

Abstract
Urothelial carcinomas are well known to feature multifocal development in the urinary tract, both synchronously and asynchronously. This phenomenon can be explained by either seeding of cancer cells in the urinary tract or field cancerization. As there are two characteristic morphological patterns of urothelial carinomas, papillary and nodular, published papers were here reviewed to understand the development and progression of urothelial carcinoma regarding multifocality due to seeding or field changes with reference to the type of urothelial carcinoma. From animal experiments using rats, mice and dogs treated with N‐butyl‐N‐(4‐hydoroxybutyl) nitrosamine, and from pathological observation of human cystectomy specimens on step‐sectioning and molecular analysis, nodular carcinomas appear to either develop via papillary carcinomas or de novo. Clinical aspects of multifocal tumor development are outside of the scope of this review, although an understanding of the mechanisms underlying multifocality and the papillary/nodular morphological relationship is important to determine follow‐up strategies for patients treated for primary urothelial carcinomas and for reconstruction of the urinary tract after cystectomy. (Cancer Sci 2006; 97: 821–828)
The urothelium (also known as the transitional cell epithelium) covers the luminal surface of almost the entire urinary tract, extending from the renal pelvis, through the ureter and bladder, to the proximal urethra. Typically it is composed of three to seven layers of cells, that is, basal cells, intermediate cells and superficial cells. The superficial cells are large and binucleated, with a flat characteristic shape leading to the term ‘umbrella cell’. Their luminal surfaces are covered with an asymmetric unit membrane, which functions as the permeability barrier between the urine and blood vessels in the urothelium.( 1 )
Worldwide, there are approximately 336 000 new cases of urothelial carcinoma and 132 000 deaths annually.( 2 ) The majority of lesions are bladder carcinomas, and urothelial carcinomas of the renal pelvis and ureter account for only approximately 7% of the total.( 3 ) In Japan and western countries, more than 90% of bladder carcinomas are urothelial carcinomas, and squamous cell carcinomas and adenocarcinomas are rare. Risk factors include tobacco smoking, occupational exposure to aromatic amines, consumption of arsenic‐laced water, radiation therapy of neighboring organs and chemotherapeutic drugs such as alkylating agents. In contrast, in Egypt, where the dominant etiology is schistosomiasis infection, squamous cell carcinomas are the most prevalent bladder carcinoma. Here the focus of attention is urothelial carcinoma in its papillary and nodular forms. Squamous cell carcinoma and the clinical relevance of metaplasia will not be covered.
Multifocal development of urothelial carcinomas
The clinical aspects of multiple urothelial carcinomas need to be emphasized:( 4 )
-
1
There are patients in which lesions in the renal pelvis, ureter and bladder are observed simultaneously (1, 2).
-
2
When standard nephrectomy is performed for renal pelvic and ureteral carcinomas, approximately one‐third of the lower ureter is left intact. In this remaining ureter, urothelial carcinomas develop subsequently in 20–50% of the patients (Fig. 1b). Consequently, at the present time, the state of the art surgery for renal pelvic and/or ureteral carcinomas is total nephroureterectomy, including removal of a small portion of the bladder with the ureteral orifice (bladder cuff) in the affected side.
-
3
Even if total nephroureterectomy is carried out, however, 15–50% of patients exhibit subsequent carcinomas in the bladder (Fig. 1c).
-
4
When superficial papillary urothelial carcinomas of the bladder are resected transurethrally (TUR), the rate for subsequent development of urothelial carcinomas of a similar biological nature in the normal‐appearing bladder mucosa is reported to be 50–80% (Fig. 1d).
-
5
When cystoprostatectomy (i.e. removal of the bladder and prostate) is carried out for male bladder cancer patients, 4–17% incidences of urothelial carcinomas in the remaining urethra have been described( 5 ) (Fig. 1e). In female bladder cancer patients, involvement of the urethra is reported to occur in 1.4–36% of cases.( 6 )
Figure 1.
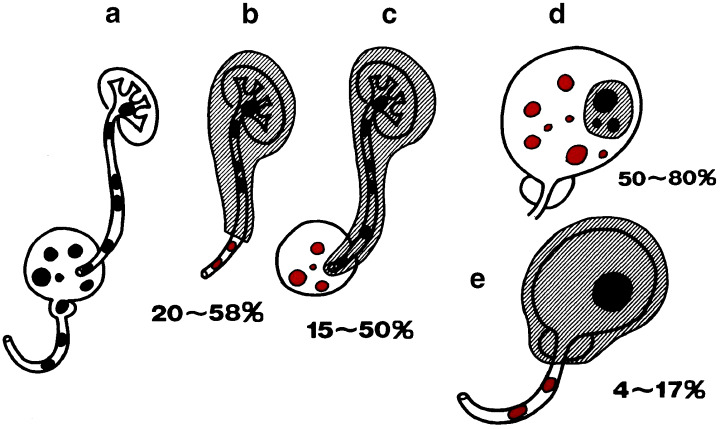
Clinical findings indicating multifocal tumor development in the urinary tract. Black lesions are original tumors and red lesions are recurrent tumors.
Figure 2.

A case of renal pelvic, ureteral and vesical carcinomas who underwent right nephroureterocystectomy.
After surgical treatment of bladder carcinomas, the incidence of subsequent upper urinary tract carcinomas (of the renal pelvis or ureter) ranges from 0.7 to 4%.( 7 ) Most of these carcinomas are diagnosed 4–6 years after the initial appearance of the bladder carcinoma.( 8 , 9 ) However, the risk is reported to increase to the level of 6–20% (15–22‐fold) if the patients suffer from vesico‐ureteral reflux.( 10 )
With upper urinary tract carcinomas, simultaneous bilateral lesions are rare, the estimated incidence being 1–5%.( 11 ) Approximately 2–3% of patients with unilateral upper urinary tract carcinoma experience subsequent contralateral upper urinary tract carcinoma.( 12 ) These clinical data should be taken into serious consideration when we decide on nephroureterectomy or cystectomy and plans for follow up after surgery, and the upper and lower urinary tract and the contralateral upper urinary tract should be assumed to constitute a single clinical unit from the renal pelvis to the urethra.
Field cancerization versus clonal expansion
To explain multifocal carcinoma development in the urinary tract, two theories have been proposed.( 13 , 14 ) The first is field cancerization, proposed in 1953 by Slaughter et al., which was based on observations of the multicentric development of cancers in the oral cavity, with the high impact of carcinogens and promoting agents being associated with some lifestyle factors.( 15 ) A similar understanding is possible for the urinary tract as the entire urothelium is exposed to carcinogens contaminating the urine. The second hypothesis is that multiple carcinomas in the urinary tract are the result of intraluminal spread from a single lesion, originating from a single transformed cell, namely seeding or implantation of cancer cells at different sites. This phenomenon is also called clonal expansion of multifocal carcinomas. Debates on multiple cancer development have been similar for cancers of the oral cavity,( 16 ) respiratory tract,( 17 ) head and neck,( 18 ) breast,( 19 ) ovary( 20 ) and cervix.( 21 ) Recently, strong molecular evidence has been presented in support of clonal expansion in the epithelium of oral cavity and respiratory tract cases.( 16 , 22 )
Many urologists and pathologists have supported the field cancerization hypothesis in the urinary tract, but recent molecular studies have also pointed to a clonal origin for most multifocal urothelial carcinomas. Various molecular analyses have been applied. Sidransky et al. investigated X‐chromosome inactivation in multiple bladder carcinomas of four female patients and proved that the same allele of the X‐chromosome was inactivated in all lesions within the single bladder.( 23 ) Subsequently, Lunec et al.( 24 ) and Habuchi et al.( 25 ) examined patients having heterotopic synchronous or recurrent urothelial carcinomas in the bladder or upper urinary tract. These carcinomas had identical mutation sites and patterns of p53 gene alteration, indicating the metachronous carcinomas to be derived from the original carcinoma cells due to clonal expansion. Habuchi also reviewed the origin of multifocal carcinomas of the bladder and upper urinary tract.( 26 ) Other genetic features that can be used to assess clonal origin are loss of heterozygosity (LOH) and microsatellite alteration patterns, both commonly used as markers of neoplasia.
Stoehr et al. analyzed primary carcinomas in 14 cystectomy specimens for p53 protein overexpression by immunohistochemistry and p53 gene mutation by genomic sequencing.( 27 ) They reported detection of p53‐mutant cells in histologically normal adjacent or remote mucosa and in preneoplastic urothelial areas in four patients with invasive bladder carcinoma, concluding extensive intraurothelial tumor cell spread.
Evidence of a monoclonal origin and intraepithelial spread has also been provided by Simon et al. from comparative genomic hybridization in 32 bladder tumors originating from six cystectomy specimens.( 28 ) Identical TP53 mutations and protein overexpression were found in tumors from the same individual, as well as in mucosal samples from the continuous areas. The sequence of genomic changes apparently acquired during progression of bladder carcinomas was highly complex and varied within each patient and from tumor to tumor. Early changes included alterations in −17p, +20p, −9p, −9q, +2q34‐qter, +12q14‐q21, +1q22‐q25, −8p22‐pter, −5q31‐qter and +17q. Subsequent tumor progression was characterized by accumulation of changes in +11q14, −21q, −5q13‐q14, +8q22, +10p, −10q22qter and −11p. Cytogenetic variety in multifocal tumors has also been described in support of intraluminal tumor seeding.( 29 )
It is conceivable that widespread p53‐mutated cells in the normal urothelium are generated in the bladder due to carcinogen exposure, and that from these, new tumors later develop with surrounding normal‐appearing mucosa having p53 mutations. However, there is no mechanistic explanation for the intraepithelial spread of carcinoma cells to remote normal‐appearing mucosa, and it is unrealistic to consider a mechanism due to cell motility.
Although the clonal theory now dominates in explanations of multifocality of urothelial carcinomas, there are also conflicting observations. Cheng et al. collected cancer cells by microdissection from 18 cystectomy specimens from female patients and analyzed the X‐chromosome‐linked human androgen receptor gene.( 30 ) Only 11 of the 18 specimens were informative, with nine exhibiting non‐random inactivation of the target locus and seven showing different patterns, indicating field change in these cases. Paiss et al. examined X‐chromosome inactivation in 45 archival or fresh frozen bladder tumors obtained from 27 female patients using a polymerase chain reaction‐based procedure.( 31 ) Polyclonal patterns were observed in 16 of the 45 tumors. Stoehr et al. examined multiple samples from four cystectomy specimens for LOH at chromosomes 8p, 9p, 9q and 17p and they observed oligoclonality in two patients.( 27 ) Thus, both hypotheses continue to be discussed, although clonal expansion by intraluminal spread of primary carcinoma appears the dominant explanation for multifocality.
Relationship between papillary carcinoma and nodular carcinoma
Urothelial carcinogenesis has been investigated in various species of animal. A particular focus has been on the histogenesis of lesions in rats treated with the carcinogen N‐butyl‐N‐(4‐hydroxybutyl) nitrosamine (BHBN).( 32 ) Normal urothelium of rats is composed of two to three layers of urothelial cells and when BHBN is given in the drinking water, the mucosal layer becomes hyperplastic at 4 weeks. If BHBN administration is stopped at this point, mild hyperplastic mucosal lesion regresses to the normal state. However, if BHBN treatment is continued, mucosal hyperplasia progresses to papillary growth and papillary carcinomas develop via papillomas (Fig. 3). Large papillary carcinomas may occasionally invade the bladder wall. Usually, papillary carcinomas are multifocal but superficial, indicating bladder carcinoma in rats to be a good model for human papillary bladder carcinoma. With progression, urinary bladders become filled with multifocal urothelial carcinomas and rats die due to massive bleeding. Papillary carcinomas are always induced in rats, irrespective of the strain of animal, the concentration of BHBN, or the carcinogen, with similar findings being reported with N‐(4‐[5‐nitro‐2‐furyl]‐2‐thizolyl)formamide and N‐methyl‐N‐nitrosourea.
Figure 3.
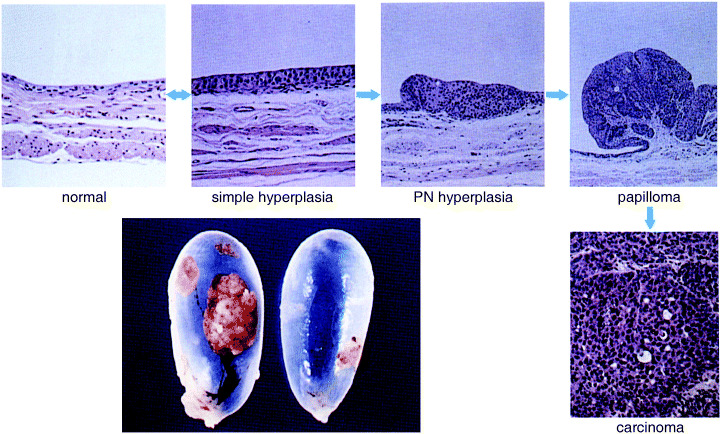
Histogenesis and progession of papillary carcinomas in rats. (Reproduced with permission from Medical view Co., T. Kakizoe, Development and Progression of Bladder Cancer, 1995.)
Urinary bladder carcinogenesis in mice treated with BHBN in the drinking water originates in the normal mucosa (composed of two to three layers of urothelium) and progresses through mild hyperplasia, dysplasia and carcinoma in situ, to form large nodular invasive carcinomas( 33 ) (Fig. 4). Bilateral ureters are frequently obstructed due to invasion of carcinomas into the bladder wall, and when advanced, mice die because of renal insufficiency due to hydronephrosis. Because of these features, the bladder carcinomas induced by BHBN in mice offer good models for human nodular invasive bladder carcinoma. Of interest, it has proven impossible to induce multiple papillary carcinomas in any strain of mouse, not with any concentration of BHBN nor any other carcinogen. Thus, there is a clear contrast between the biological and morphological characteristics of bladder carcinomas in rats and mice.
Figure 4.
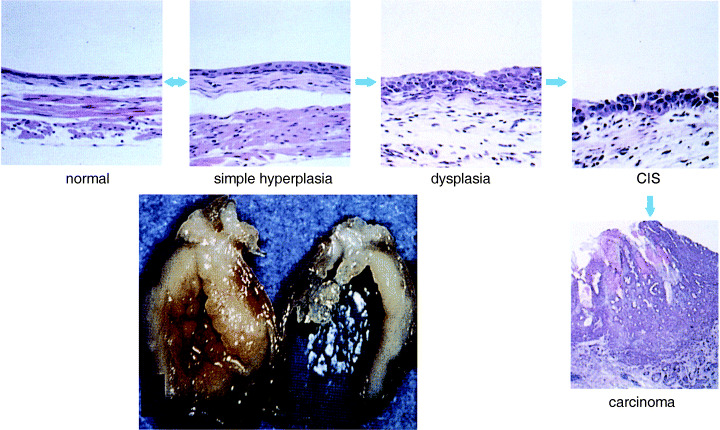
Histogenesis and progression of nodular invasive carcinoma in mice. (Reproduced with permission from Medical view Co., T. Kakizoe, Development and Progression of Bladder Cancer, 1995.)
Bladder carcinogenesis in female dogs has been studied extensively by Okajima et al. who used these animals to periodically observe the surface of the bladder epithelium directly by cystoscopy.( 34 ) They made capsules of BHBN (80–500 mg/capsule), which were administered once a day. After 4–5 years, papillary superficial bladder carcinomas were induced (Fig. 5), and when these were examined by cystoscopy without further BHBN treatment after more than 10 years, the bladders of the dogs were full of multifocal papillary carcinomas. When dogs were given 500 mg of BHBN daily, nodular invasive carcinomas were induced after approximately 1 year. These findings indicate that bladder carcinogenesis in dogs can be controlled by the concentration and period of BHBN administration in terms of the type of carcinoma (i.e. papillary superficial and nodular invasive bladder carcinoma). Thus, in the various animal species, rats, mice and dogs, the relationship between papillary and nodular carcinomas in the bladder appears to differ.
Figure 5.
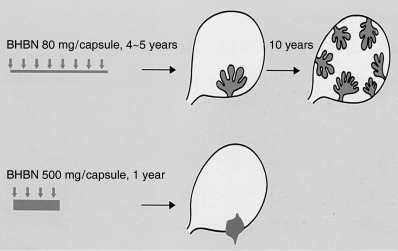
Development and progression of papillary and nodular carcinomas depending on the concentration and period of administration of N‐butyl‐N‐(4‐hydroxybutyl) nitrosamine (BHBN) in female dogs.( 34 ) (Reproduced with permission from Medical view Co., T. Kakizoe, Development and Progression of Bladder Cancer, 1995.)
Morphological and pathological characteristics of human urothelial carcinomas, mainly bladder carcinomas, are a combination of papillary (P), papillonodular (PN), nodular (N) and carcinoma in situ (C). On careful analysis of cancerous lesions and normal‐looking mucosa of 186 cystectomized specimens by step‐sectioning,( 35 ) we classified 17 as C and 80 as P and P + C. Fifty‐seven cases featured apparent early changes from P to a mixture of P and N, whereas six showed late development of N with repeated recurrence of P. The findings thus indicated some N to have developed from P as more anaplastic cell populations within a pre‐existing low‐grade lesion, whereas others arose directly de novo from C (Fig. 6). Topographic relationships between P and N in the pT3 group are illustrated in Fig. 7. Figure 8 demonstrates findings for patients having a previous history of repeated recurrence of papillary carcinomas treated by TUR. At the time of cystectomy, all the cystectomized specimens showed a variable degree of coexistence of P, N and C.
Figure 6.
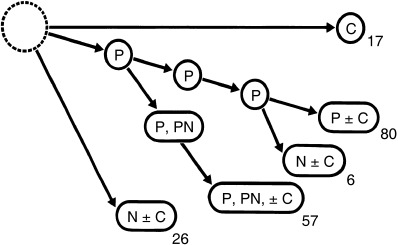
Conceptual progression routes of papillary (P), papillonodular (PN) and nodular (N) carcinoma, and carcinoma in situ (C), in 186 cystectomized specimens examined by step‐sectioning.( 35 )
Figure 7.
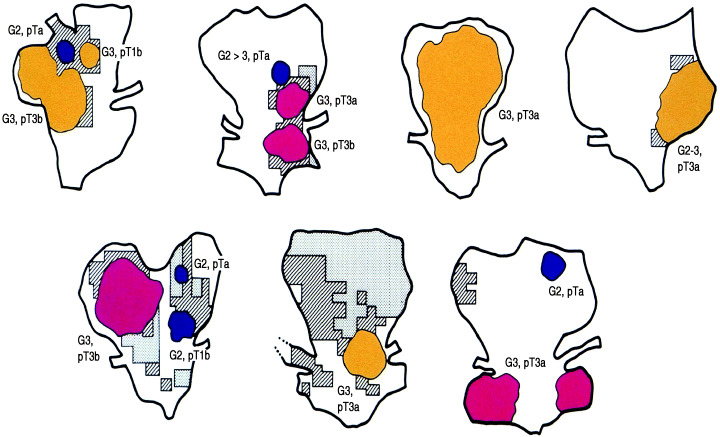
Cases of pT3 cystectomized specimens indicating coexistence of papillary (P; blue), papillonodular (PN; yellow) and nodular (N; red) carcinoma together with oblique line area (C) and shaded area (dysplasia).( 35 )
Figure 8.
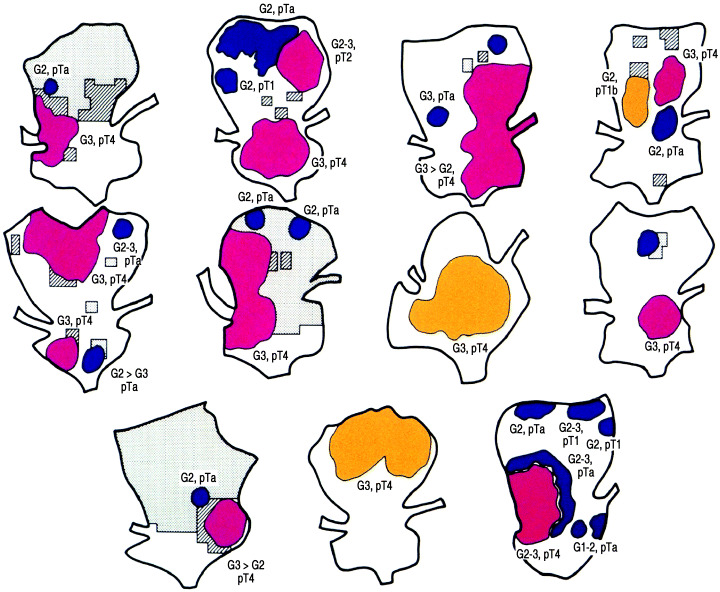
Eleven cases of cystectomized specimens having histories of multiple transurethral resection for papillary recurrent carcinomas showing variety of stages and morphology in a single bladder.( 35 )
Molecular pathways of urothelial carcinogenesis
With papillary superficial and nodular invasive carcinomas, there appear to be differences in molecular pathways as well as morphology( 36 , 37 , 38 ) (Fig. 9). The most common genetic alterations in low‐grade papillary urothelial carcinomas are LOH of chromosome 9 and activating mutations of fibroblast growth factor receptor 3 (FGFR3).( 39 , 40 ) Over 70% of low‐grade papillary carcinoma exhibit FGFR3 mutations, but only 10–20% of high‐grade invasive carcinomas have FGFR3 mutations, implying a key role for FGFR3 together with mutations of 9p and 9q, specifically for the induction of low‐grade papillary carcinomas. Invasive carcinoma is frequently associated with p53 mutations.( 41 , 42 , 43 )
Figure 9.
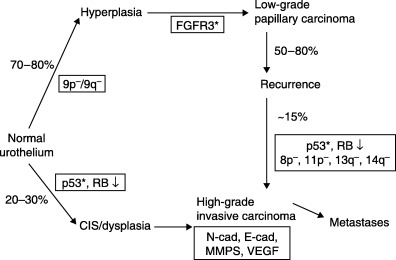
Presumed molecular pathways of development of papillary and nodular carcinoma. (Modified from Wu.( 38 )) CIS, carcinoma in situ; FGFR3, fibroblast growth factor receptor 3; MMPS, matrix metal proteinases; RB, retinoblastoma gene; VEGF, vascular endothelial growth factor.
In addition to the above‐mentioned genomic abnormalities associated with urothelial carcinoma, epigenetic alterations also occur during urothelial carcinogenesis. Almost all cells in the human body contain the same sequence of DNA, but cells in different organs during different developmental stages express different genes by epigenetic control of cellular function. This expression control of DNA is achieved by DNA methylation, chromatin structure and transcription factors. DNA can be methylated at cytosine residues adjacent to guanine residues (CpG) and CpG sites are distributed non‐randomly throughout the genome, being found as islands in the promoter and exonic regions. Inactivation of tumor suppressor genes is known to occur via promoter hypermethylation, frequently due to DNA methyltransferase 1 (DNMT1). Expression of DNMT1 is increased in tumors and even during the precancerous stages of the urothelium with the development of flat carcinomas in situ. ( 44 ) Hypermethylation in urothelial carcinogenesis has also been observed in the promoter region of the E‐cadherin gene, indicating an association with carcinoma in situ and detachment of cells or clusters of carcinoma cells in the urine.( 45 )
Development and progression of urothelial carcinoma
As is shown in Fig. 1, multifocal transitional cell carcinomas may develop in any region of the urinary tract, from the renal pelvis/ureter to the bladder/urethra.( 4 , 46 ) Whereas upper urinary tract carcinomas occur infrequently after transurethral management of bladder carcinomas, the incidence is much greater in patients with vesico‐ureteral reflux,( 10 ) suggesting seeding or implantation of primary urothelial carcinoma cells after spread via the urine rather than field cancerization. In addition, recent molecular analyses using X‐chromosome inactivation,( 23 ) p53 mutation,( 24 , 25 , 26 , 27 , 28 ) LOH( 27 , 28 ) and comparative genomic hybridization( 29 ) have provided compelling evidence that multifocal urothelial carcinomas are monoclonal in origin, despite some discrepancies.( 30 , 31 )
Comparing the basic morphological patterns of urothelial carcinomas, namely low‐grade, superficial papillary carcinomas and high‐grade, invasive nodular carcinomas, these two patterns of urothelial carcinomas are clearly separated in rats( 32 ) and mice.( 33 ) However, in dogs,( 34 ) papillary carcinomas and nodular carcinomas can both be induced, depending on the concentration and period of carcinogen administration. In humans, papillary carcinomas and nodular carcinomas may originally develop separately, but coexistence of the two types is occasionally observed in a single bladder together with dysplasia and carcinoma in situ. ( 35 ) During the process of repeated recurrence, progression from papillary carcinoma to nodular carcinoma may be observed and molecular analysis of bladder carcinogenesis indicates the presence of two pathways: LOH of 9p/9q loss and FGFR3 mutation resulting in papillary carcinoma, and if p53 mutation occurs, nodular carcinoma. If p53 mutations occur initially, nodular carcinoma develops via dysplasia and carcinoma in situ. The available data clearly indicate that multiple genetic alterations are associated with the development and progression of bladder cancer.( 36 , 37 , 38 )
In the normal‐appearing mucosa of the renal pelvis, ureter and bladder, dysplasia and carcinoma in situ may be frequently observed.( 46 ) As mucosal dysplasia is not malignant, a derivation by implantation from primary carcinomas is not conceivable. In normal‐appearing mucosa in remote areas from tumors, p53 mutation may be observed.( 27 ) Intraepithelial spread( 27 , 28 ) has been proposed as an explanation but this would appear unlikely. The phenomenon of coexistence of dysplasia, carcinoma in situ and p53 mutation in normal‐appearing mucosa can be far more readily explained by the field cancerization theory. Differences in growth patterns of papillary and nodular carcinomas, and carcinoma in situ, as well as in cellular polarity and grade of malignancy make a single origin by seeding unreasonable. Finally, on detailed analysis of recurrent patterns of papillary carcinomas after TUR with or without intravesical instillation therapy, Akaza et al. concluded that recurrence patterns are biphasic, with an initial peak due to seeding or implantation of cancer cells during therapy and a second peak due to a new tumor occurrence from a background of field changes.( 47 ) Thus, multifocal bladder recurrence of urothelial carcinomas is due to a combination of seeding and field changes. This is directly relevant to the condition for reconstruction of the urinary tract after cystectomy, inhibition and control of multiple recurrences after TUR, and the frequency and timing of follow‐up for upper tract malignancies after treatment of bladder carcinoma. However, such clinical issues will be covered elsewhere.
References
- 1. Koss LG. Tumors of the Urinary Bladder. Washington DC: Armed Forces Institute of Pathology, 1975. [Google Scholar]
- 2. Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer 2001 1998; 94: 153–6. [DOI] [PubMed] [Google Scholar]
- 3. Cancer Incidence in Sweden 1998. Stockholm, Sweden: The National Board of Health and Welfare, 1998. [Google Scholar]
- 4. Kakizoe T, Tobisu K, Tanaka Y et al. Development of multiple transitional cell carcinomas in the urinary tract. Jpn J Clin Oncol 1991; 21: 110–14. [DOI] [PubMed] [Google Scholar]
- 5. Studer UE, Hautmann RE, Hohenfellner M et al. Indication for continent diversion after cystectomy and factors affecting long‐term result. Urol Oncol 1998; 4: 172–80. [DOI] [PubMed] [Google Scholar]
- 6. Coloby P, Kakizoe T, Tobisu K, Sakamoto M. Urethral involvement in female bladder cancer patients: mapping of 47 consecutive cysto‐urethrectomy specimens. J Urol 1994; 152: 1438–42. [DOI] [PubMed] [Google Scholar]
- 7. Kirkali Z, Tuzel E. Transitional cell carcinoma of the ureter and renal pellvis. Crit Rev Oncol Hematol 2003; 47: 155–60. [DOI] [PubMed] [Google Scholar]
- 8. Palon J, Farina LA, Villavicencio H, Vicente J. Upper tranct urothelial tumor after transurethral resection for bladder tumor. Eur Urol 1992; 21: 110–14. [DOI] [PubMed] [Google Scholar]
- 9. Canales BK, Anderson JK, Premoli J, Slaton JW. Risk factors for upper tract recurrence in patients undergoing long‐term surveillance for stage Ta bladder cancer. J Urol 2006; 175: 74–7. [DOI] [PubMed] [Google Scholar]
- 10. De Torres Mateos JA, Banus Gassol JM, Palou Redorta J, Morote Robles J. Vesicorenal reflux and upper urinary tract transitional cell carcinoma after transurethral resection of recurrent superficial bladder carcinoma. J Urol 1987; 138: 49–51. [DOI] [PubMed] [Google Scholar]
- 11. Kang CH, Yu TJ, Hsieh HH, Yang JW, Shu K, Huang CC. The development of bladder tumors and contralateral upper urinary tract tumors after primary transitional cell carcinoma of the upper urinary tract. Cancer 2003; 98: 1620–6. [DOI] [PubMed] [Google Scholar]
- 12. Charbit L, Gendrean MC, Mee S, Cukier J. Tumors of the upper urinary tract: 10 years of experience. J Urol 1991; 146: 1243–6. [DOI] [PubMed] [Google Scholar]
- 13. Harris AL, Neal DE. Bladder cancer‐field versus clonal origin. N Engl J Medical 1992; 326: 759–61. [DOI] [PubMed] [Google Scholar]
- 14. Carcia SB, Park HS, Novelli M, Wright NA. Field cancerization, clonality, and epithelial stem cells: the spread of mutated clones in epithelial sheets. J Pathol 1999; 187: 61–81. [DOI] [PubMed] [Google Scholar]
- 15. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953; 6: 963–8. [DOI] [PubMed] [Google Scholar]
- 16. Scholes AG, Woolgar JA, Boyle MA, Brown JS. Synchronous oral carcinomas: independent or common clonal origin? Cancer Res 1998; 58: 2003–6. [PubMed] [Google Scholar]
- 17. Sozzi G, Miozzo M, Pastorinou U et al. Genetic evidence for an independent origin of multiple preneoplastic and neoplastic lung lesions. Cancer Res 1995; 55: 135–40. [PubMed] [Google Scholar]
- 18. Bedi GC, Westra WH, Gabrielson E, Koch W, Sindransky D. Multiple head and neck tumors: evidence for a common clonal origin. Cancer Res 1996; 56: 2484–7. [PubMed] [Google Scholar]
- 19. Noguchi S, Aihara T, Koyama H, Motomura K, Inaji H, Imaoka S. Discrimination between multicentric and multifocal carcinomas of the breast through clonal analysis. Cancer 1994; 74: 872–7. [DOI] [PubMed] [Google Scholar]
- 20. Abeln EC, Kuipers‐Dijkshoorn NJ, Berns EM, Henzen‐Logmans SC, Fleuren GJ, Cornelisse CJ. Molecular genetic evidence for unifocal origin of advanced epithelial ovarian cancer and for minor clonal divergence. Br J Cancer 1995; 71: 1330–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larson AA, Liao SY, Stanbridge EJ, Cavenee WK, Hampton GM. Genetic alterations accumulate during cervical tumorigenesis and indicate a common origin for multifocal lesions. Cancer Res 1997; 57: 4171–6. [PubMed] [Google Scholar]
- 22. Franklin WA, Gazdar AF, Haney J, Wistuba II. Widely dispersed p53 mutation in respiratory epithelium: A novel mechanism for field carcinogenesis. J Clin Invest 1997; 100: 2133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sidransky D, Frost P, Von Eschenbach A, Oyasu R, Preisinger AC, Vogelstein B. Clonal origin of bladder cancer. N Engl J Med 1992; 326: 737–40. [DOI] [PubMed] [Google Scholar]
- 24. Lunec J, Challen C, Wright C, Mellon K, Neal DE. c‐erb B‐2 amplification and identical p53 mutations in concomitant transitional carcinomas of renal pelvis and urinary baldder. Lancet 1992; 339: 439–40. [DOI] [PubMed] [Google Scholar]
- 25. Habuchi T, Takahashi R, Yamada H, Kakehi Y, Sugiyama T, Yoshida O. Metachronous multifocal development of urothelial cancers by intraluminal seeding. Lancet 1993; 342: 1087–8. [DOI] [PubMed] [Google Scholar]
- 26. Habuchi T. Origin of multifocal carcinomas of the bladder and upper urinary tract: molecular analysis and clinical implications. Int J Urol 2005; 12: 709–16. [DOI] [PubMed] [Google Scholar]
- 27. Stoehr R, Knuechel R, Boecker J et al. Histologic–genetic mapping by allele‐specific PCR reveals intraurothelial spread of p53 mutant tumor clones. Lab Invest 2002; 82: 1553–61. [DOI] [PubMed] [Google Scholar]
- 28. Simon R, Eltze E, Schaefer KL et al. Cytogenetic analysis of multifocal bladder cancer supports a monoclonal origin and intraepithelial spread of tumor cells. Cancer Res 2001; 61: 355–62. [PubMed] [Google Scholar]
- 29. Elmula IF, Gorunova L, Mandahl N et al. Cytogenetic monoclonality in multifocal uroepithelial carcinomas: evidence of intraluminal tumor seeding. Br J Cancer 1999; 81: 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng L, Gu J, Ulbright T et al. Precise microdissection of human bladder carcinomas reveals divergent tumor subclones in the same tumor. Caner 2002; 94: 104–10. [DOI] [PubMed] [Google Scholar]
- 31. Paiss T, Wohr G, Hautmann RE et al. Some tumors of the bladder are polyclonal in origin. J Urol 2002; 167: 718–23. [DOI] [PubMed] [Google Scholar]
- 32. Ito N. Early changes caused by N‐buthyl‐N‐(4‐hydroxybuthyl) nitrosamine in the bladder epithelium of different animal species. Cancer Res 1976; 36: 2528–31. [PubMed] [Google Scholar]
- 33. Ohtani M, Kakizoe T, Sato S, Sugimura T, Fukushima S. Strain differences in mice with invasive bladder carcinomas induced by N‐butyl‐N‐(4‐hydroxybutyl) nitrosamine. J Cancer Res Clin Oncol 1986; 112: 107–10. [DOI] [PubMed] [Google Scholar]
- 34. Okajima E, Hiramatsu T, Motomiya Y et al. Urinary bladder tumors induced by N‐butyl‐N‐(4‐hydroxybutyl) nitrosamine in dogs. Cancer Res 1981; 41: 1958–65. [PubMed] [Google Scholar]
- 35. Kakizoe T, Tobisu K, Takai K, Tanaka Y, Kishi K, Teshima S. Relationship between papillary and nodular transitional cell carcinoma in the human urinary bladder. Cancer Res 1988; 48: 2293–303. [PubMed] [Google Scholar]
- 36. Spruck CH 3rd, Ohneseit PF, Gonzales‐Zulueta M et al. Two molecular pathways to transitional cell carcinomas of the bladder. Cancer Res 1994; 54: 784–8. [PubMed] [Google Scholar]
- 37. Woff EM, Liang G, Jones PA. Mechanisms of disease: genetic and epigenetic alterations that drive bladder cancer. Nature Clin Pract Urol 2005; 2: 502–10. [DOI] [PubMed] [Google Scholar]
- 38. Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nature Rev Cancer 2005; 5: 713–25. [DOI] [PubMed] [Google Scholar]
- 39. Cappellen D, De Oliveria C, Ricol D et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Naturel Genet 1999; 23: 18–20. [DOI] [PubMed] [Google Scholar]
- 40. Rieger‐Christ KM, Mourtzinos A, Lee PJ et al. Identification of fibroblast growth factor receptor 3 mutations in urine sediment DNA samples complements cytology in bladder tumor detection. Cancer 2003; 98: 737–44. [DOI] [PubMed] [Google Scholar]
- 41. Fujimoto K, Yamada Y, Okajima E et al. Frequent association of p53 gene mutation in invasive bladder cancer. Cancer Res 1992; 52: 1393–8. [PubMed] [Google Scholar]
- 42. Hartmann A, Schlake G, Zaak D et al. Occurrence of chromosome 9 and p53 alterations in multifocal dysplasia and carcinoma in situ of human urinary bladder. Cancer Res 2002; 62: 809–18. [PubMed] [Google Scholar]
- 43. Stein JP, Ginsberg DA, Grossfeld GD et al. Effect of p21WAF1/CIP1 expression on tumor progression in bladder cancer. J Natl Cancer Inst 1998; 90: 1072–9. [DOI] [PubMed] [Google Scholar]
- 44. Nakagawa T, Kanai Y, Saito Y, Kitamura T, Kakizoe T, Hirohashi S. Increased DNA methyltransferase 1 protein expression in human transitional cell carcinoma of the bladder. J Urol 2003; 170: 2463–6. [DOI] [PubMed] [Google Scholar]
- 45. Horikawa Y, Sugano K, Shigyo M et al. Hypermethylation of an E‐cadherin (CDH1) promoter region in high grade transitional cell carcinoma of the bladder comprising carcinoma in situ . J Urol 2003; 169: 1541–5. [DOI] [PubMed] [Google Scholar]
- 46. Kakizoe T, Fujita J, Murase T, Matsumoto K, Kishi K. Transitional cell carcinoma of the bladder in patients with renal pelvic and ureteral cancer. J Urol 1980; 124: 17–19. [DOI] [PubMed] [Google Scholar]
- 47. Akaza H, Kurth KH, Hinotsu S et al. Intravesical chemotherapy and immunotherapy for superficial tumors: basic mechanism of action and future direction. Urol Oncol 1998; 4: 121–9. [DOI] [PubMed] [Google Scholar]