injected through the caudal vein at a concentration of 60 mg/m3 on day 1 and continued for 5 days. On day 8, the animals were killed by cervical dislocation. The exact length and width of tumors were measured before clot implantation and after mouse death. Tumor size S = LW 2/2, where L is tumor length and W is tumor width) and the relative growth rate of the tumor (TS = (S 2 − S 1)/S 1, where S 1 is tumor size before clot implantation and S 2 is the tumor size after mouse death) were used to evaluate the drug sensitivity of cell lines to ADM. The experiment was carried out along established, institutional animal welfare guidelines concordant with NIH species criteria.
Flow cytometry assay. Flow cytometry was used to quantitatively detect the apoptotic rate. Cells (1 × 106) were plated into 10‐cm tissue culture dishes 1 day before the treatment, and were then treated with ADM. After the treatment, floating and attached cells were harvested, washed with PBS, fixed in 70% ethanol overnight at 4°C, and stained with propidium iodide (50 mg/mL). The sub‐G1 peak (DNA content less than 2N) was measured with FACScan Flow Cytometry (Becton Dickinson Labware, Franklin Lakes, NJ, USA) and analyzed using Cell Quest software (Becton Dickinson, San Jose, CA, USA).
Caspase activity assay. Cells were collected and resuspended in ice‐cold lysis buffer containing 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.1%3‐([3‐cholamidopropyl]‐dimethylammonio]‐2‐hydroxy‐1‐propanesulfonic acid (CHAPS), 2 mM dithiothreitol (DTT), and 0.1 mM EDTA for 15 min on ice and centrifuged at 12 000 g for 15 min at 4°C. The supernatants (cytosolic extract) were collected as the samples for caspase activity detection. The samples were incubated with 500 mM caspase substrates in 100 mL caspase activity assay buffer (50 mM HEPES [pH7.4], 100 mM NaCl, 0.1% CHAPS, 10 mM DTT, 0.1 mM EDTA, and 10% glycerol) for 4 h at 37°C. Optical density was measured at 405 nm.
Statistical analysis. Statistics were calculated using SPSS software (Chicago, IL, USA). The results are presented as mean ± SEM. ANOVA, Student’s t‐test analysis, and Dunnett’s multiple comparison tests were used to compare mean values. Pearson’s correlation coefficient was used to determine whether two prognosis‐related factors were correlated with each other over all cases. A P‐value of less than 0.05 was defined as statistical significance.
Results
Overexpression of SATB1 in multidrug‐resistant breast cancer cells. The expression levels of SATB1 in MCF7 and MCF7/ADR were determined by quantitative RT‐PCR and western blot analysis, respectively. It was confirmed that SATB1 had higher mRNA and protein expression in MCF7/ADR than in MCF7 cells (Fig. 1a,b). Moreover, as shown in Figure 1(c), relative luciferase units from pGL3‐751‐luc showed and kept higher transcriptional activity in MCF7/ADR cells than the other two vectors pGL3‐2946‐luc and pGL3‐1718‐luc (P < 0.05), while relative luciferase units from three vectors showed no obvious activity in MCF7 cells. These results demonstrated that the SATB1 promoter drives gene expression with cell specificity (MCF7/ADR cells only) and its core promoter region may be located within the region −751 to −9 bp of the 5′ untranslated region.
Figure 1.
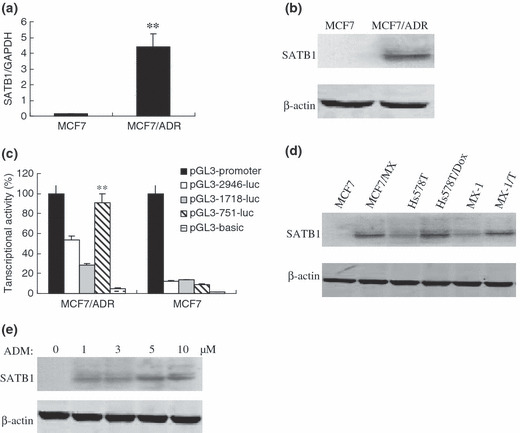
The overexpression of special AT‐rich sequence binding protein (SATB) 1 in multidrug resistance (MDR) breast cancer cells. SATB1 expression at the (a) mRNA and (b) protein levels in MCF7/ADR cells was assessed by real‐time PCR and western blot analysis respectively. (c) Relative promoter activity of pGL3‐2946‐luc, pGL3‐1718‐luc, and pGL3‐751‐luc in MCF7 and MCF7/ADR cells. Expression levels of SATB1 in different (d) MDR breast cancer cell lines and (e) MDR sublines of MCF7 were determined by immunoblotting. β‐Actin was used as an internal control. Bars represent the mean of triplicate samples; error bars represent standard deviation. Data are representative of three independent experiments. MCF7/MX, MCF7/mitoxantrone; Hs578T/Dox, Hs578T/doxorubicin; MX‐1/T, MX‐1/Taxol. **P < 0.05 versus corresponding controls cells.
SATB1 was also found to be highly expressed in the other multidrug‐resistant breast carcinoma cell lines MCF7/MX, Hs578T/Dox, and MX‐1/T compared with their parental sensitive control cells (Fig. 1d). We evaluated SATB1 expression levels in the MDR variants of MCF7 cultured continuously in gradually increasing doses of ADM up to 10 μM. When compared with their parental line, the MDR variants were 10‐ to 200‐fold more resistant to ADM (data not shown). We found that the level of SATB1 expression was closely correlated with the degree of ADM resistance (Fig. 1e). These results indicate that SATB1 may promote the MDR phenotype in breast cancers.
Involvement of SATB1 in MDR of breast carcinoma. To determine whether SATB1 is involved in MDR of breast carcinoma, the in vitro effects of P‐gp substrates and non‐P‐gp substrates on the growth of MCF7‐SATB1 and MCF7/ADR‐SATB1RNAi were evaluated by MTT assay. As shown in Table 1, SATB1 had different effects on drug sensitivity, depending on the drug used. MCF7‐SATB1 cells showed a >10‐fold increased resistance to the P‐gp‐related drugs ADM, Toxel, and vincristine (VCR), and a >3‐fold increased resistance to the P‐gp‐non‐related drugs Methotrexate (MTX) and 5‐fluorouracil (5‐FU) compared with MCF7‐cont cells (P < 0.05). MCF7/ADR‐SATB1RNAi showed significantly increased sensitivity to these drugs compared with MCF7/ADR‐cont (P < 0.05). All these data suggest that SATB1 might confer MDR phenotypes on breast cancer cells by both P‐gp‐related and P‐gp‐non‐related pathways. However, SATB1 did not change the sensitivity of MCF7‐SATB1 and MCF7/ADR‐SATB1RNAi cells to the P‐gp‐non‐related drugs bleomycin (BLM) and cisplatin (CDDP) (P > 0.05).
Table 1.
IC50 of P‐gp‐related drugs and P‐gp‐non‐related drugs in special AT‐rich sequence binding protein 1‐expressing cells
| Cell line | P‐gp‐related drugs (μg/mL) | P‐gp‐non‐related drugs (μg/mL) | |||||
|---|---|---|---|---|---|---|---|
| Adriamycin | Toxel | VCR | BLM | MTX | 5‐FU | CDDP | |
| MCF7 | 0.38 ± 0.02 | 0.12 ± 0.01 | 0.42 ± 0.03 | 0.51 ± 0.04 | 0.19 ± 0.02 | 4.78 ± 0.62 | 34.56 ± 4.13 |
| MCF7‐con | 0.42 ± 0.03 | 0.16 ± 0.01 | 0.31 ± 0.04 | 0.60 ± 0.04 | 0.16 ± 0.01 | 5.10 ± 0.48 | 31.29 ± 3.21 |
| MCF7‐SATB1 | 24.11 ± 2.15* | 1.61 ± 0.17* | 9.81 ± 0.85* | 0.53 ± 0.06 | 1.02 ± 0.22* | 16.20 ± 2.33* | 31.56 ± 4.76 |
| MCF7/ADR | 31.05 ± 3.87 | 0.49 ± 0.05 | 12.95 ± 2.13 | 0.49 ± 0.02 | 0.81 ± 0.01 | 18.79 ± 2.15 | 30.56 ± 3.29 |
| MCF7/ADR‐con | 32.04 ± 3.11 | 0.40 ± 0.03 | 12.35 ± 1.52 | 0.55 ± 0.07 | 0.83 ± 0.06 | 16.28 ± 2.51 | 31.57 ± 3.89 |
| MCF7/ADR‐SATB1RNAi | 4.58 ± 0.51* | 0.21 ± 0.01* | 2.10 ± 0.30* | 0.41 ± 0.05 | 0.22 ± 0.02* | 6.15 ± 0.72* | 33.10 ± 3.23 |
*P < 0.05 versus control cells. IC50 values were expressed in μg/mL and were evaluated as reported in Materials and Methods. Standard deviations for all of the experiments carried out in triplicate were less than 5%. P‐gp, P‐glycoprotein; VCR, vincristine; MTX, Methotrexate; 5‐FU, 5‐Fluorouracil; BLM, bleomycin; CDDP, cisplatin.
We also investigated whether SATB1 is required for the MDR phenotypes of breast cancer cells in vivo by expressing shRNA to knock down SATB1 expression. As shown in Figure 2(a), SATB1 expression was reduced by 70% in SATB1 shRNA‐expressing cells. SATB1 expression remained unaltered in MCF7/ADR cells expressing a control shRNA whose sequence did not match any known human gene. The relative inhibitory rates of different concentrations of ADM to MCF7/ADR‐shRNASATB1 cells were all higher than those to MCF7/ADR‐con (Fig. 2b), suggesting that suppression of SATB1 expression could reverse ADM resistance of MCF7/ADR cells. We then determined the in vivo reactivity of SATB1‐related transfectants to ADM. Figure 2(c) shows a strong tumor growth‐inhibitory effect of ADM treatment in MCF7‐con and MCF7/ADR‐shRNASATB1 tumors (P < 0.05), suggesting that in vivo implantations of MCF7‐SATB1 and MCF7/ADR‐con were more resistant to ADM compared with MCF7‐cont and MCF7/ADR‐shRNASATB1, respectively.
Figure 2.
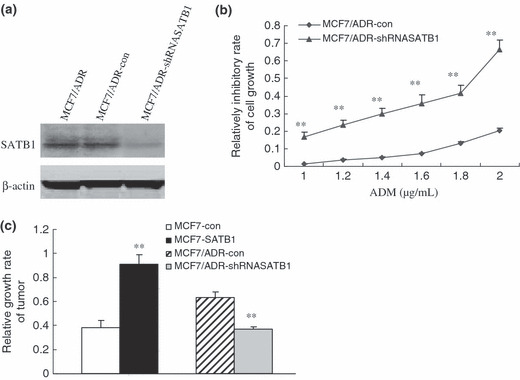
Modulation of multidrug resistance (MDR) phenotype by special AT‐rich sequence binding protein (SATB) 1 in vitro and in vivo. (a) After MCF7/ADR cells were transfected with vectors expressing shRNASATB1, SATB1 expression levels were assessed by western blot analysis. β‐Actin was used as an internal control. (b) Inhibitory effects of various concentrations of adriamycin (ADM) on MCF7/ADR‐shRNASATB1 cells were evaluated by MTT assays. (c) After in vivo ADM treatment for 8 days, the exact length and width of implanted tumors were measured and the relative growth rate was determined. Bars represent the mean of triplicate samples; error bars represent standard deviation. Data are representative of three independent experiments. **P < 0.05 versus corresponding controls cells.
Regulatory effects on classical MDR molecules in SATB1‐related transfectants. To study the possible molecular mechanisms involved in SATB1‐related MDR of breast cancer, expression levels of the three classical MDR molecules P‐gp, MRP1, and BCRP were examined in SATB1‐related transfectants. P‐gp expression levels in MCF7‐SATB1 and MCF7/ADR‐cont cells were higher than those in MCF7‐cont and MCF7/ADR‐SATB1RNAi cells, respectively. Although MRP1 and BCRP expression levels in MCF7/ADR‐derived cell lines were much higher than those in MCF7‐derived cell lines, the change of cellular SATB1 expression did not significantly affect the expression of MRP1 and BCRP (Fig. 3a). We next did quantitative RT‐PCR to detect the levels of MDR1 mRNA in SATB1 transfectants. As shown in Figure 3(b), MDR1 mRNA from the MCF7‐SATB1 and MCF7/ADR‐SATB1RNAi cells was 13.2‐fold higher and 6.8‐fold lower than that in the corresponding control cells, indicating that P‐gp expression was regulated by SATB1 at both the mRNA and protein levels. All of these results demonstrated that P‐gp might mediate the SATB1‐related MDR of breast cancers.
Figure 3.
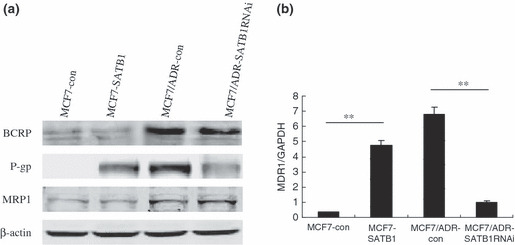
Induction of P‐glycoprotein (P‐gp) expression by special AT‐rich sequence binding protein (SATB) 1 in breast cancer cells. After introducing SATB1 into MCF7 cells, the expression level of breast cancer resistant protein (BCRP), P‐gp, and multidrug resistant protein (MRP) 1 in SATB1‐related transfectants was determined by (a) western blot analysis and (b) multidrug resistance (MDR) 1 mRNA level was evaluated by quantitative RT‐PCR. β‐Actin was used as an internal control. Bars represent the mean of triplicate samples; error bars represent standard deviation. Data are representative of three independent experiments. **P < 0.05 versus corresponding controls cells.
Role of P‐gp function in SATB1‐related MDR. In order to examine the phenotype associated with P‐gp expression, intracellular ADM accumulation was assessed by means of laser cytometry. As summarized in Table 2, intracellular ADM accumulation in MCF7/Adr‐con cells was significantly less than that in MCF7‐con cells. Both MCF7‐SATB1 and MCF7/ADR‐con cells have significantly lower intracellular ADM concentrations than do MCF7‐con and MCF7/ADR‐SATB1RNAi cells, respectively, when incubated with ADM, indicating functional expression of P‐gp.
Table 2.
Intracellular concentrations of ADM in sequence binding protein 1‐related transfectants as determined by laser cytometry
| Cell line | ADM concentration (μM) in transfectants |
|---|---|
| MCF7‐con | 772 ± 6.85 |
| MCF7‐SATB1 | 271 ± 2.11 |
| MCF7/ADR‐con | 243 ± 2.58 |
| MCF7/ADR‐SATB1RNAi | 665 ± 6.09* |
*P < 0.05 versus control cells. Standard deviations for all of the experiments carried out in triplicate were less than 5%. ADM, adriamycin.
To further investigate the possible role of P‐gp in SATB1‐related MDR, we incubated MCF7‐SATB1 cells with P‐gp mAb, which neutralizes the transporter function of P‐gp, for 3.5 h prior to treatment with ADM (P‐gp substrate) and MTX (non‐P‐gp substrate). The results of the MTT assay showed that P‐gp mAb could partially reverse ADM resistance, but exerted no effects on MTX resistance in MCF7‐SATB1 cells (Table 3), suggesting that drug resistance of the cells could be modulated by P‐gp inhibitor. Thus, P‐gp, the classical MDR‐related molecule, plays an important role in SATB1‐related MDR.
Table 3.
Effects of the P‐glycoprotein inhibitor on sequence binding protein 1‐mediated multidrug resistance
| Cell line | IC50 value (μg/mL) | |
|---|---|---|
| Adriamycin | MTX | |
| MCF7 | 0.43 ± 0.03 | 0.16 ± 0.01 |
| MCF7‐con | 0.39 ± 0.03 | 0.22 ± 0.03 |
| MCF7‐SATB1 | 22.59 ± 3.11 | 1.15 ± 0.25 |
| MCF7‐SATB1 + P‐gp mAb | 9.37 ± 0.87* | 0.98 ± 0.08 |
*P < 0.05 versus control cells. IC50 values were expressed in μg/mL and were evaluated as reported in Materials and Methods. Standard deviations for all of the experiments carried out in triplicate were <5%. MTX, Methotrexate.
Correlations of SATB1 and P‐gp expression in 60 breast cancer cases. The correlation between SATB1 and P‐gp expression was evaluated in breast cancer specimens. First, we analyzed SATB1 expression in 60 primary breast carcinomas. The number of cases scoring 0, 1, and 2 was 10, 14, and 36, respectively. As shown in Figure 4, the immunostaining of SATB1 was highly correlated with that of P‐gp in most breast carcinoma tissues. Moreover, Pearson’s correlation coefficient between SATB1 and P‐gp was 0.870, P < 0.001 (Table 4), suggesting a close relationship between the two variables.
Figure 4.
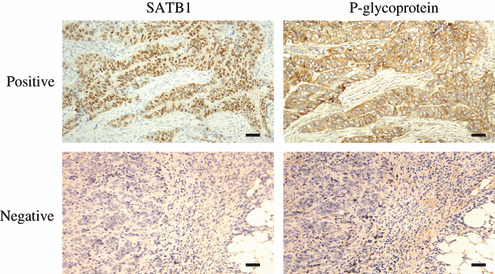
Representative immunostaining of special AT‐rich sequence binding protein (SATB) 1 and P‐glycoprotein (P‐gp) in human breast carcinoma samples. Immunohistochemical analyses showed expression of SATB1 parallel with that of P‐gp in most breast carcinomas. The immunoreactivity was assessed on the basis of intensity and the proportion of positive‐staining cells. The positive staining of tumor cells was expressed as yellow‐brown particles with weak to moderate–strong intensity. SATB1 was readily detected in the nucleus, whereas P‐gp was detected in the cytoplasm and on the cytomembrane. Scale bar = 20 μm.
Table 4.
Correlation of SATB 1 expression with P‐glycoprotein (n =60)
| SATB1 | P‐glycoprotein | ||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |
| 0 | 9 | 1 | 0 | 0 | 0 |
| 1 | 2 | 10 | 2 | 0 | 0 |
| 2 | 0 | 1 | 5 | 12 | 18 |
| γ | 0.87 | ||||
| P‐value | <0.001 | ||||
γ, Pearson’s correlation coefficient. SATB, sequence binding protein.
Effects of SATB1 on cell apoptosis induced by ADM. The suppression of apoptosis is another important mechanism of MDR. As SATB1 cleavage is apoptosis specific, we studied the cleavage kinetics of SATB1 in SATB1‐related transfectants treated with ADM. SATB1 migrated at a molecular weight 103 kDa without cleavage in SDS‐PAGE in both MCF7‐SATB1 and MCF7/ADR‐con cells. However, in MCF7/ADR‐SATB1RNAi cells, cleavage of SATB1 occurred 36 h after treatment with ADM and progressed with ongoing apoptosis, leading to substantial conversion of a native 103‐kDa protein to a 70‐kDa cleavage polypeptide (Fig. 5a). Correspondingly, ADM greatly induced the apoptosis rate of MCF7‐con and MCF7/ADRSATB1RNAi cells as determined by flow cytometric assays (Fig. 5b). The results of the caspase activity assays also suggested that compared with control cells, the ADM‐induced caspase activity was largely reduced in MCF7 cells with ectopic SATB1 expression, but significantly higher in SATB1‐depleted MCF7/ADR cells (Fig. 5c). These results suggest a causative linkage between SATB1 and the anti‐apoptotic effects.
Figure 5.
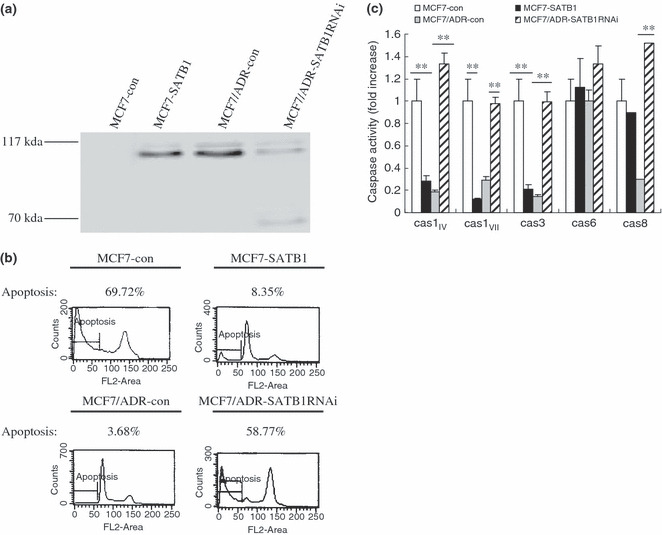
Effects of special AT‐rich sequence binding protein (SATB) 1 expression on drug‐induced apoptotic cell death. Cells were treated with adriamycin (5 μM) for 36 h. (a) Cell lysates from the treated cells were separated by 8% SDS–PAGE, blotted, and probed with anti‐SATB1 polyclonal antibody. Molecular weight in kDa is indicated on the left. Apoptosis and caspase activity were determined by (b) flow cytometry and (c) caspase activity assay respectively. Caspase activity in MCF7‐con was set as 1. Bars represent the mean of triplicate samples; error bars represent standard deviation. Data are representative of three independent experiments. **P < 0.05 versus corresponding controls cells.
Discussion
SATB1 is not expressed in all cells and it seems particularly important in cells that must change their function – as many progenitor cells do, including the thymocytes that turn into T cells, and as cancerous cells must do to turn into metastatic cells.( 15 ) In the present work, SATB1 was found to be highly expressed in multidrug‐resistant breast carcinoma cell lines and tissues with P‐gp overexpression. SATB1 not only conferred P‐gp‐related drug resistance but also P‐gp‐non‐related drug resistance on MCF7 cells. Furthermore, the suppression of SATB1 significantly increased the chemosensitivity to chemotherapeutic drugs in vitro and in vivo, thereby drawing the conclusion that SATB1 could promote a drug‐resistance ability of breast carcinomas. This is the first report that directly links SATB1 with a MDR phenotype. As emerging evidence has suggested the two phenotypes of malignant tumors, MDR and tumor metastasis, are functionally associated with ( 16 , 17 ) each other, we hypothesized that SATB1 may assume a dual role, as it had intrinsic stimulative effects on tumor metastasis as well as increasing resistance to chemotherapeutic agents.
Overexpression of P‐gp, encoded by the MDR1 gene, is known to be the major molecular mechanism mediating MDR to efflux pump for various anticancer drugs.( 18 ) As the MDR phenotype of the SATB1‐overexpressing cell lines could be partially reversed by disrupting P‐gp pump function (anti‐P‐gp mAb), the high level of P‐gp expression caused by SATB1 must be an important mechanism involved in SATB1‐related MDR. As a nuclear architectural protein, SATB1 folds and remodels the chromatin (the intertwined DNA and proteins that form chromosomes) into new shapes, bringing even distant parts of the genome together for coordinated control of gene expression and regulation.( 1 , 2 , 6 ) Han et al. showed that in metastatic breast cancer cells, SATB1 controls expression of over 1000 genes affecting cell adhesion, cell signaling, cell‐cycle regulation, and other functions.( 9 ) Among the important genes regulated by SATB1, a few of them, such as ErbB2 and β‐catenin, have been identified to play a role in regulating MDR. The work of Misra et al. suggested that ErbB2, phosphoinositide 3‐kinase, and hyaluronan form a positive feedback loop that strongly amplifies MDR1 expression and regulates drug resistance in cancer cells.( 19 ) Also, the regulation of P‐gp expression can be influenced by β‐catenin signaling.( 20 ) Our data confirmed that SATB1 could significantly elevate the level of ErbB2 (data not shown), which may be due to P‐gp upregulation in cell lines in which SATB1 is highly expressed. The regulation of MDR1 expression in tumor cells involves a complex interplay of multiple factors. Apart from ErbB2, other pathways mediating SATB1‐induced P‐gp expression should not be excluded.
Prevention of apoptosis as a mechanism of MDR has been widely accepted.( 21 ) In the present study, SATB1 suppression in MCF7/ADR cells via RNAi rendered the cells more sensitive to drug treatment and more prone to drug‐induced apoptosis. During early apoptosis, DNA fragmentation is initiated and is preceded by the degradation of nuclear proteins.( 22 ) The specific cleavage of SATB1 relates to the detachment of the protein from DNA, suggesting that the SATB1 cleavage was apoptosis specific and accompanied by collapse of the nuclear architecture.( 23 , 24 ) Because apoptosis was potently triggered in MCF7/ADR‐SATB1RNAi cells, we observed cleavage of residual SATB1 in the SATB1‐suppressed cells upon ADM treatment, whereas no similar phenomenon was found in the SATB1‐overexpressing cells. In contrast, ADM‐induced apoptosis was largely abrogated by SATB1 overexpression, which might be another mechanism contributing to SATB1‐related MDR of breast cancer.
SATB1 acts as an important player involved in EMT, which is known to occur during carcinoma progression.( 9 ) This EMT leads to enhanced motility and invasiveness in many cell types, and is often considered a prerequisite for conferring cancer developmental and malignant phenotypes.( 25 ) The introduction of ectopic SATB1 induces a marked change in the cellular morphology and promotes the acquisition of further aggressive phenotypes, characterized by MDR and enhanced invasive potential. Therefore, we inferred that upregulation of SATB1 in MDR tumor cells plays a key role in the multiphase evolution from a benign to an invasive malignant tumor, that not only accelerates the course of metastasis, but also effectively evades the cytotoxic attack of the drugs.
In summary, our data make evident the relationships between SATB1 and MDR, contribute to a better understanding of how SATB1 regulates the MDR phenotype in breast cancer cells, and suggest the potential inhibitive points that may lead to improved effectiveness of clinical chemotherapy. Further study of the biological functions of SATB1 will be of great help in understanding the mechanisms of occurrence and development of clinical breast carcinomas.
Disclosure Statement
We have no conflict of interest.
Acknowledgements
This work was supported by grants from the National Nature Science Foundation of China (no. 30870972 and no. 30872971). We thank members of our laboratory for helpful discussions.
References
- 1. Alvarez JD, Yasui DH, Niida H, Joh T, Loh DY, Kohwi‐Shigematsu T. The MAR‐binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T‐cell development. Genes Dev 2000; 14: 521–35. [PMC free article] [PubMed] [Google Scholar]
- 2. Yasui D, Miyano M, Cai S, Varga‐Weisz P, Kohwi‐Shigematsu T. SATB1 targets chromatin remodeling to regulate genes over long distances. Nature 2002; 419: 641–5. [DOI] [PubMed] [Google Scholar]
- 3. Kohwi‐Shigematsu T, DeBelle I, Dickinson LA, Galande S, Kohwi Y. Identification of base‐unpairing region‐binding proteins and characterization of their in vivo binding sequences. Methods Cell Biol 1998; 53: 323–54. [DOI] [PubMed] [Google Scholar]
- 4. Cai S, Han HJ, Kohwi‐Shigematsu T. Tissue‐specific nuclear architecture and gene expression regulated by SATB1. Nat Genet 2003; 34: 42–51. [DOI] [PubMed] [Google Scholar]
- 5. Kumar PP, Bischof O, Purbey PK et al. Functional interaction between PML and SATB1 regulates chromatin‐loop architecture and transcription of the MHC class I locus. Nat Cell Biol 2007; 9: 45–56. [DOI] [PubMed] [Google Scholar]
- 6. Cai S, Lee CC, Kohwi‐Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet 2006; 38: 1278–88. [DOI] [PubMed] [Google Scholar]
- 7. Rygiel AM, Davelaar AL, Milano F, Fockens P, Krishnadath KK. SATB1 influences expression of cytokines in colorectal cell lines and is associated with a more aggressive phenotype. AGA abstracts 2009; S2050. [Google Scholar]
- 8. Agrelo R, Souabni A, Novatchkova M et al. SATB1 defines the developmental context for gene silencing by Xist in lymphoma and embryonic cells. Dev Cell 2009; 16: 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han HJ, Russo J, Kohwi Y, Kohwi‐Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumor growth and metastasis. Nature 2008; 452: 187–93. [DOI] [PubMed] [Google Scholar]
- 10. Mirski SE, Gerlach JH, Cole SP. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin. Cancer Res 1987; 47: 2594–8. [PubMed] [Google Scholar]
- 11. Li QQ, Wang WJ, Xu JD et al. Up‐regulation of CD147 and matrix metalloproteinase‐2, ‐9 induced by P‐glycoprotein substrates in multidrug resistant breast cancer cells. Cancer Sci 2007; 98: 1767–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Remmele W, Stegner HE. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER‐ICA) in breast cancer tissue. Pathologe 1987; 8: 138–40. [PubMed] [Google Scholar]
- 13. Li QQ, Wang WJ, Xu JD et al. Involvement of CD147 in regulation of multidrug resistance to P‐gp substrate drugs and in vitro invasion in breast cancer cells. Cancer Sci 2007; 98: 1064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suonio E, Lipponen P, Maenpaa J, Syrjanen K, Kangas L, Tuomisto L. Mitotic index in the subrenal capsule assay as an indicator of the chemosensitivity of ovarian cancer. Cancer Chemother Pharmacol 1997; 41: 15–21. [DOI] [PubMed] [Google Scholar]
- 15. Zheng J. Is SATB1 a master regulator in breast cancer growth and metastasis? Womens Health 2008; 4: 329–32. [DOI] [PubMed] [Google Scholar]
- 16. Kerbel RS, Korczak B, Lagarde A. Growth dominance of the metastatic cancer cell: cellular and molecular aspects. Adv Cancer Res 1990; 5: 87–132. [DOI] [PubMed] [Google Scholar]
- 17. Su ZZ, Austin VN, Zimmer SG, Fisher PB. Defining the critical gene expression changes associated with expression and suppression of the tumorigenic and metastatic phenotype in Ha‐ras‐transformed cloned rat embryo fibroblast cells. Oncogene 1993; 8: 1211–19. [PubMed] [Google Scholar]
- 18. Ambudkar SV, Kimchi‐Sarfaty C, Sauna ZE, Gottesman MM. P‐glycoprotein: from genomics to mechanism. Oncogene 2003; 22: 7468–85. [DOI] [PubMed] [Google Scholar]
- 19. Misra S, Ghatak S, Toole BP. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3‐kinase, and ErbB2. J Biol Chem 2005; 280: 20310–15. [DOI] [PubMed] [Google Scholar]
- 20. Lim JC, Kania KD, Wijesuriya H et al. Activation of β‐catenin signaling by GSK‐3 inhibition increases p‐glycoprotein expression in brain endothelial cells. J Neurochem 2008; 106: 1855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reed JC. Bcl‐2: prevention of apoptosis as a mechanism of drug resistance. Hematol Oncol Clin North Am 1995; 9: 451–73. [PubMed] [Google Scholar]
- 22. Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell‐free mitotic extracts: a model system for analysis of the active phase of apoptosis. J Cell Biol 1993; 123: 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dickinson LA, Joh T, Kohwi Y, Kohwi‐Shigematsu T. A tissue‐specific MAR/SAR DNA‐binding protein with unusual binding site recognition. Cell 1992; 70: 631–45. [DOI] [PubMed] [Google Scholar]
- 24. Sun Y, Wang T, Su Y et al. The behavior of SATB1, a MAR‐binding protein, in response to apoptosis stimulation. Cell Biol Int 2006; 30: 244–7. [DOI] [PubMed] [Google Scholar]
- 25. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]