

Abstract
Many studies on carcinogenesis carried out early in the last century are united on the consensus that cancer is a genetic disease. Cancer cells typically display gene dysfunction and endogenous or exogenous insults resulting in gene dysfunction are often carcinogenic. Recent advances in stem cell biology added the new concept that cancer originates from a single cancer‐initiating cell. To understand the molecular basis of carcinogenesis from the beginning to the full acquirement of malignancy, factors concerned with carcinogenesis were categorized into three groups: those guarding and stabilizing genomes, those regulating cell proliferation, and those conferring resistance to various micro‐environmental stresses. One example of particular interest is the Keap1‐Nrf2 system since, according to recent studies, it has turned out to be ambivalent. Nrf2 heterodimerizes with small Maf protein to strongly activate transcription through the Maf recognition element (MARE) and Keap1 is an inhibitory regulator of Nrf2. The genes regulated by Nrf2 are very important for cellular protection of the genome from xenobiotic and oxidative stresses and, consequently, for preventing carcinogenesis. This implies that enhancing Nrf2 activity is a promising method for thwarting cancer. On the contrary, the constitutive activation of Nrf2 due to mutations in the keap1 gene is characteristically observed in lung cancer cells, suggesting that induced expression of Nrf2 target genes favors the prevalence of cancer cells. (Cancer Sci 2007; 98: 135–139)
*Abbreviations used:
- APC
adenomatosis polyposis coli
- ARE
antioxidant responsive element
- ARK5
AMP‐activated protein kinase‐related protein kinase 5
- ATF
activating transcription factor
- ATM
gene, ataxia telangiectasia mutated gene
- bZip
basic region‐leucine zipper
- BTB
domain, Bric‐a‐brac, Tramtrack, Broad‐complex
- CCR1
chemokine receptor‐1
- CRE
cyclic AMP‐responsive element
- C‐MARE
CRE‐type MARE
- CNC
family, Cap ′n′ collar family
- CREB
CRE‐binding protein
- DGR
domain, double glycine repeat domain of Keap1
- EGFR
epidermal growth factor receptor
- EpRE
electrophile response elements
- HIF‐1
hypoxia‐inducible factor‐1
- Keap1
Kelch‐like ECH associating protein 1
- MARE
Maf recognition element
- MUTYH
mutY homolog
- NER
factors, nucleotide excision repair factors
- NF‐E2
nuclear factor‐erythroid 2
- Nrf1
NF‐E2‐related factor 1
- Nrf2
NF‐E2‐related factor 2
- Nrf3
NF‐E2‐related factor 3
- OGG1
8‐oxoguanine DNA glycosylase 1
- PTEN
phosphatase and tensin homolog
- ROS
reactive oxygen species
- Runx1
runt‐related transcription factor 1
- siRNA
small interfering RNA
- SNP
single nucleotide polymorphism
- TRE
TPA‐responsive element
- T‐MARE
TRE‐type MARE
- VHL
factor, Von Hippel‐Lindau factor.
Chemical carcinogenesis and the tumor virus are the two major streams of cancer research dating back to the early 20th century. In 1915, Yamagiwa showed that coal tar applied experimentally to rabbit ears caused skin carcinomas. This kicked off an era of fruitful research on chemical carcinogenesis and clearly demonstrated the presence of cancer‐inducing substances or carcinogens.( 1 ) In 1910, Rous discovered a transmissible avian tumor virus, which was followed by the identification of various oncogenes and the consequent elucidation of the mechanisms of normal cell growth and proliferation.( 2 ) One important message that emerged from latter studies is that cancer is a genetic disease, and so former studies revealed the necessity of genome safeguards against noxious substances to prevent carcinogenesis.( 3 ) Therefore, the multistep process of cancer development is defined as the gradual accumulation of gene dysfunction and the subsequent gradual increase of malignancy.
A new trend arising from stem cell biology has greatly advanced during the last decade and joined with cancer research to establish the concept that most cancers are derived from a single cancer stem cell, which is characterized by the ability to self‐renew and generate an aberrantly large number of cancer cells.( 4 , 5 ) Cancer stem cells, although displaying grave gene dysfunction, are considered to undergo processes that are analogous to the self‐renewal and differentiation of normal stem cells.
v‐Maf is an oncogene isolated from the avian retrovirus AS42, which causes musculoaponeurotic fibrosarcoma in chickens.( 6 ) The discovery of v‐maf led to the identification of its cellular counterpart c‐maf and related genes, which comprise the Maf family. The Maf family proteins, including c‐Maf, MafB, NRL, MafA, MafK, MafF and MafG, possess a unique stretch of amino acids just upstream of a typical basic region‐leucine zipper (bZip*) motif. The Maf proteins bind to the Maf recognition element (MARE) through the formation of homodimers.( 7 ) The complete consensus sequences of the MARE consist of a 13 bp palindromic T‐MARE (TGCTGAGTCAGCA) and a 14 bp C‐MARE (TGCTGAGCTCAGCA), containing a TPA‐responsive element (TRE: TGAGTCA) and a cyclic AMP‐responsive element (CRE: TGAGCTCA), respectively (Fig. 1, top).( 8 , 9 ) Thus, the Maf family proteins recognize unusually long binding sequences. The requirement of the three flanking base pairs on each side of the TRE or CRE core sequences (TGC and GCA) distinguishes this protein family from the other bZip transcription factors.
Figure 1.
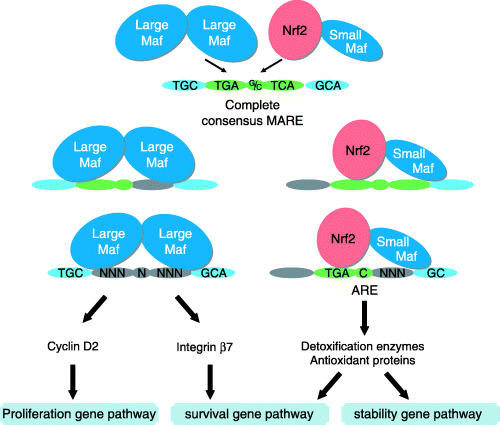
The distinct contribution of Maf‐containing dimers (Nrf2‐small Maf heterodimer and large Maf homodimer) to carcinogenesis through binding to the MARE and its related sequences. The Nrf2‐small Maf heterodimer activates transcription of detoxification enzyme genes and antioxidant responsive genes. Cyclin D2 and integrin β7 are candidate target genes of c‐Maf, one of the large Maf proteins. The Nrf2‐small Maf heterodimer tends to require a conserved core region (shown in green) of the MARE, while the large Maf homodimer prefers to bind to the site where a flanking region (shown in blue) is conserved.
By virtue of its containing a TRE or CRE sequence, the MARE is likely to be shared by Maf, Jun/Fos, and the CREB/ATF (CRE‐binding protein/activating transcription factor) family.( 8 , 9 ) In addition, heterodimers of small Maf and CNC (Cap′n′collar) family proteins bind to the MARE and its related sequences. MafK, MafG and MafF constitute the small Maf proteins. The four members of the bZip CNC family are NF‐E2 (nuclear factor‐erythroid 2) p45, Nrf1 (NF‐E2‐related factor 1), Nrf2 (NF‐E2‐related factor 2), and Nrf3 (NF‐E2‐related factor 3).( 7 ) Hence, an interesting feature of the MARE and its related sequences is the ability to interact with many kinds of homodimers and heterodimers consisting of various combinations of b‐Zip factors. In this review, we especially elaborate on the contribution of transcriptional regulation through the MARE to the development of cancer.
Three classes of genes involved in carcinogenesis
We surmise that genes involved in carcinogenesis can be classified into three groups: those maintaining genome integrity and stability (stability genes), those promoting or inhibiting cell proliferation (proliferation genes), and those conferring resistance against environmental stresses, such as exposure to oxidative stress, hypoxia and anticancer drugs, for survival (survival genes).
Stability genes are involved in the DNA repair process, such as ATM (ataxia telangiectasia mutated gene),( 10 ) MUTYH (mutY homolog),( 11 ) OGG1 (8‐oxoguanine DNA glycosylase 1),( 12 ) and NER (nucleotide excision repair) factors,( 13 ) and the maintenance of chromosomal stability. Thus, the products of stability genes act to prevent the accumulation of gene dysfunctions by inhibiting mutagenesis and chromosomal rearrangements. The malfunctioning of these genes accelerates the acquisition of malignant properties. Cancer prevention must therefore aim at increasing the activity of stability genes.
Proliferation genes include typical proto‐oncogenes and tumor suppressor genes, such as EGFR (epidermal growth factor receptor),( 14 ) Runx1 (runt‐related transcription factor 1),( 15 ) PTEN (phosphatase and tensin homolog),( 16 ) and APC (adenomatosis polyposis coli).( 17 ) They are principally involved in the regulation of cell proliferation through various pathways, and mutations in these genes found in cancer cells usually favor cell proliferation.( 3 ) The dysfunction of proliferation genes is responsible for the enhanced proliferation potential of cancer cells. Thus, proper regulation of proliferation genes is important for cancer prevention.
Survival genes are important for cancer cells to overcome various stresses, such as oxidative stress, hypoxia and the toxicity of anticancer drugs. For instance, VHL (Von Hippel‐Lindau factor), a repressor of the hypoxic response, is often mutated in clear cell renal cell carcinoma. Consequently, HIF‐1α (hypoxia‐inducible factor‐1α) and HIF‐2α, important transcriptional activators in hypoxic responses, is constitutively activated.( 18 ) Enhanced HIF‐1α/HIF‐2α activity promotes tumor angiogenesis, which is supportive of tumor growth. Antagonizing the activity of survival genes should therefore serve as an effective strategy for cancer therapy.
The Keap1‐Nrf2 system regulates detoxification and the oxidative stress response.
Upon exposure to xenobiotic or oxidative stresses, Nrf2 coordinately regulates the inducible expression of cytoprotective genes through cis‐acting antioxidant responsive elements (AREs) or electrophile response elements (EpREs), which are highly similar to the MARE (Fig. 1).( 19 , 20 , 21 , 22 ) Nrf2 and one of the small Maf proteins form a heterodimer that binds to these cis‐acting elements.( 23 , 24 ) The cytoplasmic protein Keap1 (Kelch‐like ECH associating protein 1) represses the activity of Nrf2 during unstressed conditions by mediating the ubiquitination and subsequent proteasomal degradation of Nrf2 (Fig. 2, upper panel).( 25 , 26 ) Upon exposure to electrophilic reagents or oxidative stresses, the ubiquitin ligase activity of Keap1 is inhibited and Nrf2 is consequently stabilized and translocated into the nucleus (Fig. 2, lower panel). When the mouse keap1 gene was disrupted, constitutive activation of Nrf2 and induction of Nrf2 target genes were observed, which proves that Keap1 plays an indispensable role as an inhibitory regulator of Nrf2 in vivo.( 27 ) More precise molecular mechanisms underlying the Keap1‐Nrf2 system have been described in detail elsewhere.( 28 , 29 )
Figure 2.
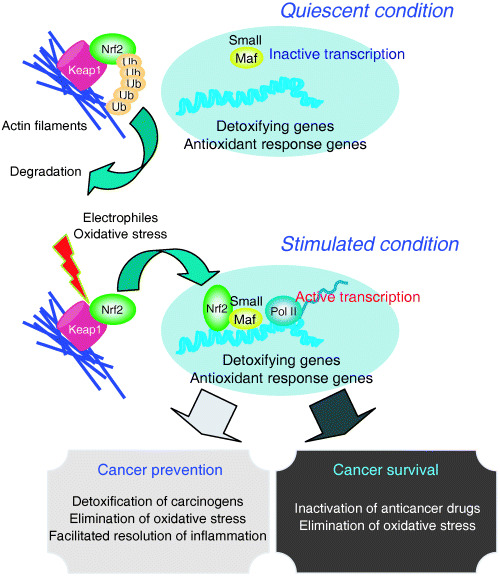
The Keap1‐Nrf2 system under quiescent and stimulated conditions. Stabilized Nrf2 is translocated into nuclei and activates transcription. The Nrf2 target genes encode critical effectors for cancer prevention, which at the same time are effective regulators of cancer cell survival.
Ambivalence of the Keap1‐Nrf2 regulatory pathway in carcinogenesis
The Keap1‐Nrf2 pathway has been shown to contribute to two different aspects of carcinogenesis. Nrf2‐deficient mice are susceptible to a variety of xenobiotic or oxidative insults, clearly indicating the critical contribution of detoxification enzymes and antioxidant proteins regulated by Nrf2 to cellular protection against such insults.( 30 , 31 ) Indeed, nrf2‐null mice had a significantly higher burden of stomach cancers after treatment with benzo[a]pyrene than wild‐type mice.( 32 ) Nrf2‐deficient mice were also susceptible to the urinary bladder‐specific carcinogen N‐nitrosobutyl(4‐hydroxybutyl)amine.( 33 ) In the absence of Nrf2, oltipraz (a member of dithiolethiones with antioxidative and cancer chemoprotective properties) was ineffective in both cases of carcinogen treatment, indicating the critical importance of Nrf2 activity in chemoprotection.( 32 , 33 ) ARE‐mediated gene expression in keratinocytes was shown to be important for protection against topically applied benzo[a]pyrene, especially at the early stage of carcinogenesis.( 34 ) A deficiency in Nrf2 exacerbates DNA adduct formation following exposure to diesel exhaust,( 35 ) thus Nrf2 functions as a stability gene to protect genome integrity by inducing enzymes that eliminate genotoxic substances (Fig. 2).
Not only exogenous carcinogens, but also endogenous substances under certain conditions, serve as important cancer‐inducing substances. Many researchers noticed that malignant cancers are frequently preceded by chronic inflammation, an observation dating back to Virchow's hypothesis proposed in the 19th century that cancers occur at the site of chronic inflammation.( 36 ) With plenty of our current knowledge on cancer biology, causal links between chronic inflammation and carcinogenesis have been intensively analyzed and discussed since the last decade. Inflammatory cells infiltrating the site of recurrent and persistent inflammation are believed to produce excessive amounts of reactive oxygen species (ROS) and nitrogen species. This induces DNA damage and releases an abundance of cytokines and growth factors that promote cell proliferation.( 37 ) An important input to the resolution of inflammation by Nrf2 has been suggested by the following observations. Carrageenan (one of the sulfated polysaccharides extracted from seaweeds)‐induced pleurisy was more persistent and resolution was delayed in the absence of Nrf2.( 38 ) Elastase‐provoked emphysema was also markedly exacerbated in nrf2‐null mice.( 39 ) Therefore, once inflammation is provoked, Nrf2 normalizes the environment and achieves early resolution such that the genome is protected from oxidative damage, aberrant cell proliferation is avoided and carcinogenesis is inhibited.
Based on the repressive effect of Keap1 on Nrf2 activity, the application of small interfering RNA (siRNA) against Keap1 was suggested to enhance a cellular cancer chemopreventive phenotype.( 40 ) Germ line mutation of the keap1 gene results in lethality around weaning due to obstructive lesions caused by hyperkeratotic outgrowth of the esophageal and forestomach epithelial cells.( 27 ) Therefore, conditional disruption of the keap1 gene was performed instead.( 41 ) Hepatocyte‐specific keap1 disruption causes constitutive elevation of detoxifying enzyme genes and confers resistance against acute drug toxicity. This implies that keap1 gene deletion is desirable for preventing carcinogenesis by promoting the elimination of carcinogens.( 41 )
However, the simple paradigm that enhanced Nrf2 activity prevents cancer, as summarized in Fig. 2, was challenged by a surprising result obtained from analysis of single nucleotide polymorphisms (SNPs) in human cancer cells. Two kinds of SNPs resulting in amino acid substitutions in Keap1 were identified in a small‐cell lung carcinoma cell line and in a case of lung cancer.( 42 ) The former was a homozygous mutation, while the latter was a somatic mutation found only in cancer cells. Both mutations affected the amino acid sequence of the Keap1 DGR (double glycine repeat or Kelch) domain responsible for interacting with Nrf2, such that the mutant Keap1 molecules did not effectively repress the activity of Nrf2 due to their reduced ability to bind to Nrf2. Consequently, Nrf2 activity was constitutively increased in the cancer cells. This result suggests that constitutive activation of Nrf2 due to Keap1 dysfunction assists in the survival of cancer cells in defiance of various adverse conditions, indicating that the Keap1‐Nrf2 pathway functions as a survival gene (Fig. 2). Many detoxification enzymes and some of the transporters involved in drug resistance are regulated by Nrf2.( 43 ) By way of these Keap1 mutations, the evolution of cancer cells might be reasonably promoted to acquire more malignant or drug‐resistant phenotypes. These observations suggest the importance of the stress response of a cancer cell, which seems to support the recently presumed analogy of the life of a cancer cell to that of a normal cell.( 4 , 5 ) A cancer cell proliferates and differentiates by conquering miscellaneous micro‐environmental stresses.
The distinct pathway to carcinogenesis originated from variations in Maf recognition elements.
The MARE is a consensus binding sequence of Maf family proteins whose original member c‐Maf was identified as a cellular counterpart of the oncoprotein v‐Maf. The striking similarity between the ARE and the MARE led us to identify an Nrf2‐small Maf heterodimer as a trans‐acting factor of the ARE (Fig. 1).( 7 ) Since the ARE sequence is involved in the canonical MARE sequence, the presence of cross talk between Nrf2‐small Maf heterodimers and various Maf homodimers was speculated. A surface plasmon resonance (SPR)‐micro‐array method, where the Maf family was represented by MafG, was used to analyze the binding affinities between various MARE‐related sequences and Nrf2‐small Maf heterodimers or Maf‐Maf homodimers. This comprehensive analysis demonstrated that the high‐affinity binding sequences of Nrf2‐small Maf heterodimers substantially overlap with those of the Maf homodimers.( 44 ) However, the specificities of the Nrf2‐small Maf heterodimers and Maf homodimers clearly differ from each other (Fig. 1).( 44 ) ARE sequences in the regulatory regions of endogenous Nrf2 target genes were found to be characteristic of a high‐affinity binding site for Nrf2‐small Maf heterodimers, whereas MARE sequences in the regulatory regions of endogenous Maf target genes possess the characteristics of a high‐affinity binding site for Maf homodimers. This implies that there is no significant cross talk between these two kinds of dimers as far as currently known target genes are concerned.
Consistent with these results, c‐Maf has been implicated to contribute to carcinogenesis through a different pathway than that used by Nrf2. An elevated expression of c‐Maf was observed in nearly 50% of multiple myelomas and 60% of angioimmunoblastic T‐cell lymphomas.( 45 , 46 ) Genes encoding cyclin D2, integrin β7, CCR1 (chemokine receptor‐1), and ARK5 (AMP‐activated protein kinase‐related protein kinase 5) were induced in these lymphoma cells and are good candidate target genes for c‐Maf. These reports suggest that c‐Maf directly enhances cell proliferation through activation of the cyclin D2 gene and facilitates cell adhesion to the bone marrow matrix by activating the integrin β7 gene, which probably aids the proliferation of lymphoma cells. Cyclin D2 has been categorized as a proliferation gene and integrin β7 could be classified as a survival gene. Therefore, c‐Maf contributes to carcinogenesis as an upstream regulator of a proliferation gene and a survival gene.
Whether these two types of dimers, that is, Nrf2‐small Maf heterodimers and Maf homodimers, actually share any target genes remains to be elucidated. A complete consensus sequence of the MARE, which should serve as a platform for any cross talk, is unexpectedly rare in endogenous regulatory regions, while suboptimal sequences with a few base substitutions are more frequently encountered. The suboptimal sequences often display lower affinities than the complete consensus MARE and show preferences for either type of dimer.( 44 ) In order to establish independence between the two systems regulated by each dimer, affinity might have given way to specificity during the evolution of cis‐regulatory elements.
Perspective
It has been widely accepted that increased Nrf2 activity is advantageous for cancer chemoprevention.( 47 ) Plant‐derived Nrf2 inducers, including sulforaphane in broccoli sprouts,( 48 ) 6‐methylsulfinylhexyl isothiocyanate in Japanese horseradish,( 49 ) and curcumin in turmeric powder,( 50 ) are expected to maintain good health when incorporated into the daily diet. The Keap1‐Nrf2 pathway is a good target of intervention for the prevention of cancer. Mutations in the keap1 gene abrogate its function in lung cancer cells, strongly suggesting that the Keap1‐Nrf2 pathway is also a good target for cancer therapy.
Recent structural analyses revealed that one Nrf2 protein binds to two molecules of Keap1 that have formed a homodimer through their BTB (Bric‐a‐brac, Tramtrack, Broad‐complex) domains.( 42 , 51 ) We and other groups showed that electrophilic reagents directly modify the cysteine residues of Keap1. Based on these results, one plausible model is that Nrf2 is held by a Keap1 dimer at two sites, one with high‐ and one with low‐binding affinity, and ubiquitinated under quiescent conditions. However, under stressed conditions, the low‐affinity binding site between Nrf2 and Keap1 is disrupted due to chemical modification of the Keap1 molecule and results in the stabilization of Nrf2. More precise mechanisms explaining the halt of Keap1‐mediated Nrf2 degradation upon exposure to electrophilic stress should provide critical information for the development of Nrf2 inducers and repressors.
Among the transcriptional regulations carried out through MARE‐related sequences, ARE‐mediated transcriptional activation is critical for our defense mechanism as well as for that of a cancer cell, whereas Maf homodimer‐specific activation mediates different arrays of gene induction. We recently found that the DNA recognition specificity of the Nrf2‐small Maf heterodimer is determined by a single amino acid in the basic region of Nrf2 (unpublished observation). When this single amino acid was swapped between Nrf2 and MafG, the DNA binding specificity of the Nrf2‐MafG heterodimer and that of the MafG homodimer were interchanged. Seemingly subtle variations in MARE‐related sequences found in the regulatory regions of various genes actually represent functionally distinct sequences, each of which requires a specific mode of DNA recognition. Thus, one intriguing question is whether a target gene common to the two types of dimers, if any, contributes to carcinogenesis, and if so, how. Further elucidation of the whole view on MARE‐dependent transcription and that of its related sequences will provide a leading prototype of transcriptional regulatory mechanisms mediated by dimers formed from multiple combinations of cognate transcription factors.
Acknowledgments
We thank Dr Takashi Tsuruo for encouraging us to work on this review and Dr Fumiki Katsuoka, Ms. Mariko Takayama and Momoko Kimura for intensive discussion and critical reading. This work was supported in part by grants from Japan Science and Technology Corporation‐ERATO Environmental Response Project and the Ministry of Education, Science, Sports and Culture.
References
- 1. Huff J. Long‐term chemical carcinogenesis bioassays predict human cancer hazards: issues, controversies, and uncertainties. Ann NY Acad Sci 1999; 895: 56 – 79. [DOI] [PubMed] [Google Scholar]
- 2. Heather L, Van E. Peyton Rous: father of the tumor virus. J Exp Med 2005; 201: 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vogelstein B, Kinzler KW. Cancer genes and the pathway they control. Nat Med 2004; 10: 789 – 99. [DOI] [PubMed] [Google Scholar]
- 4. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105 – 11. [DOI] [PubMed] [Google Scholar]
- 5. Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJA. The origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer 2005; 5: 899 – 904. [DOI] [PubMed] [Google Scholar]
- 6. Nishizawa M, Kataoka K, Goto N, Fujiwara KT, Kawai S. v‐maf, a viral oncogene that encodes a leucine zipper motif. Proc Natl Acad Sci USA 1989; 86: 7711 – 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2001; 294: 1 – 12. [DOI] [PubMed] [Google Scholar]
- 8. Kataoka K, Noda M, Nishizawa M. Maf nuclear oncoprotein recognizes sequences related to an AP‐1 site and forms heterodimer with both Fos and Jun. Mol Cell Biol 1994; 14: 700 – 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kerppola TK, Curran T. A conserved region adjacent to the basic domain is required for recognition of an extended DNA binding site by Maf/Nrl family proteins. Oncogene 1994; 9: 3149 – 58. [PubMed] [Google Scholar]
- 10. Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. ATM signaling and genomic stability in response to DNA damage. Mutat Res 2005; 569: 123 – 32. [DOI] [PubMed] [Google Scholar]
- 11. Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol Chem 2006; 387: 373 – 9. [DOI] [PubMed] [Google Scholar]
- 12. Arai T, Kelly VP, Minowa O, Noda T, Nishimura S. The study using wild‐type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8‐hydroxyguanine in kidney DNA. Toxicology 2006; 221: 179 – 86. [DOI] [PubMed] [Google Scholar]
- 13. De Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev 1999; 13: 768 – 85. [DOI] [PubMed] [Google Scholar]
- 14. Normanno N, De Luca A, Bianco C et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006; 366: 2 – 16. [DOI] [PubMed] [Google Scholar]
- 15. Yamagata T, Maki K, Mitani K. Runx1/AML1 in normal and abnormal hematopoiesis. Int J Hematol 2005; 82: 1 – 8. [DOI] [PubMed] [Google Scholar]
- 16. Lian Z, Di Cristofano A. Class reunion: PTEN joins the nuclear crew. Oncogene 2005; 24: 7394 – 400. [DOI] [PubMed] [Google Scholar]
- 17. Taketo MM. Mouse models of gastrointestinal tumors. Cancer Sci 2006; 97: 355 – 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maxwell PH. The HIF pathway in cancer. Semin Cell Dev Biol 2005; 16: 523 – 30. [DOI] [PubMed] [Google Scholar]
- 19. Itoh K, Chiba T, Takahashi S et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997; 236: 313 – 22. [DOI] [PubMed] [Google Scholar]
- 20. Ishii T, Itoh K, Takahashi S et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress‐inducible genes in macrophages. J Biol Chem 2000; 275: 16023 – 9. [DOI] [PubMed] [Google Scholar]
- 21. Rushmore TH, Morton MR, Pickett CB. Transcriptional regulation of the rat NAD (P) H. quinone reductase gene. J Biol Chem 1991; 266: 11632 – 9. 1646813 [Google Scholar]
- 22. Friling RS, Bensimon A, Tichauer Y, Daniel V. Xenobiotic‐inducible expression of murine glutathione S‐transferase Ya subunit gene is controlled by an electrophile‐responsive element. Proc Natl Acad Sci USA 1990; 87: 6258 – 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1‐Nrf2 regulatory pathway. Proc Natl Acad Sci USA 2004; 101: 6379 – 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element‐dependent genes. Mol Cell Biol 2005; 25: 8044 – 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Itoh K, Wakabayashi N, Katoh Y et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino‐terminal Neh2 domain. Genes Dev 1999; 13: 76 – 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi A, Kang MI, Okawa H et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3‐based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 2004; 24: 7130 – 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wakabayashi N, Itoh K, Wakabayashi J et al. Keap1‐null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 2003; 35: 238 – 45. [DOI] [PubMed] [Google Scholar]
- 28. Kobayashi M, Yamamoto M. Nrf2‐Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Advan Enzyme Regul in press. [DOI] [PubMed]
- 29. Tong KI, Kobayashi A, Yamamoto M. Two‐site substrate recognition model for the Keap1‐Nrf2 system: a hinge and a latch? Biol Chem in press. [DOI] [PubMed]
- 30. Motohashi H, Yamamoto M. Nrf2‐Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 2004; 10: 549 – 57. [DOI] [PubMed] [Google Scholar]
- 31. Dinkova‐Kostova AT, Holtzclaw WD, Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry 2005; 44: 6889 – 99. [DOI] [PubMed] [Google Scholar]
- 32. Ramos‐Gomez M, Kwak MK, Dolan PM et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor‐deficient mice. Proc Natl Acad Sci USA 2001; 98: 3410 – 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iida K, Itoh K, Kumagai Y et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 2004; 64: 6424 – 31. [DOI] [PubMed] [Google Scholar]
- 34. Auf Dem Keller U, Huber M, Beyer TA et al. Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol Cell Biol 2006; 26: 3773 – 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol 2001; 173: 154 – 60. [DOI] [PubMed] [Google Scholar]
- 36. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001; 357: 539 – 45. [DOI] [PubMed] [Google Scholar]
- 37. Schottenfeld D, Beebe‐Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin 2006; 56: 69 – 83. [DOI] [PubMed] [Google Scholar]
- 38. Itoh K, Mochizuki M, Ishii Y et al. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15‐deoxy‐Delta (12,14)‐prostaglandin j (2). Mol Cell Biol 2004: 24: 36 – 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ishii Y, Itoh K, Morishima Y et al. Transcription factor Nrf2 plays a pivotal role in protection against elastase‐induced pulmonary inflammation and emphysema. J Immunol 2005; 175: 6968 – 75. [DOI] [PubMed] [Google Scholar]
- 40. Devling TW, Lindsay CD, McLellan LI, McMahon M, Hayes JD. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc Natl Acad Sci USA 2005; 102: 7280 – 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. Hepatocyte‐specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun 2006; 339: 79 – 88. [DOI] [PubMed] [Google Scholar]
- 42. Padmanabhan B, Tong KI, Ohta T et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 2006; 21: 689 – 700. [DOI] [PubMed] [Google Scholar]
- 43. Maher JM, Cheng X, Slitt AL, Dieter MZ, Klaassen CD. Induction of the multidrug resistance‐associated protein family of transporters by chemical activators of receptor‐mediated pathways in mouse liver. Drug Metab Dispos 2005; 33: 956 – 62. [DOI] [PubMed] [Google Scholar]
- 44. Yamamoto T, Kyo M, Kamiya T et al. Predictive base substitution rules that determine the binding and transcriptional specificity of Maf recognition elements. Genes Cells 2006; 11: 575 – 91. [DOI] [PubMed] [Google Scholar]
- 45. Hurt EM, Wiestner A, Rosenwald A et al. Overexpression of c‐maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004; 5: 191 – 9. [DOI] [PubMed] [Google Scholar]
- 46. Morito N, Yoh K, Fujioka Y et al. Overexpression of c‐Maf contributes to T‐cell lymphoma in both mice and human. Cancer Res 2006; 66: 812 – 9. [DOI] [PubMed] [Google Scholar]
- 47. Kwak MK, Wakabayashi N, Kensler TW. Chemoprevention through the Keap1‐Nrf2 signaling pathway by phase 2 enzyme inducers. Mutat Res 2004; 555: 133 – 48. [DOI] [PubMed] [Google Scholar]
- 48. Fahey JW, Haristoy X, Dolan PM et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic‐resistant strains of Helicobacter pylori and prevents benzo[a]pyrene‐induced stomach tumors. Proc Natl Acad Sci USA 2002; 99: 7610 – 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morimitsu Y, Nakagawa Y, Hayashi K et al. A sulforaphane analogue that potently activates the Nrf2‐dependent detoxification pathway. J Biol Chem 2002; 277: 3456 – 63. [DOI] [PubMed] [Google Scholar]
- 50. Balogun E, Hoque M, Gong P et al. Curcumin activates the haem oxygenase‐1 gene via regulation of Nrf2 and the antioxidant‐responsive element. Biochem J 2003; 371: 887 – 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two‐site molecular recognition model. Mol Cell Biol 2006; 26: 2887 – 900. [DOI] [PMC free article] [PubMed] [Google Scholar]