

Abstract
Intermediate filaments (IF) form the structural framework of the cytoskeleton. Although histopathological detection of IF proteins is utilized for examining cancer specimens as reliable markers, the molecular mechanisms by which IF are involved in the biology of cancer cells are still unclear. We found that site‐specific phosphorylation of IF proteins induces the disassembly of filament structures. To further dissect the in vivo spatiotemporal dynamics of IF phosphorylation, we developed site‐ and phosphorylation state‐specific antibodies. Using these antibodies, we detected kinase activities that specifically phosphorylate type III IF, including vimentin, glial fibrillary acidic protein and desmin, during mitosis. Cdk1 phosphorylates vimentin‐Ser55 from prometaphase to metaphase, leading to the recruitment of Polo‐like kinase 1 (Plk1) to vimentin. Upon binding to Phospho‐Ser55 of vimentin, Plk1 is activated, and then phosphorylates vimentin‐Ser82. During cytokinesis, Rho‐kinase and Aurora‐B specifically phosphorylate IF at the cleavage furrow. IF phosphorylation by Cdk1, Plk1, Rho‐kinase and Aurora‐B plays an important role in the local IF breakdown, and is essential for the efficient segregation of IF networks into daughter cells. As another part of our research on IF, we have set out to find the binding partners with simple epithelial keratin 8/18. We identified tumor necrosis factor receptor type 1‐associated death domain protein (TRADD) as a keratin 18‐binding protein. Together with data from other laboratories, it is proposed that simple epithelial keratins may play a role in modulating the response to some apoptotic signals. Elucidation of the precise molecular functions of IF is expected to improve our understanding of tumor development, invasion and metastasis. (Cancer Sci 2006; 97: 167–174)
Intermediate filaments (IF) form the cytoskeletal framework in the cytoplasm of various eukaryotic cells, and are also present in the nuclear envelope as the major component of nuclear lamina.( 1 , 2 ) Although structural components of other major cytoskeletal proteins (e.g. actin and tubulin) are highly conserved in different cell types, constituent proteins of IF show intriguing molecular diversities and are expressed in tissue‐specific programs during development and differentiation. Therefore, the types of IF present in cells are supposed to be related to their physiological functions. This feature is relevant to the association of mutations in human IF genes with a variety of tissue‐specific disorders.( 3 , 4 , 5 ) In addition, IF proteins can serve as markers of the tissue origin of tumors (keratins define epithelial tissues, whereas vimentin defines mesenchymal origin).( 4 , 6 ) In some cancers, particularly malignant melanoma and breast carcinoma, there is a strong indication that vimentin and keratin IF are coexpressed, thus presenting as a dedifferentiated or interconverted (between epithelial and mesenchymal) phenotype.( 7 ) Epithelial–mesenchymal transition (EMT) is implicated in tumor progression, and the expression pattern of keratin and vimentin is one of the reliable markers for EMT. Although accumulating data suggest that IF proteins may play an important role in cancer development, the molecular mechanisms by which IF are involved in the biology of cancer cells are largely unknown and should be elucidated.
The intracellular organization of IF networks is under the control of protein kinases and phosphatases (Fig. 1).( 8 , 9 , 10 ) Nuclear lamin is phosphorylated by Cdk1/Cdc2 kinase, leading to breakdown of the nuclear envelope, a key event in prometaphase.( 11 , 12 ) Cytoplasmic IF are also reorganized dramatically during mitosis, and this reorganization is considered to be controlled by IF protein phosphorylation. Using site‐ and phosphorylation state‐specific antibodies, which recognize a phosphorylated serine/threonine residue and its flanking sequence,( 13 , 14 , 15 ) we detected kinase activities that specifically phosphorylate IF at the cleavage furrow during cytokinesis, and called these kinases cleavage furrow (CF) kinases.( 16 , 17 , 18 , 19 ) The analysis of in vivo phosphorylation sites on IF led to the identification of Rho‐kinase and Aurora‐B as CF kinases.( 20 , 21 , 22 , 23 , 24 , 25 , 26 ) Recently, we found that Polo‐like kinase 1 (Plk1) binds to vimentin‐Ser55 phosphorylated by Cdk1 and then phosphorylates vimentin‐Ser82 during mitosis.( 27 )
Figure 1.
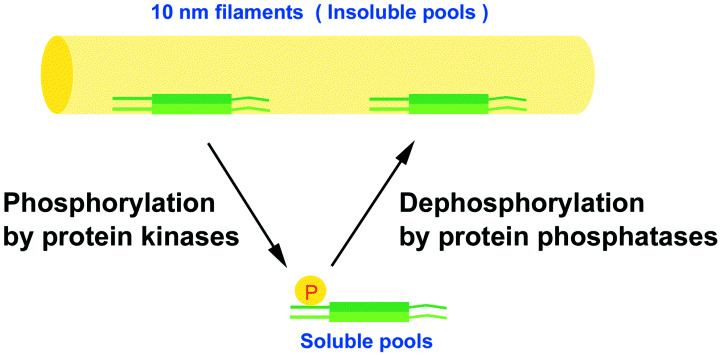
Regulation of intermediate filament (IF) organization by phosphorylation. The site‐specific phosphorylation in the amino‐terminal head domain of IF proteins induces the disassembly of IF. The balance between IF phosphorylation by protein kinases and dephosphorylation by protein phosphatases controls the continuous exchange of IF subunits between a soluble pool and polymerized IF.
The present review first provides an overview of the regulation of IF organization by phosphorylation and the development of site‐ and phosphorylation state‐specific antibodies. We then focus on the spatiotemporal regulation of type III IF by site‐specific phosphorylation during mitosis. In the final part of this review, we briefly discuss the recent research on simple epithelial keratin 8/18‐binding proteins, which provide us with new insights for understanding the physiological functions of IF.
Site‐specific phosphorylation induces the disassembly of IF in vitro
The correlation between increase in IF phosphorylation and filament reorganization observed in mitotic cells had led to the notion that organization of IF may be regulated by phosphorylation.( 28 , 29 ) The first direct evidence that the organization of IF is regulated by phosphorylation was obtained in in vitro studies using vimentin.( 8 ) Vimentin filaments reconstituted in vitro underwent complete disassembly when phosphorylated by purified cAMP‐dependent protein kinase (A kinase) or protein kinase C (C kinase). Subsequently, similar in vitro disassembly induced by phosphorylation was noted for almost all major IF proteins, such as vimentin,( 8 , 30 , 31 , 32 , 33 ) glial fibrillary acidic protein (GFAP),( 34 ) desmin,( 35 ) keratin( 36 ) and lamin.( 11 , 12 ) IF proteins are composed of an amino‐terminal head, a central rod and a carboxy‐terminal tail.( 1 ) Most of the phosphorylation sites of IF that induce disassembly of IF are located in the amino‐terminal non‐α‐helical head domain, indicating that amino‐terminal domain‐specific and site‐specific phosphorylation of IF induce disassembly of the filament structure, in vitro.( 8 , 9 , 31 , 37 )
Development of site‐ and phosphorylation state‐specific antibodies
Protein phosphorylation acts as the main molecular switch in intracellular signal transduction. There have been several commonly used methods to assist in investigating kinase activities. However, the data obtained by classic techniques, such as isotope labeling and biochemical cell fractionation, do not provide important ‘in vivo’ spatiotemporal information on ‘active’ kinases (phosphorylation/dephosphorylation state of target proteins). During the course of our research on IF, we attempted to monitor in vivo spatiotemporal changes of the phosphorylation and dephosphorylation of various IF proteins by kinases and phosphatases. For this, in 1990 we devised a method and procedures to develop site‐ and phosphorylation state‐specific antibodies.( 16 , 17 , 38 , 39 ) The most important advantage of these antibodies is that a phosphorylation site can be predesigned as an epitope by immunizing animals with a synthetic peptide containing a phosphorylated amino acid residue.( 13 , 14 , 15 ) Because the site‐specific phosphorylation of various kinases often reflects their catalytically activated states, site‐ and phosphorylation state‐specific antibodies against these sites are useful for monitoring in vivo kinase activities spatially and temporally.( 15 ) Many site‐ and phosphorylation state‐specific antibodies are now commercially available and utilized widely to analyze the phosphorylation of many proteins in various cellular events, including cell cycle progression, protein degradation, transcriptional control and cellular transformation. In addition, these antibodies have further facilitated the development of new kinase inhibitors that will be used for the treatment of human diseases.
Site‐specific phosphorylation of type III IF during mitosis
Using these site‐ and phosphorylation state‐specific antibodies, we studied in vivo phosphorylation states of type III IF, including GFAP expressed in glial cells, vimentin in mesenchymal cells and most types of cultured cells, and desmin in muscle cells. The first evidence was obtained for four distinct sites on the amino‐terminal head domain of GFAP.( 16 , 18 ) During mitosis, Ser8 phosphorylation of GFAP in glial cells began in the prometaphase, remained until metaphase, and declined gradually thereafter. The phosphorylation was observed diffusely throughout the cytoplasm. In contrast, the phosphorylation of residues Thr7, Ser13 and Ser38 was spatially and temporally distinct, increasing in anaphase, maintained until telophase, and decreasing at the exit of mitosis, and this phosphorylation was localized specifically at the CF. These observations suggested that GFAP was phosphorylated by at least two different kinases, which were activated during early mitosis and at the CF.
Cdk1/Cdc2 kinase is known to be activated specifically during early mitosis,( 40 ) and was found to phosphorylate GFAP only at Ser8 in vitro.( 41 ) As vimentin‐Ser55 was phosphorylated specifically by Cdk1 among known IF kinases in vitro,( 33 , 42 ) we further produced a site‐ and phosphorylation state‐specific antibody for this site. Ser55 on vimentin was also phosphorylated in various types of cells only during the early mitotic phase. A chromatographical analysis of mitotic cell lysates revealed a single peak of Ser55 kinase activity that was identical to Cdk1.( 43 ) Together with the data obtained by a tryptic peptide analysis,( 32 ) these observations strongly suggested that Cdk1 was responsible for the in vivo vimentin‐Ser55 phosphorylation specifically during early mitosis (Fig. 2).
Figure 2.

Phosphorylation sites in the mouse vimentin head domain. Multiple serine residues are phosphorylated by cAMP‐dependent protein kinase (A kinase), protein kinase C (C kinase), Ca2+/calmodulin‐dependent protein kinase II (CaM kinase II), p21‐activated kinase (PAK), Cdk1/Cdc2 kinase, Rho‐kinase, Aurora‐B and Polo‐like kinase 1 (Plk1). Yellow circles indicate the important in vivo phosphorylation sites during mitosis.
In contrast, there was another protein kinase, which phosphorylated Thr7, Ser13 and Ser38 on GFAP specifically at the CF during cytokinesis. Because this kinase seemed to be activated specifically at the CF during cytokinesis, we named it CF kinase.( 16 , 18 ) This CF kinase activity was observed not only in astroglial cells but also in several cultured cells in which GFAP is expressed ectopically.( 19 ) These findings indicated that the activation of CF kinase occurred in a wide range of cell types, suggesting its important role in cytokinesis. We postulated the possible involvement of Rho in the regulation of the CF kinase activity. Rho‐kinase,( 44 ) a Rho target, was revealed to phosphorylate GFAP at Thr7, Ser13 and Ser38 but not at Ser8, in vitro.( 20 ) The in vitro Rho‐kinase phosphorylation sites (Ser38 and Ser71) on vimentin were also shown to be phosphorylated at the CF during cytokinesis (Fig. 2).( 21 , 45 ) All accumulating data on the substrate specificity were consistent with the idea that Rho‐kinase was responsible for CF kinase activity. To determine the biological significance of the CF‐specific IF phosphorylation, we produced a mutant GFAP in which the Rho‐kinase/CF kinase phosphorylation sites (Thr7, Ser13 and Ser38) were changed to alanine residues, and introduced this mutant GFAP into type III IF‐negative T24 cells.( 22 ) The expression of this mutant GFAP impaired the cytokinetic segregation of GFAP filaments, resulting in the formation of an unusually long bridge‐like structure (referred to as an IF bridge) between unseparated postmitotic cells. The expression of wild‐type GFAP or mutants (in which three Ser/Thr residues other than those at the Rho‐kinase/CF kinase sites were altered) showed no remarkable phenotype.( 22 ) These observations suggested that the phosphorylation of IF proteins by Rho‐kinase/CF kinase was required for the local IF breakdown and the separation of IF into daughter cells. Based on these results, we concluded that Rho‐kinase was one of the CF kinases.
To further elucidate the CF‐specific phosphorylation of type III IF, we constructed a series of vimentin mutants in which the phosphorylation sites for one or more known vimentin kinase were changed to alanine. We transiently expressed these mutants in T24 cells, and observed the formation of the IF bridge.( 46 ) We found that only 3.2% of cells that were transfected with vimentin mutated at all sites for known vimentin kinases (Rho‐kinase, protein kinase C, CaM kinase II and Cdk1) formed IF bridges.( 46 ) This suggested the existence of an unknown protein kinase(s) that may phosphorylate vimentin at a site(s) other than known phosphorylation sites and may be responsible for vimentin filament separation. We produced another set of vimentin mutants in which known vimentin kinase sites and an additional serine residue were altered. Among these mutations, the vimentin mutation at Ser72 in addition to mutations at sites phosphorylated by known vimentin kinases induced a remarkable phenotype in terms of filament separation. Many cells transfected with this mutant (60.2% cells relative to the total number of transfected postmitotic cells) formed IF bridges between daughter cells. We then produced a site‐ and phosphorylation state‐specific antibody for vimentin‐Ser72 (YG72), and demonstrated that vimentin‐Ser72 was phosphorylated specifically at the CF during cytokinesis. These results indicated that an unidentified vimentin‐Ser72 kinase(s) may play important roles in vimentin separation through site‐ or domain‐specific vimentin phosphorylation.( 46 ) We asked if Aurora‐B, which belongs to the Aurora kinase family regulating key cell‐cycle processes,( 47 ) could be a vimentin‐Ser72 kinase. We observed that the intracellular distribution of Aurora‐B overlapped with that of vimentin phosphorylated at Ser72.( 24 ) The inhibition of endogenous Aurora‐B by the expression of its dominant‐negative mutant reduced the vimentin‐Ser72 phosphorylation at undetectable levels. By identifying eight Aurora‐B phosphorylation sites on vimentin in vitro, we demonstrated that vimentin‐Ser72 was an in vitro phosphorylation site of Aurora‐B (Fig. 2).( 24 ) Mutations at Aurora‐B phosphorylation sites on vimentin induced the impaired segregation of vimentin filaments in postmitotic cells, and thus Ser72 was found to be one of the important phosphorylation sites responsible for vimentin IF bridge‐phenotype formation.( 24 ) Mutations at both the Aurora‐B and Rho‐kinase sites showed synergistic effects on the IF bridge formation. Aurora‐B phosphorylated GFAP at Thr7, Ser13 and Ser38, which were the same sites as those phosphorylated by CF kinase and Rho‐kinase.( 25 ) Taken together, these results suggested that Aurora‐B may regulate CF‐specific phosphorylation and segregation of IF coordinately with Rho‐kinase during cytokinesis.( 24 , 25 )
In the course of the analysis of Aurora‐B, we found that human Aurora‐B was phosphorylated at Thr232 through the interaction with inner centromere protein, in vivo.( 48 ) The phosphorylation of Thr232 occurred by means of an autophosphorylation mechanism, which was indispensable for the Aurora‐B kinase activity. The activation of Aurora‐B spatiotemporally correlated with the site‐specific phosphorylation of its physiological substrates, histone H3 and vimentin. Overexpression of the TA mutant of Aurora‐B, in which Thr232 was changed to alanine, frequently induced multinuclearity in cells. To further investigate the mechanism by which the impairment of Aurora‐B activity resulted in the formation of multinucleate cells, we examined in detail the phenotype of the overexpression of vimentin mutants, in which phosphorylation sites by Aurora‐B were changed to alanine or glycine. The overexpression of the vimentin mutant in T24 cells caused not only inhibition of vimentin filament segregation into daughter cells but also multinuclearity. This suggested that the formation of multinucleate cells expressing the TA mutant of Aurora‐B may partly result from disorganization of vimentin filament segregation in the cytokinetic processes.( 48 )
As described above, Cdk1 also phosphorylates vimentin from prometaphase to metaphase, but its significance remained unknown. Recently, we demonstrated a direct interaction between Plk1 and vimentin‐Ser55 phosphorylated by Cdk1, an event that led to Plk1 activation.( 27 ) Activated Plk1 induced the phosphorylation of vimentin‐Ser82, which was elevated from metaphase and maintained until the end of mitosis. Mutational analyses revealed that Plk1‐induced vimentin‐Ser82 phosphorylation played an important role in vimentin filament segregation, coordinately with Rho‐kinase and Aurora‐B (Fig. 3).( 27 )
Figure 3.
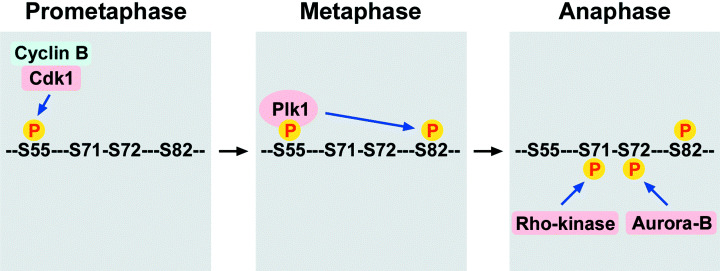
Cooperative phosphorylation of vimentin by mitotic kinases. Cdk1 phosphorylates vimentin‐Ser55 from prometaphase to metaphase. Polo‐like kinase 1 (Plk1) binds to vimentin‐Ser55 phosphorylated by Cdk1, leading to Plk1 activation and phosphorylation at Ser82 by Plk1. During cytokinesis, Ser71 and Ser72 are specifically phosphorylated by Rho‐kinase and Aurora‐B, respectively. These phosphorylations play an important role in vimentin filament segregation.
Mitotic checkpoint defects promote aneuploidy and tumorigenesis.( 49 ) Polyploid cells are formed during development in otherwise diploid organisms, and also arise during a variety of pathological conditions.( 50 ) It was suggested that genetic instability in tetraploid cells might provide a route to aneuploidy and thereby contribute to the development of cancer.( 50 ) As described earlier, the overexpression of vimentin mutants, in which phosphorylation sites by Rho‐kinase, Aurora‐B, Cdk1 and Plk1 are changed to alanine or glycine, in T24 cells causes multinuclearity.( 27 , 48 ) It is tempting to speculate that impairment of IF phosphorylation by the abnormal regulation of these mitotic kinases would contribute to the generation of tetraploid cells that have two nuclei, leading to subsequent apoptotic cell death in normal cells. The loss of tetraploidy checkpoint and a subsequent catastrophic mitosis in these cells might result in the generation of aneuploid cells, leading to cancer development (Fig. 4). This possible route from tetraploidy to aneuploidy, which is caused by the impairment of IF phoshorylation in cancer cells, should be examined in the future.
Figure 4.
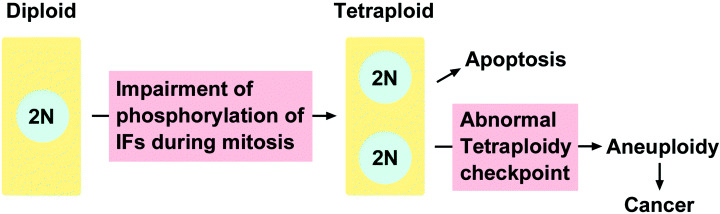
A potential route to generate aneuploid cells. During mitosis, the phosphorylation of intermediate filaments (IF) by mitotic kinases is impaired, leading to the generation of tetraploid cells. If these cells lose the tetraploidy checkpoint, they will be transformed into aneuploid cells.
New insights for biological functions of IF
Intermediate filament proteins exert their functions through interactions with a variety of structural and non‐structural proteins (Table 1).( 1 , 2 , 5 , 51 ) A principal function of IF is considered to be the protection of cells from mechanical stress, which is accomplished by the association with cell adhesion machinery, hemidesmosome and desmosome.( 52 , 53 ) To dissect the role of IF in cancer development, we set out to search for the binding partners of keratin 8/18. Keratin 8 and 18 form obligate non‐covalent heteropolymers and constitute the major components of the IF of simple or single‐layered epithelia, as found in the gastrointestinal tract, liver, exocrine pancreas and mammary gland, from which many carcinomas arise.( 54 ) Gene‐targeting techniques have been used to elucidate the function of keratin 8/18. Keratin 8 knockout mice in one strain died at around day 12 from undetermined tissue damage,( 55 ) whereas in a different strain they survived to adulthood but colorectal hyperplasia and inflammation were present.( 56 ) Intoxicated keratin 8 knockout mice suffer from extensive porphyria and progressive toxic liver damage without formation of Mallory bodies, suggesting that keratin 8 plays a protective role in certain types of toxic liver injury.( 57 ) Keratin 18 null mice were fertile and had a normal life span, whereas old keratin 18 null mice developed a distinct liver pathology with abnormal hepatocytes containing keratin 8‐positive aggregates that resembled Mallory bodies seen in human livers with alcoholic hepatitis.( 58 ) Human keratin 8/18 mutations are reported as risk factors for developing liver disease of multiple etiologies.( 4 ) In addition, missense mutations in the keratin 8 gene were reported in a subset of patients with inflammatory bowel disease (Crohn's disease and ulcerative colitis).( 59 )
Table 1.
Intermediate filament (IF)‐associated proteins
| Protein name | IF partners | Notable features |
|---|---|---|
| Cell adhesion and cytoskeleton | ||
| Plectin | V, D, G, NF, K, L‐B | Formation of hemidesmosomes |
| BPAG1 | K, NF | Formation of hemidesmosomes |
| Desmoplakin | K, V | Formation of desmosomes |
| Envoplakin | K | Localized in the cornified envelope of keratinocytes |
| Periplakin | K | Localized in the cornified envelope of keratinocytes |
| Plakophilin | K | Formation of desmosomes |
| Pinin | K | Formation of desmosomes |
| Filaggrin | K, V, D | Packing of keratin filaments |
| Trichohyalin | K | Cross‐linking in the hair follicle |
| Fimbrin | V | Adhesion‐dependent formation of fimbrin‐vimentin complex |
| PLIC | V | Anchoring vimentin to the plasma membrane |
| FTCD | V | Integrates the Golgi compartment with IF |
| Trichoplein | K | Trichohyalin and plectin homology domain |
| Fbf‐1 | K | Trichohyalin and plectin homology domain |
| Molecular chaperones | ||
| αB‐crystallin | G, V, K | Mutated in desmin‐related myopathy |
| Hsp27 | G, V, K | Management of the interaction between IF |
| Hsp/c70 | K8 | Direct association in an ATP‐dependent manner |
| Hsp90 | K | Co‐localization |
| Grp78 | K8 | Direct association in an ATP‐dependent fashion |
| Mrj | K18 | Recruitment of Hsp/c70 to K8/18 |
| Cell signaling | ||
| PKC | V, K | Association and phosphorylation |
| PKB | K10 | Sequestration of PKCζ and PKB by K10 |
| JNK | K8 | K8 is a target for JNK in Fas receptor‐mediated signaling |
| PKN | NF, V | Association and phosphorylation |
| Cdk5 | NF, N | Association and phosphorylation |
| Plk1 | V | Binds to vimentin phosphorylated at Ser55 by Cdk1 |
| 14‐3‐3 | K, V | Binds to K18 phosphorylated at Ser33 |
| TNFR2 | K8/18 | Keratin‐dependent resistance to TNF‐induced apoptosis |
| TRADD | K18, K14 | K18 sequesters TRADD to attenuate TNF‐induced apoptosis |
| DEDD | K18 | Regulates degradation of K8/18 filaments in apoptosis |
| c‐FLIP | K8/18 | K8/18 modulates c‐Flip/ERK1/2 antiapoptotic signaling |
| Hamartin | NF‐L | Hamartin is a protein encoded by TSC1 |
| Lamin‐associated | ||
| Lamin‐B receptor | L‐B | Membrane‐embedded enzyme that binds B‐type lamins |
| Emerin | L‐A, L‐B | Mutated in Emery–Dreifuss muscular dystrophy |
| Others | ||
| SNAP23 | V | Possible implication of vimentin in membrane traffic |
| VIP54 | V | Codistribution with vimentin |
D, desmin; G, glial fibrillary acidic protein; K, keratin; L, lamin; N, nestin; NF, neurofilament; V, vimentin.
Oshima and colleagues addressed a new, fascinating function of keratin 8/18, and explained the phenotypes of keratin 8 or keratin 18 knockout mice.( 60 ) They stated that normal and malignant epithelial cells deficient in keratin 8 and keratin 18 are approximately 100 times more sensitive to tumor necrosis factor (TNF)‐induced cell death. Keratin 8 and keratin 18 both bind to the cytoplasmic domain of tumor necrosis factor receptor (TNFR)2 and moderate the effects of TNF. This diminution may be a fundamental function of keratin 8/18 seen in liver regeneration, inflammatory bowel disease, hepatotoxin sensitivity, and the persistent expression of these keratins in many carcinomas.( 60 , 61 ) We discovered an important molecular mechanism by which epithelial cells can be resistant to TNF‐induced cytotoxicity.( 62 ) We found a direct association of keratin 18 with TNFR1‐associated death domain protein (TRADD), an indispensable adaptor molecule for TNFR1 signaling. Thus, keratin 18 may sequester TRADD to attenuate the interaction of TRADD with activated TNFR1, leading to diminution of TNF‐induced apoptosis.( 62 ) Keratin 14–TRADD interaction was postulated to be important for the pathogenesis of epidermolysis bullosa simplex.( 63 ) In addition, Gilbert et al. reported that keratin 8/18 provides resistance to Fas‐mediated apoptosis and that this protection occurs through a modulation of Fas targeting to the cell surface.( 64 ) Other adaptor molecules in apoptotic signaling, including death effector domain containing DNA binding protein and cellular FLICE inhibitory protein,( 65 , 66 , 67 ) were also reported to be associated with keratin 8/18 (Table 1). Together with these reports described above, it is proposed that simple epithelial keratins may play a role in modulating the response to some apoptotic signals.( 59 , 61 )
In addition to TRADD, we have thus far identified Mrj and trichoplein as keratin 8/18‐binding proteins (Table 1).( 68 , 69 ) Mrj, a member of the DnaJ/Heat shock protein (Hsp) 40 family of proteins, binds directly to keratin 18. Mrj may play an important role in the regulation of keratin 8/18 filament organization as a keratin 18‐specific cochaperone to work together with Hsp/c70.( 68 ) Trichoplein has a domain that shows a low degree of sequence similarity to trichohyalin and plectin, designated as the trichohyalin/plectin homology domain (TPHD).( 69 ) Trichoplein may be involved in organization of the apical network of keratin filaments and desmosomes in simple epithelial cells. We have also identified Fas binding factor‐1, which has TPHD, as a keratin‐binding protein (M. Sugimoto and M. Inagaki, unpublished observation).
The identification of IF‐binding proteins and lessons from transgenic mouse models and human diseases have provided us with clues about the functions of IF proteins. An attractive concept drawn from these studies is that IF may affect cell growth and cell death through interactions with a variety of non‐structural proteins, including kinases and adaptors for cell signaling.( 1 , 2 , 5 , 51 , 59 , 61 ) It is thus plausible that IF may play profound roles in cancer development, invasion and metastasis.
Conclusions
The site‐specific phosphorylation of IF proteins alters their filament structures. We have identified a number of in vitro phosphorylation sites of IF proteins by various kinases. Recent analyses using the site‐ and phosphorylation state‐specific antibodies demonstrated that several mitotic kinases, including Cdk1, Plk1, Rho‐kinase and Aurora‐B, phosphorylate IF proteins in a spatiotemporally regulated manner, leading to the efficient separation of IF to daughter cells. It is possible that the impairment of IF phosphorylation by mitotic kinases might result in the formation of multinucleated cells. The identification of novel IF‐binding proteins has provided us with some keys to open the door to the dissection of the molecular functions of IF. Further elucidation of the crosstalk between IF and intracellular signaling pathways is expected to improve our understanding of tumor development, invasion and metastasis and to find new therapeutic strategies for cancers.
During the course of our research on IF, in 1990 we devised a method and procedures to develop site‐ and phosphorylation state‐specific antibodies. These antibodies have facilitated understanding of the cytoskeletal organization, signal transduction and transcriptional mechanisms as well as clinical diseases, and will be further utilized for developing new drugs.
Acknowledgments
We regret that we were unable to cite many other excellent works in detail and that Table 1 did not contain all of the IF‐binding proteins. We are grateful to past and present members of the Inagaki laboratory for their essential contributions. This work was supported in part by grants‐in‐aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by a grant‐in‐aid for the Third Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labor, and Welfare of Japan, by a grant from The Naito Foundation, and by a grant from the Uehara Memorial Foundation.
References
- 1. Herrmann H, Hesse M, Reichenzeller M, Aebi U, Magin TM. Functional complexity of intermediate filament cytoskeletons: from structure to assembly to gene ablation. Int Rev Cytol 2003; 223: 83–175. [DOI] [PubMed] [Google Scholar]
- 2. Coulombe PA, Wong P. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat Cell Biol 2004; 6: 699–706. [DOI] [PubMed] [Google Scholar]
- 3. Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science 1998; 279: 514–19. [DOI] [PubMed] [Google Scholar]
- 4. Omary MB, Coulombe PA, McLean WHI. Intermediate filament proteins and their associated diseases. N Engl J Med 2004; 351: 2087–100. [DOI] [PubMed] [Google Scholar]
- 5. Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol 2005; 6: 21–31. [DOI] [PubMed] [Google Scholar]
- 6. Moll R. Cytokeratins as markers of differentiation in the diagnosis of epithelial tumors. Subcell Biochem 1998; 31: 205–62. [PubMed] [Google Scholar]
- 7. Hendrix MJC, Seftor EA, Chu YH, Trevor KT, Seftor REB. Role of intermediate filaments in migration, invasion and metastasis. Cancer Metastasis Rev 1996; 15: 507–25. [DOI] [PubMed] [Google Scholar]
- 8. Inagaki M, Nishi Y, Nishizawa K, Matsuyama M, Sato C. Site‐specific phosphorylation induces disassembly of vimentin filaments in vitro . Nature 1987; 328: 649–52. [DOI] [PubMed] [Google Scholar]
- 9. Inagaki M, Matsuoka Y, Tsujimura K et al. Dynamic property of intermediate filaments: Regulation by phosphorylation. Bioessays 1996; 18: 481–7. [Google Scholar]
- 10. Chang L, Goldman RD. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol 2004; 5: 601–13. [DOI] [PubMed] [Google Scholar]
- 11. Peter M, Nakagawa J, Dorée M, Labbé JC, Nigg EA. In vitro disassembly of the nuclear lamina and M phase‐specific phosphorylation of lamins by cdc2 kinase. Cell 1990; 61: 591–602. [DOI] [PubMed] [Google Scholar]
- 12. Dessev G, Iovcheva‐Dessev C, Bischoff JR, Beach D, Goldman R. A complex containing p34cdc2 and cyclin B phosphorylates the nuclear lamin and disassembles nuclei of clam oocytes in vitro . J Cell Biol 1991; 112: 523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inagaki N, Ito M, Nakano T, Inagaki M. Spatiotemporal distribution of protein kinase and phosphatase activities. Trends Biochem Sci 1994; 19: 448–52. [DOI] [PubMed] [Google Scholar]
- 14. Inagaki M, Inagaki N, Takahashi T, Takai Y. Phosphorylation‐dependent control of structures of intermediate filaments: a novel approach using site‐ and phosphorylation state‐specific antibodies. J Biochem 1997; 121: 407–14. [DOI] [PubMed] [Google Scholar]
- 15. Nagata K, Izawa I, Inagaki M. A decade of site‐ and phosphorylation state‐specific antibodies: recent advances in studies of spatiotemporal protein phosphorylation. Genes Cells 2001; 6: 653–64. [DOI] [PubMed] [Google Scholar]
- 16. Nishizawa K, Yano T, Shibata M et al. Specific localization of phospho‐intermediate filament protein in the constricted area of dividing cells. J Biol Chem 1991; 266: 3074–9. [PubMed] [Google Scholar]
- 17. Yano T, Taura C, Shibata M et al. A monoclonal antibody to the phosphorylated form of glial fibrillary acidic protein: application to a non‐radioactive method for measuring protein kinase activities. Biochem Biophys Res Commun 1991; 175: 1144–51. [DOI] [PubMed] [Google Scholar]
- 18. Matsuoka Y, Nishizawa K, Yano T et al. Two different protein kinases act on a different time schedule as glial filament kinase during mitosis. EMBO J 1992; 11: 2895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sekimata M, Tsujimura K, Tanaka J, Takeuchi Y, Inagaki N, Inagaki M. Detection of protein kinase activity specifically activated at metaphase–anaphase transition. J Cell Biol 1996; 132: 635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kosako H, Amano M, Yanagida M et al. Phosphorylation of glial fibrillary acidic protein at the same sites by cleavage furrow kinase and Rho‐associated kinase. J Biol Chem 1997; 272: 10 333–6. [DOI] [PubMed] [Google Scholar]
- 21. Goto H, Kosako H, Tanabe K et al. Phosphorylation of vimentin by Rho‐associated kinase at a unique amino‐terminal site that is specifically phosphorylated during cytokinesis. J Biol Chem 1998; 273: 11 728–36. [DOI] [PubMed] [Google Scholar]
- 22. Yasui Y, Amano M, Nagata K et al. Roles of Rho‐associated kinase in cytokinesis: Mutations in Rho‐associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol 1998; 143: 1249–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inada H, Togashi H, Nakamura Y, Kaibuchi K, Nagata K, Inagaki M. Balance between activities of Rho‐kinase and type 1 protein phosphatase modulates turnover of phosphorylation and dynamics of desmin/vimentin filaments. J Biol Chem 1999; 274: 34 932–9. [DOI] [PubMed] [Google Scholar]
- 24. Goto H, Yasui Y, Kawajiri A et al. Aurora‐B regulates the cleavage furrow‐specific vimentin phosphorylation in the cytokinetic process. J Biol Chem 2003; 278: 8526–30. [DOI] [PubMed] [Google Scholar]
- 25. Kawajiri A, Yasui Y, Goto H et al. Functional significance of the specific sites phosphorylated in desmin at cleavage furrow: Aurora‐B may phosphorylate and regulate type III intermediate filaments during cytokinesis coordinately with Rho‐kinase. Mol Biol Cell 2003; 14: 1489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yokoyama T, Goto H, Izawa I, Mizutani H, Inagaki M. Aurora‐B and Rho‐kinase/ROCK, the two cleavage furrow kinases, independently regulate the progression of cytokinesis: possible existence of a novel cleavage furrow kinase phosphorylates ezrin/radixin/moesin (ERM). Genes Cells 2005; 10: 127–37. [DOI] [PubMed] [Google Scholar]
- 27. Yamaguchi T, Goto H, Yokoyama T et al. Phosphorylation by Cdk1 induces Plk1‐mediated vimentin phosphorylation during mitosis. J Cell Biol 2005; 171: 431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans RM, Fink LM. An alteration in the phosphorylation of vimentin‐type intermediate filaments is associated with mitosis in cultured mammalian cells. Cell 1982; 29: 43–52. [DOI] [PubMed] [Google Scholar]
- 29. Celis JE, Lasen PM, Fey SJ, Celis A. Phosphorylation of keratin and vimentin polypeptides in normal and transformed mitotic human epithelial amnion cells: behavior of keratin and vimentin filaments during mitosis. J Cell Biol 1983; 97: 1429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans RM. Cyclic AMP‐dependent protein kinase‐induced vimentin filament disassembly involves modification of N‐terminal of intermediate filaments subunits. FEBS Lett 1988; 234: 73–8. [DOI] [PubMed] [Google Scholar]
- 31. Ando S, Tanabe K, Gonda Y, Sato C, Inagaki M. Domain‐ and sequence‐specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry 1989; 28: 2974–9. [DOI] [PubMed] [Google Scholar]
- 32. Chou YH, Bischoff JR, Beach D, Goldman RD. Intermediate filament reorganization during mitosis is mediated by p34cdc2 phosphorylation of vimentin. Cell 1990; 62: 1063–71. [DOI] [PubMed] [Google Scholar]
- 33. Kusubata M, Tokui T, Matsuoka Y et al. p13SUC1 suppresses the catalytic function of p34cdc2 kinase for intermediate filament proteins, in vitro . J Biol Chem 1992; 267: 20 937–42. [PubMed] [Google Scholar]
- 34. Inagaki M, Gonda Y, Nishizawa K et al. Phosphorylation sites linked to glial filament disassembly in vitro locate in a non‐α‐helical head domain. J Biol Chem 1990; 265: 4722–9. [PubMed] [Google Scholar]
- 35. Inagaki M, Gonda Y, Matsuyama M, Nishizawa K, Nishi Y, Sato C. Intermediate filament reconstitution in vitro. The role of phosphorylation on the assembly–disassembly of desmin. J Biol Chem 1988; 263: 5970–8. [PubMed] [Google Scholar]
- 36. Yano T, Tokui T, Nishi Y et al. Phosphorylation of keratin intermediate filaments by protein kinase C, by calmodulin‐dependent protein kinase and by cAMP‐dependent protein kinase. Eur J Biochem 1991; 197: 281–90. [DOI] [PubMed] [Google Scholar]
- 37. Inagaki M, Takahara H, Nishi Y, Sugawara K, Sato C. Ca2+‐dependent deimination‐induced disassembly of intermediate filaments involves specific modification of the amino‐terminal head domain. J Biol Chem 1989; 264: 18 119–27. [PubMed] [Google Scholar]
- 38. Nishizawa K, Yano T, Shibata M, Takahashi T, Inagaki M. Distribution of phospho‐glial fibrillary acidic protein in astro glial cells. Cell Struct Funct 1990; 15: 497. [Google Scholar]
- 39. Yano T, Taura C, Hirono Y et al. Specific antibodies to phospho‐glial fibrillary acidic protein: production and characterization. Cell Struct Funct 1990; 15: 497. [Google Scholar]
- 40. Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem 1992; 61: 441–70. [DOI] [PubMed] [Google Scholar]
- 41. Tsujimura K, Tanaka J, Ando S et al. Identification of phosphorylation sites on glial fibrillary acidic protein for cdc2 kinase and Ca2+‐calmodulin‐dependent protein kinase II. J Biochem 1994; 116: 426–34. [DOI] [PubMed] [Google Scholar]
- 42. Chou YH, Ngai KL, Goldman R. The regulation of intermediate filament reorganization in mitosis: p34cdc2 phosphorylates vimentin at a unique N‐terminal site. J Biol Chem 1991; 266: 7325–8. [PubMed] [Google Scholar]
- 43. Tsujimura K, Ogawara M, Takeuchi Y, Imajoh‐Ohmi S, Ha MH, Inagaki M. Visualization and function of vimentin phosphorylation by cdc2 kinase during mitosis. J Biol Chem 1994; 269: 31 097–106. [PubMed] [Google Scholar]
- 44. Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by Rho family GTPases in mammalian cells. Annu Rev Biochem 1999; 68: 459–86. [DOI] [PubMed] [Google Scholar]
- 45. Kosako H, Goto H, Yanagida M et al. Specific accumulation of Rho‐associated kinase at the cleavage furrow during cytokinesis: cleavage furrow‐specific phosphorylation of intermediate filaments. Oncogene 1999; 18: 2783–8. [DOI] [PubMed] [Google Scholar]
- 46. Yasui Y, Goto H, Matsui S et al. Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene 2001; 20: 2868–76. [DOI] [PubMed] [Google Scholar]
- 47. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4: 842–54. [DOI] [PubMed] [Google Scholar]
- 48. Yasui Y, Urano T, Kawajiri A et al. Autophosphorylation of a newly identified site of Aurora‐B is indispensable for cytokinesis. J Biol Chem 2004; 279: 12 997–3003. [DOI] [PubMed] [Google Scholar]
- 49. Kops GJPL, Weaver BAA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 2005; 5: 773–85. [DOI] [PubMed] [Google Scholar]
- 50. Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004; 5: 45–54. [DOI] [PubMed] [Google Scholar]
- 51. Coulombe PA, Omary MB. ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol 2002; 14: 110–22. [DOI] [PubMed] [Google Scholar]
- 52. Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci 1998; 111: 2477–86. [DOI] [PubMed] [Google Scholar]
- 53. Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol 2000; 1: 208–16. [DOI] [PubMed] [Google Scholar]
- 54. Oshima RG, Baribault H, Caulín C. Oncogenic regulation and function of keratins 8 and 18. Cancer Metastasis Rev 1996; 15: 445–71. [DOI] [PubMed] [Google Scholar]
- 55. Baribault H, Price J, Miyai K, Oshima RG. Mid‐gestational lethality in mice lacking keratin 8. Genes Dev 1993; 7: 1191–202. [DOI] [PubMed] [Google Scholar]
- 56. Baribault H, Penner J, Iozzo RV, Wilson‐Heiner M. Colorectal hyperplasia and inflammation in keratin 8‐deficient FVB/N mice. Genes Dev 1994; 8: 2964–73. [DOI] [PubMed] [Google Scholar]
- 57. Zatloukal K, Stumptner C, Lehner M et al. Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am J Pathol 2000; 156: 1263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Magin TM, Schröder R, Leitgeb S et al. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver‐specific keratin 8‐positive aggregates. J Cell Biol 1998; 140: 1441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Owens DW, Lane EB. The quest for the function of simple epithelial keratins. Bioessays 2003; 25: 748–58. [DOI] [PubMed] [Google Scholar]
- 60. Caulin C, Ware CF, Magin TM, Oshima RG. Keratin‐dependent, epithelial resistance to tumor necrosis factor‐induced apoptosis. J Cell Biol 2000; 149: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oshima RG. Apoptosis and keratin intermediate filaments. Cell Death Differ 2002; 9: 486–92. [DOI] [PubMed] [Google Scholar]
- 62. Inada H, Izawa I, Nishizawa M et al. Keratin attenuates tumor necrosis factor‐induced cytotoxicity through association with TRADD. J Cell Biol 2001; 155: 415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yoneda K, Furukawa T, Zheng YJ et al. An autocrine/paracrine loop linking keratin 14 aggregates to tumor necrosis factor α‐mediated cytotoxicity in a keratinocyte model of epidermolysis bullosa simplex. J Biol Chem 2004; 279: 7296–303. [DOI] [PubMed] [Google Scholar]
- 64. Gilbert S, Loranger A, Daigle N, Marceau N. Simple epithelium keratins 8 and 18 provide resistance to Fas‐mediated apoptosis. The protection occurs through a receptor‐targeting modulation. J Cell Biol 2001; 154: 763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee JC, Schickling O, Stegh AH et al. DEDD regulates degradation of intermediate filaments during apoptosis. J Cell Biol 2002; 158: 1051–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dinsdale D, Lee JC, Dewson G, Cohen GM, Peter ME. Intermediate filaments control the intracellular distribution of caspases during apoptosis. Am J Pathol 2004; 164: 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gilbert S, Loranger A, Marceau N. Keratins modulate c‐FLIP/extracellular signal‐regulated kinase 1 and 2 antiapoptotic signaling in simple epithelial cells. Mol Cell Biol 2004; 24: 7072–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Izawa I, Nishizawa M, Ohtakara K, Ohtsuka K, Inada H, Inagaki M. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J Biol Chem 2000; 275: 34 521–7. [DOI] [PubMed] [Google Scholar]
- 69. Nishizawa M, Izawa I, Inoko A et al. Identification of trichoplein, a novel keratin filament‐binding protein. J Cell Sci 2005; 118: 1081–90. [DOI] [PubMed] [Google Scholar]