

Abstract
Norcantharidin has been used as an efficacious anticancer drug in China for many years, but its true mechanism remains poorly understood. Intriguingly, in an in vitro series study of anticancer drugs, we found that norcantharidin can effectively inhibit epithelial tumor cells from expressing integrin αvβ6. Our previous studies have confirmed that integrin αvβ6 is closely relevant to malignant epithelial cell tumor biology behavior, and it can promote cancer cells to invade and metastasize through a special αvβ6–extracellular signal‐related kinase (ERK) direct signaling pathway. In this study, we investigated the relationship between the norcantharidin anticancer mechanism and integrin αvβ6. After HT‐29 colon cancer cells were treated with norcantharidin, cell apoptosis increased remarkably. The expression of αvβ6 and the amount of p‐ERK decreased substantially; simultaneously, the linkage between αvβ6 and ERK was barely detectable. However, the expression of other integrins and the levels of mitogen‐activated protein kinase hardly changed. On these grounds, we presumed that norcantharidin induced HT‐29 colon cancer cell apoptosis through the αvβ6–ERK signaling pathway. This finding elicited a novel strategy for targeting the whole αvβ6–ERK signal pathway, rather than simply blocking the combining site of αvβ6–ERK in colon cancer treatment. (Cancer Sci 2009; 100: 2302–2308)
Cantharidin is an efficacious antitumor drug extracted from blister beetles (Mylabris phalerata Pall). It has been used in China for over 2000 years. As cantharidin generates significant side effects, such as urinary system toxicity,( 1 ) it has been restrained in clinical treatment. Recently, congener drugs with less adverse reactions have been synthesized, such as N‐hydroxycantharidimide and sodium cantharidinate. A new kind of congener drug, norcantharidin (NCTD) is an ademethylated form of cantharidin that is synthesized using furane and maleic anhydride through additive reactions. With the removal of two methyl groups, symptoms of urinary system toxicity caused by cantharidin disappear, while its anticancer activity remains. Many papers have shown that NCTD can inhibit the proliferation of several tumor cell lines in vivo and in vitro,( 2 , 3 ) but with different mechanisms.
Our studies of integrin αvβ6 in the past confirmed that αvβ6 can enhance the invasion metastasis of tumor cells and inhibit apoptosis,( 4 , 5 , 6 , 7 , 8 ) and our group was the first to verify the existence of a special αvβ6–ERK direct signaling pathway in epithelial cancer cells through which integrin αvβ6 effects these crucial roles in tumor cells.( 9 , 10 , 11 , 12 ) In the present study, the human colon carcinoma HT‐29 cell line was used to determine the effects of NCTD on cell proliferation and apoptosis in vitro and to investigate whether the αvβ6 and αvβ6–ERK signaling pathway participates in NCTD anticancer mechanisms.
Materials and Methods
Cell line and culture conditions. The human colon cancer cell line HT‐29 was obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA) and maintained as monolayers in standard medium comprising Dulbecco’s modified Eagle’s medium (4.5 g/L of glucose) (Sigma, St. Louis, MO, USA) containing 10% heat‐inactivated fetal calf serum (FCS) (Sigma) and supplemented with 20 mM HEPES, 100 IU/mL penicillin, and 100 μg/mL streptomycin (Merck, Darmstadt, Germany). The cells were incubated at 37°C, 5% CO2, and saturated humidity. Cells in the exponential phase of growth at 2 × 105 cells/mL were exposed to various doses of NCTD for different time intervals. The negative control culture was left untreated.
Antibodies and reagents. The mouse‐antihuman monoclonal antibody L230 and R6G9 (IgG2a) respectively directed to the extracellular domain of the human integrin αv subunit and subunit β6, the function‐blocking antibody 10D5 (IgG2a) against αvβ6, P1F6 (IgG1) against αvβ5, and LM609 (IgG1) against αvβ3, were all obtained from Chemicon International (Temecula, CA, USA). Mouse immunoglobulins, IgG2a and IgG1 were acquired from Dako (Copenhagen, Denmark). Antibodies against ERK, phosphorylated ERK (p‐ERK), JNK, phosphorylated JNK (p‐JNK), p38, and phosphorylated p38 (p‐p38) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Purified gelatinases were prepared from medium conditioned by cultured human fibroblasts. Gelatin Sepharose 4B beads for affinity concentration of gelatinases from cell lysates were purchased from Amersham Pharmacia (Sydney, Australia). Reagents for SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and molecular weight markers were obtained from Bio‐Rad Laboratories (Hercules, CA, USA), and the Hoechst 33258 Cell Apoptosis Assay Kit was from Keygentec (Nanjing, China). NCTD of analytical grade purity was obtained from the Beijing Fourth Pharmaceutical Works, Beijing, China.
Cell proliferation assay. Cell viability was determined by measuring cellular metabolism using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. In brief, cells were plated at 5000 cells/well in 96‐well tissue culture plates. After treatment, MTT was added to each well at a final concentration of 5 mg/mL, and the cells were incubated for 4 h at 37°C. The medium was then removed, and the cells were dissolved with dimethyl sulfoxide (Sigma). Absorbance was measured at 570 nm (referenced to 650 nm) in a microplate reader (Bio‐Tech Instruments, Winooski, VT, USA).
Zymography. Gelatinase B (MMP‐9) and MMP‐3 were analyzed using SDS‐substrate gels by adding gelatin (0.1 mg/mL final concentration) to the 10% acrylamide separating gel. Tumor‐conditioned medium (TCM) collected under serum‐free conditions was mixed with substrate gel sample buffer (10% SDS, 50% glycerol, 25 mM Tris–HCl [pH 6.8], and 0.1% bromophenol blue), and 70 μL loaded onto the gel without prior boiling. Following electrophoresis, the gels were washed twice in 2.5% Triton X‐100 for 30 min at room temperature to remove the SDS. The gels were then incubated at 37°C overnight in substrate buffer containing 50 mM Tris–HCl and 5 mM CaCl2 (pH 8.0). The gels were stained with 0.15% Coomassie blue R250 in 50% methanol and 10% glacial acetic acid for 20 min at room temperature, and de‐stained in the same solution without Coomassie blue. Gelatin‐degrading enzymes were identified as clear bands against the blue background of stained gel.
MMP activity assay. MMP‐9 and MMP‐3 levels in TCM obtained from NCTD‐treated and ‐untreated HT‐29 cells were assayed using the Biotrak MMP‐9 and MMP‐3 activity assay system, respectively (Amersham, Aylesbury, UK). This assay measures total MMP levels (inactive pro‐enzyme activated artificially plus endogenous active enzyme forms), and MMP secretion is calculated on a per‐cell basis.
Detection of apoptosis by Hoechst 33258. After the cells had been treated with NCTD (60 μmol/L) for 12 h, the slides were washed twice with iced PBS and then fixed with 4% paraformaldehyde for 10 min at 4°C after being washed twice with PBS. Then, Hoechst 33258 fluorescent dye was added to the slides, and they were incubated for 10 min at room temperature. The slides were then washed twice with PBS and examined under a fluorescence microscope. Apoptotic features were assessed by observing chromatin condensation and fragments stained by Hoechst 33258. In each case, 10 random fields and more than 500 cells were counted.
FACScan analyses. Monolayer cultures of HT‐29 were treated with various doses of NCTD, harvested with 20 mM EDTA and then blocked with goat serum at 4°C for 10 min. The cells were washed once with PBS, incubated with primary antibody (R6G9, P1F6, or LM609) for 30 min at 4°C, and then washed twice with PBS. The cells were then stained with secondary antibody conjugated with phycoerythrin for 20 min at 4°C, washed twice with PBS, and re‐suspended in 0.5 ml PBS prior to FACScan analysis.
Western blotting. HT‐29 cells were treated with various doses of NCTD for 12, 24, 36, and 48 h. Both adherent and floating cells were collected and frozen at −80°C. To detect the levels of αv, β6, β5, β3, ERK, p‐ERK, JNK, p‐JNK, p38, and p‐p38, TCM from those cells was concentrated 50‐fold; the protein content was measured using the bicinchoninic acid (BCA) protein assay reagent and 10 μg was electrophoresed on a 12.5% SDS‐PAGE gel under non‐reducing conditions. Prior to loading the sample on the gel, the protein loads were equalized, and the electrophoresed proteins were transferred to nitrocellulose membranes. An equivalent protein loading for each lane was reconfirmed by staining the nitrocellulose membrane with Ponceau using the 36‐kDa GAPDH band present in the insulin selerons acid, and transferrin (ITS) supplement as a reference marker. Membranes were then probed with primary polyclonal antibody against αv, β6, β5, β3, ERK, p‐ERK, JNK, p‐JNK, p38, and p‐p38, followed by peroxidase‐labeled secondary antibodies. Western blots were visualized using the enhanced chemiluminescence detection system (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions.
Co‐immunoprecipitation. Lysates of equal cell numbers from NCTD‐treated group and control group cultures were incubated with the integrin αvβ6 monoclonal antibody R6G9 overnight at 4°C. Lysates were then centrifuged at 10 000 g for 10 min and pre‐cleared with rabbit antimouse (RAM) immunoglobulin coupled to Sepharose‐4B beads for 2 h. The immunoprecipitated proteins were washed three times with RIPA buffer and then analyzed by 7.5% SDS–PAGE under non‐reducing conditions. The mouse‐antihuman monoclonal R6G9, 6G11 (Santa Cruz Biotechnology) which recognizes both phosphorylated and non‐phosphorylated ERK2, and anti‐ERK mAb (12D4; Santa Cruz Biotechnology) against phosphorylated forms of ERK1/2, were used to detect the levels of β6, ERK2, and p‐ERK2 on nitrocellulose membranes, respectively.
Results
Inhibition of HT‐29 cell proliferation by NCTD treatment. The MTT assay showed that after treatment with NCTD (20, 40, 60, 80, 100, and 120 μmol/L) for 12, 24, 36, 48, 60, and 72 h, the number of viable cells markedly decreased in a dose‐ and time‐dependent manner (Fig. 1).
Figure 1.
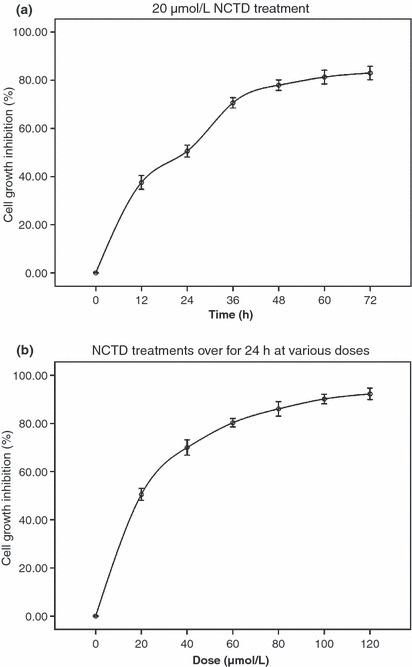
Dose‐ and time‐dependent inhibition of HT‐29 cell proliferation by norcantharidin (NCTD) treatment. (a) HT‐29 cells were treated with 20 μmol/L NCTD for various time periods; (b) HT‐29 cells were treated with various doses of NCTD for 24 h. The data are representative of three similar experiments.
NCTD suppresses HT‐29 cell secretion and activity of gelatinase B. In order to investigate whether NCTD interferes with the secretion and activity of gelatinase B, a zymography assay and MMPs activity were done. Figure 2a,b show that after HT‐29 cells were treated with 20, 40, and 60 μmol/L NCTD for 12 h, the secretion and activity of MMP‐9 and MMP‐3 decreased substantially in a dose‐dependent manner. In Figure 2c, three inhibition rate curves indicate that the MMP‐3 activity inhibition rate curve almost parallels with HT‐29 cell growth inhibition rate curve, but the MMP‐9 activity inhibition rate was obviously lower than MMP‐3 activity and the cell growth inhibition rate.
Figure 2.
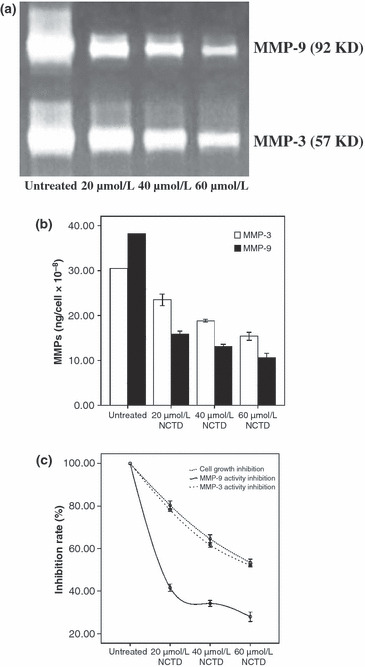
Suppression of gelatinase B secretion and activity in HT‐29 colon cancer cells treated with norcantharidin (NCTD). (a) Zymography assay showed the secretion of MMP‐9 and MMP‐3 without or with NCTD treatment; (b) MMP activity assay showed the activity of MMP‐9 and MMP‐3 without or with NCTD treatment; (c) inhibition rate curves respectively indicate the MMP‐9, MMP‐3 activity inhibition rate and HT‐29 cell growth inhibition rate.
NCTD induces HT‐29 cell apoptosis. We treated HT‐29 cells with 60 μmol/L NCTD for 12 h to determine whether it could induce apoptosis. The cells were examined using fluorescence microscopy and Hoechst 33258 staining. We found significant morphological changes in the cells’ nuclear chromatin (Fig. 3). In the control group, the nuclei were stained light, bright blue, and the color was homogeneous. In the NCTD‐treated group, the blue emission in the apoptotic cells was much brighter. The condensed chromatin could also be found in parts of the treated cells, some of them forming the structure of apoptotic bodies, a classic characteristic of apoptotic cells. The percentage of apoptotic cells in the counted fields in the control group was 1.45 ± 0.67%, while in the NCTD‐treated group it was 35.60 ± 1.84%. The apoptotic rate in the NCTD‐treated cells was significantly different from the control cells (P < 0.01).
Figure 3.

Certification of norcantharidin (NCTD)‐induced apoptosis in HT‐29 cells through detection by Hoechst 33258. (a) Control group HT‐29 cells not treated with NCTD. (b) HT‐29 cells treated with 60 μmol/L NCTD for 12 h. (c) The cell apoptosis rate between the control group and the NCTD group (* P < 0.01).
NCTD reduces the expression of αvβ6 in HT‐29 cells. FACScan data for αvβ6, αvβ5, and αvβ3 expression are shown in Figure 4a. The integrin αvβ6 was expressed at very high, high, moderate, or low levels with median intensity fluorescence values of 116, 87, 56, and 30, respectively, either without or with NCTD at a concentration of 20, 40, or 60 μmol/L for 12 h (P < 0.01). In contrast, in the control group the αvβ5 and αvβ3 expression did not substantially change (P > 0.05). Under the same conditions, Western blotting assay also confirmed this result. In Figure 4b, only the amount of β6 decreased accompanyed by the NCTD dose increase, but the amount of αv, β5, and β3 did not obviously change over time.
Figure 4.
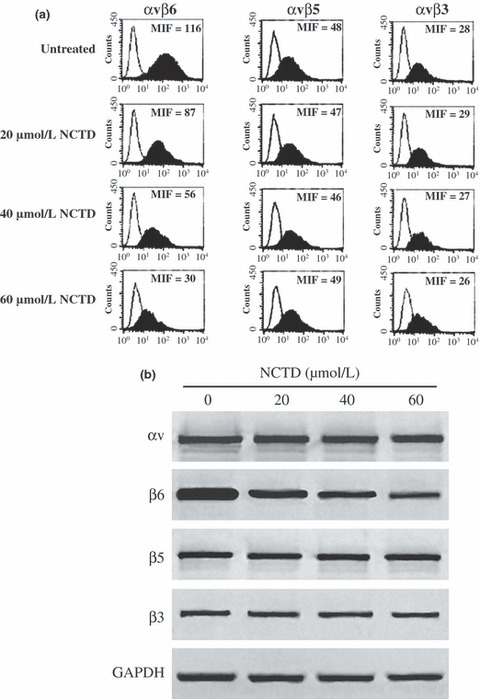
FACScan and Western blotting assay showed the suppression of β6‐subunit expression with norcantharidin (NCTD) addition in HT‐29 cells. (a) FACScan analyses of β‐integrin‐subunit expression in HT‐29 cells with or without NCTD treatment. (b) The Western blotting assay showed the amount of αv, β6, β5, and β3 in HT‐29 cells treated with or without NCTD. Data shown are representative of at least three similar experiments.
Effect of function‐blocking antibodies on NCTD‐treated HT‐29 cells. The function‐blocking antibodies against αvβ6, αvβ5, and αvβ3 were used to investigate their effect on NCTD‐treated HT‐29 cells. The IgG2a and IgG1 were used as negative controls. The cells were first treated for 2 h with the antibodies 10D5 (100 μg/mL), F1P6 (10 μg/mL), LM609 (10 μg/mL), IgG2a (100 μg/mL), and IgG1 (100 μg/mL). Next, the HT‐29 cells were incubated without or with 20 μmol/L NCTD for 12 h. The inhibition rate of HT‐29 cells is shown in Figure 5. The αvβ6‐function‐blocking antibody, 10D5, could inhibit HT‐29 cell growth (P * < 0.01 vs other sole antibodies), and this suppressing effect was obviously stronger when taken with 10D5 and NCTD together (P ** < 0.01 vs other antibodies + NCTD). The other antibodies, F1P6 and LM609, did not (P > 0.05).
Figure 5.
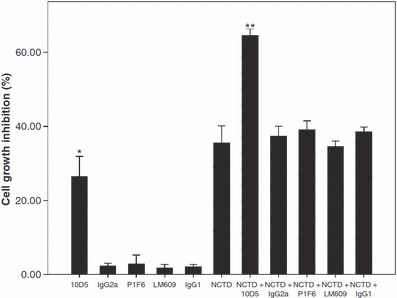
Cell inhibition assays were used to assess the effect of function‐blocking antibodies against αvβ6, αvβ5, and αvβ3 on HT‐29 cells treated with or without norcantharidin (NCTD). 10D5 alone could inhibit HT‐29 cell growth (P * < 0.01 vs other sole antibodies), and this suppressing effect was strengthened when combining 10D5 with NCTD (P ** < 0.01 vs other antibodies + NCTD).
MAPK expression in NCTD‐treated HT‐29 cells. In order to avoid the severe cytotoxicity produced by high doses of NCTD, we treated HT‐29 cells with low and moderate doses of NCTD (20, 40, and 60 μmol/L) for 24, 36, and 48 h to observe the changes in the MAPK levels. The amount of ERK, JNK, p‐JNK, p38, and p‐P38 did not change substantially over time. However, as the NCTD dose increased, the p‐ERK level decreased (Fig. 6).
Figure 6.
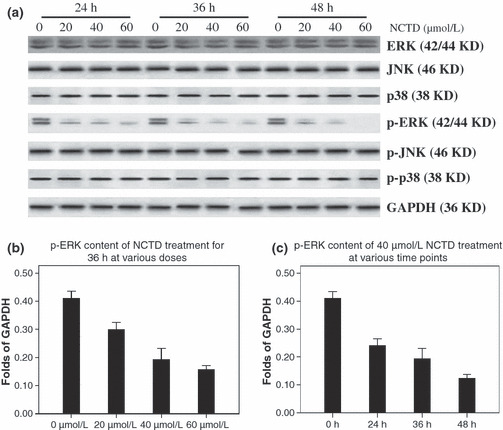
Norcantharidin (NCTD) substantially decreased the level of p‐ERK. (a) Western blotting for MAPK and p‐MAPK after various doses of NCTD (0, 20, 40, 60 μmol/L) on HT‐29 cells for 24, 36, and 48 h. (b,c) The level of p‐ERK decreased substantially with the addition of NCTD in a dose‐ and time‐dependent manner. Identical results were obtained in three independent experiments.
Effect of MAPK inhibitors on NCTD‐treated HT‐29 cells. In order to explore the exact role of MAPK in NCTD‐treated HT29 cells, the ERK inhibitor PD98059, JNK inhibitor SP600125, and p38 inhibitor SB203580 were used. (PD98059 is a direct inhibitor of MEK. As MEK was the kinase of ERK, PD98059 was used as an inhibitor of ERK in our study.) First, HT‐29 cells were treated with MAPK inhibitors PD98059 (20 μmol/L), SP600125 (40 μmol/L), and SB203580 (40 μmol/L) for 2 h. Next, the cells were incubated with 20 or 60 μmol/L NCTD for 12, 24, and 36 h. The ERK inhibitor PD98059 substantially increased the inhibition of NCTD at all time points (P < 0.01 vs the control), but the JNK inhibitor SP600125 and the p38 inhibitor SB203580 did not have an effect (P > 0.05 vs the control) (Fig. 7).
Figure 7.
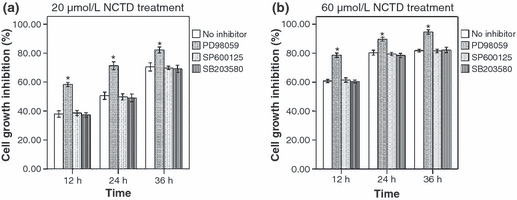
ERK inhibitor PD98059 reinforces the cell inhibition induced by norcantharidin (NCTD). (a,b) MTT analyses of HT‐29 cell‐proliferation inhibition when treated with various MAPK inhibitors and 20 or 60 μmol/L NCTD. The data from three separate experiments are expressed as mean ± SE.
NCTD interferes with ERK binding to αvβ6. We have already confirmed that the cytoplasmic domain of integrin αvβ6 only bound to ERK2 in cells, and this linkage was the basis of the configuration of the αvβ6–ERK signaling pathway.( 9 ) Only with an integrated configuration can the pathway transfer the growth‐enhancing signal to cancer cells. The co‐immunoprecipitation assay was taken to investigate whether NCTD could affect the association of αvβ6 and ERK2. After HT‐29 cells were treated with 40 μmol/L NCTD for 24 h, the association between αvβ6 and ERK2 (both phosphorylated‐ERK2 and non‐phosphorylated‐ERK2) was barely detectable (Fig. 8).
Figure 8.
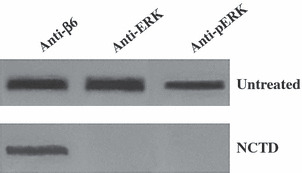
Norcantharidin (NCTD) interferes with αvβ6–ERK linkage formation. αvβ6 immunoprecipitants (mAb R6G9) from HT‐29 cell lysates treated or untreated with NCTD were immunoblotted with anti‐β6, anti‐ERK, and anti‐pERK monoclonal antibodies. (Upper) HT‐29 cells untreated with NCTD. (Lower) HT‐29 cells treated with 40 μmol/L NCTD for 24 h.
Discussion
Primary carcinoma of the colon is one of the most common digestive tract malignant tumors and is the second leading cause of cancer death in developed countries.( 13 ) The best curative therapy for colon cancer is surgical resection. Nevertheless, as its clinical symptoms are neither special nor typical, most patients with this disease are diagnosed at an advanced and unresectable stage. The 5‐year survival rate of colon cancer is still below 50%;( 14 ) worse still, the 5‐year survival rate in patients with metastatic colon cancer is less than 10%.( 15 ) Many types of alternative and palliative treatments, such as chemotherapy and radiotherapy, have come into use, including some Chinese traditional medicine. NCTD has been proven to be an ideal choice, and it can substantially prolong patients’ survival time with rare side effects.( 16 , 17 ) Unfortunately, the exact mechanism responsible for NCTD’s treatment of colon cancer has not been thoroughly explained.
Apoptosis is a crucial mechanism for carcinoma cells to prevent cancer cell invasion and metastasis, yet cancer cells frequently escape from apoptosis.( 18 ) Our previous study confirmed that αvβ6 plays an important role in inhibiting apoptosis in colon cancer cells.( 8 )
αvβ6 is a special subtype of integrin that is expressed in epithelial cells only. In normal epithelial cells, the expression of αvβ6 is rare and can hardly be detected,( 19 ) but it increases substantially in response to injury and/or inflammation, or in epithelial tumors.( 20 ) The de novo expression of αvβ6 integrin has been shown to modulate several processes in colon carcinoma cells, including cell adhesion and spreading on fibronectin, proliferation within collagen gels, tumor growth, cell apoptosis, and MMP secretion.( 4 , 5 ) Bates et al. ( 21 ) have suggested that the β6 integrin is a prognostic indicator of aggressive colon cancer, and αvβ6 would be a useful target at which to direct early therapy to prevent the spread of cancer. The same results were found by our previous studies in gastric carcinoma.( 22 )
MAPKs, including ERK, JNK, and p38, belong to a family of serine–threonine kinases integral to a number of major cell‐proliferation signal pathways.( 23 ) The ERK pathway plays a critical role in the modulation of cell proliferation, and several key growth factors and proto‐oncogenes transduce the signals that promote growth and differentiation through this kinase cascade.( 24 ) Aberrant activation of the ERK pathway is a feature of many human cancers.( 25 ) All the kinases participating in the ERK pathway offer opportunities as targets for cancer treatment, and a number of prospective therapeutics targeted to specific components of the ERK pathway have entered clinical trials recently.( 26 )
We have previously shown a direct linkage between ERK2 and the cytoplasmic domain of β6, and delineated the binding domain for ERK2 within the cytoplasmic tail of the β6‐integrin subunit.( 9 ) Through this physical interaction, integrin αvβ6 transmits a growth‐enhancing signal to cancer cells. In vivo studies of colon cancer xenografts expressing a β6‐mutant lacking the ERK2 binding domain indicate that deletion of the β6‐ERK2 binding motif greatly compromises tumor growth. Taken together, these results suggest that tumor growth is, at least in part, dependent on direct αvβ6–ERK binding.
Taking the above‐mentioned factors into account, we investigated the effect of NCTD treatment on apoptosis, integrin expression, and the phosphorylation of MAPKs. We found that NCTD inhibited HT‐29 cell growth in a time‐ and dose‐dependent manner. This result is compatible with other congener studies on various types of cancer cells( 27 , 28 , 29 ) that demonstrated NCTD is a protein phosphatase type 2A inhibitor.( 30 ) In zymography and MMP activity assay, we choose MMP‐3 as a negative control because both Haga( 31 ) and Nah( 32 ) indicated that the secretion of MMP‐3 may be regulated through the JNK signaling pathway. These assays showed that NCTD can substantially suppress the secretion and activity of gelatinase B. It is believed that these effects were mainly dependent on the suppression of living cell, rather than the loss of cell viability induced by NCTD, because the MMP‐9 activity inhibition rate was obviously lower than the MMP‐3 activity and cell growth inhibition rate, both due to the effect of NCTD toxicity. This result is also similar to congener research.( 2 ) Meanwhile, Hoechst 33258 staining confirmed that the apoptosis of NCTD‐treated HT‐29 cells increased substantially in contrast to the control group. Similar research results have also been shown for SAS (human oral cancer),( 33 ) A375‐S2 (human melanoma),( 34 ) and Hela (human cervical) cells.( 35 ) The FACScan and Western blotting experiment demonstrated that the expression of αvβ6 decreased with increasing doses of NCTD. As integrins are a kind of cell membrane surface adhesion molecule, their low expression will weaken the adhesion of cells to the extracellular matrix; thus increasing cell apoptosis. The decreased expression of αvβ6 definitely increased cell apoptosis as observed in our study. Combining the αvβ6 function‐blocking antibody 10D5 with NCTD could effectively inhibit HT‐29 cell proliferation. Western blotting indicated that only p‐ERK levels changed substantially when cells were treated with NCTD, and they were negatively correlated with the dose of NCTD. ERK, JNK, p‐JNK, p38, and p‐p38 levels did not change in the NCTD dose groups. Meanwhile, we also found that only the ERK inhibitor PD98059 could further promote cell apoptosis, while the other MAPK inhibitors cannot. These results are not consistent with another study,( 36 ) in which NCTD as a kind of protein phosphatase type 2A inhibitor could increase MAPK phosphorylation levels. We presume that this discrepancy may be due to the difference between the cell‐lines involved. In our present study, the HT‐29 colon cancer cell line is a kind of cell line which highly expresses integrin αvβ6, but other tumor cells lack integrin αvβ6 expression. As an inhibitor of PP2A, NCTD can restrain one or more types of MAPK dephosphorylation. Based on the above‐mentioned points, we think it is in the dominant status that NCTD can decrease αvβ6 expression and inhibit ERK phosphorylation in HT‐29 cells. Although we have acquired different experimental results, we believe that this is reasonable.
In light of the data and our previous results, we propose that NCTD primarily decreases the expression of αvβ6 and then interferes with the phosphorylation of ERK. Meanwhile, the expression of αvβ6 is too low to form the αvβ6–ERK direct linkage. As a result, the signal transmission is disturbed, and the carcinogenic effect of αvβ6 is blocked. Compared to our previously designed low‐molecular‐weight peptides that simply competitive bind the αvβ6–ERK interaction site (data not shown), NCTD can produce multiple inhibition effects in the αvβ6–ERK signaling pathway with a better anticancer effect. At the same time, NCTD only affects tumor cells with high selectivity. Meanwhile, we also admit that there may be another signaling pathway participating in the NCTD anticancer mechanism of inducing cell apoptosis, because some cancer cells do not express αvβ6; this needs further investigation. In addition, this study also confirms that drugs targeting integrin αvβ6 or the αvβ6–ERK direct signaling pathway are potential remedies for epithelial cell cancers expressing αvβ6.
Acknowledgments
This study was supported by two research grants from the National Natural Sciences Foundation of China (no. 30570833 and no. 30872460), one research grant from the Chinese Ministry of Education (no. 20060422048), and one research grant from the Shandong Provincial Natural Sciences Foundation (no. Y2005C42).
References
- 1. Zhang L, Sun X, Zhang ZR. An investigation on liver: targeting microemulsions of norcantharidin. Drug Deliv 2005; 12: 289–295. [DOI] [PubMed] [Google Scholar]
- 2. Fan YZ, Fu JY, Zhao ZM, Chen CQ. Influence of norcantharidin on proliferation, proliferation‐related gene proteins proliferating cell nuclear antigen and Ki‐67 of human gallbladder carcinoma GBC‐SD cells. Hepatobiliary Pancreat Dis Int 2004; 3: 603–607. [PubMed] [Google Scholar]
- 3. An WW, Wang MW, Tashiro S, Onodera S, Ikejima T. Mitogen‐activated protein kinase‐dependent apoptosis in norcan‐tharidin‐treated A375‐S2 cells is proceeded by the activation of protein kinase C. Chin Med J 2005; 118: 198–203. [PubMed] [Google Scholar]
- 4. Niu J, Gu X, Turton J, Meldrum C, Howard EW, Agrez M. Integrin‐mediated signalling of gelatinase B secretion in colon cancer cells. Biochem Biophys Res Commun 1998; 249: 287–291. [DOI] [PubMed] [Google Scholar]
- 5. Agrez M, Gu XH, Turton J. The alpha v beta 6 integrin induces gelatinase B secretion in colon cancer cells. Int J Cancer 1999; 81: 90–97. [DOI] [PubMed] [Google Scholar]
- 6. Niu J, Gu X, Ahmed N et al. The alphaVbeta6 integrin regulates its own expression with cell crowding: implications for tumour progression. Int J Cancer 2001; 92: 40–48. [DOI] [PubMed] [Google Scholar]
- 7. Yang GY, Xu KS, Pan ZQ et al. Integrin alpha v beta 6 mediates the potential for colon cancer cells to colonize in and metastasize to the liver. Cancer Sci 2008; 99: 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao‐Yang Z, Ke‐Sen X, Qing‐Si H et al. Signaling and regulatory mechanisms of integrin alphavbeta6 on the apoptosis of colon cancer cells. Cancer Lett 2008; 266: 209–215. [DOI] [PubMed] [Google Scholar]
- 9. Ahmed N, Niu J, Dorahy DJ et al. Direct integrin αvβ6‐ERK binding: implications for tumor growth. Oncogene 2002; 21: 1370–1380. [DOI] [PubMed] [Google Scholar]
- 10. Niu J, Dorahy DJ, Gu X et al. Integrin expression in colon cancer cells is regulated by the cytoplasmic domain of the beta6 integrin subunit. Int J Cancer 2002; 99: 529–537. [DOI] [PubMed] [Google Scholar]
- 11. Gu X, Niu J, Dorahy DJ, Scott R, Agrez MV. Integrin alpha (v) beta6‐associated ERK2 mediates MMP‐9 secretion in colon cancer cells. Br J Cancer 2002; 87: 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J, Zhang Z, Xu K et al. Suppression of integrin alphaupsilonbeta6 by RNA interference in colon cancer cells inhibits extracellular matrix degradation through the MAPK pathway. Int J Cancer 2008; 123: 1311–7. [DOI] [PubMed] [Google Scholar]
- 13. De Gramont A, Figer A, Seymour M et al. Leucovorin and fluorouracil with or without oxaliplatin as first‐line treatment in advanced colorectal cancer. J Clin Oncol 2000; 18: 2938–2947. [DOI] [PubMed] [Google Scholar]
- 14. Rayter Z, Leicester RJ, Mansi JL. Adjuvant chemotherapy for colorectal cancer. Ann R Coll Surg Engl 1995; 77: 81–84. [PMC free article] [PubMed] [Google Scholar]
- 15. Jemal A, Murray T, Ward E. Cancer statistics, 2005. CA Cancer J Clin 2005; 55: 10–30. [DOI] [PubMed] [Google Scholar]
- 16. Wang GS. Medical uses of mylabris in ancient China and recent studies. J Ethnopharmacol 1989; 26: 147–162. [DOI] [PubMed] [Google Scholar]
- 17. Fang Y, Tian SL, Li KO, Zhao SW, Wang ZY. Studies on antitumor agents II: synthesis and anticancer activity of dehydrogenated carbon cyclic analogs of norcantharidin. Acta Pharmacol Sin 1993; 28: 931–935. [PubMed] [Google Scholar]
- 18. Meredith JE Jr, Schwartz M. Integrins, adhesion and apoptosis. Trends Cell Biol 1997; 7: 146–150. [DOI] [PubMed] [Google Scholar]
- 19. Breuss JM, Gillett N, Lu L, Sheppard D, Pytela R. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissue. J Histochem Cytochem 1993; 41: 1521–1527. [DOI] [PubMed] [Google Scholar]
- 20. Breuss JM, Gallo J, DeLisser HM et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci 1995; 108: 2241–2251. [DOI] [PubMed] [Google Scholar]
- 21. Bates RC, Bellovin DI, Brown C et al. Transcriptional activation of integrin beta6 during the epithelial‐mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest 2005; 115: 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang ZY, Xu KS, Wang JS et al. Integrin alphanvbeta6 acts as a prognostic indicator in gastric carcinoma. Clin Ongol 2008; 20: 61–66. [DOI] [PubMed] [Google Scholar]
- 23. Dong C, Davis RJ, Flavell RA. Map kinases in the immune response. Annu Rev Immunol 2002; 20: 55–72. [DOI] [PubMed] [Google Scholar]
- 24. Troppmair J, Bruder JT, Munoz H et al. Mitogen‐activated protein kinase extracellular signal‐regulated protein kinase activation by oncogenes, serum, and 12‐O‐tetradecanoylphorbol‐13‐acetate requires Raf and is necessary for transformation. J Biol Chem 1994; 269: 7030–7035. [PubMed] [Google Scholar]
- 25. Hoshino R, Chatani Y, Yamori T et al. Constitutive activation of the 41‐/43‐kDa mitogen‐activated protein kinase signaling pathway in human tumors. Oncogene 1999; 18: 813–822. [DOI] [PubMed] [Google Scholar]
- 26. Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Ann Med 2006; 38: 200–211. [DOI] [PubMed] [Google Scholar]
- 27. Peng F, Wei YQ, Tian L et al. Induction of apoptosis by norcantharidin in human colorectal carcinoma cell lines: involvement of the CD95 receptor/ligand. J Cancer Res Clin Oncol 2002; 128: 223–230. [DOI] [PubMed] [Google Scholar]
- 28. YZ Fan, JY FU, ZM Zhao. Effect of norcantharidin on proliferation and invasion of human gallbladder carcinoma GBC‐SD cells. World J Gastroenterol 2005; 11: 2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen YN, Cheng CC, Chen JC, Tsauer W, Hsu SL. Norcantharidin‐induced apoptosis is via the extracellular signal‐regulated kinase and c‐Jun‐NH2‐terminal kinase signaling pathways in human hepatoma HepG2 cells. Br J Pharmacol 2003; 140: 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hart ME, Chamberlin AR, Walkom C, Sakoff JA, McCluskey A. Modified norcantharidins; synthesis, protein phosphatases 1 and 2A inhibition, and anticancer activity. Bioorg Med Chem Lett 2004; 14: 1969–1973. [DOI] [PubMed] [Google Scholar]
- 31. Haga A, Nagai H, Deyashiki Y. Autotaxin promotes the expression of matrix metalloproteinase‐3 via activation of the MAPK cascade in human fibrosarcoma HT‐1080 cells. Cancer Invest 2009; 27: 384–90. [DOI] [PubMed] [Google Scholar]
- 32. Nah SS, Choi IY, Yoo B, Kim YG, Moon HB, Lee CK. Advanced glycation end products increases matrix metalloproteinase‐1,‐3, and ‐13, and TNF‐a in human osteoarthritic chondrocytes. FEBS Lett 2007; 581: 1928–32. [DOI] [PubMed] [Google Scholar]
- 33. Kok SH, Cheng SJ, Hong CY et al. Norcantharidin‐induced apoptosis in oral cancer cells is associated with an increase of proapoptotic to antiapoptotic protein ratio. Cancer Lett 2005; 217: 43–52. [DOI] [PubMed] [Google Scholar]
- 34. An WW, Wang MW, Tashiro S, Onodera S, Ikejima T. Norcantharidin induces human melanoma A375‐S2 cell apoptosis through mitochondrial and caspase pathways. J Korean Med Sci 2004; 19: 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. An WW, Gong XF, Wang MW, Tashiro S, Onodera S, Ikejima T. Norcantharidin induces apoptosis in HeLa cells through caspase, MAPK, and mitochondrial pathways. Acta Pharmacol Sin 2004; 25: 1502–1508. [PubMed] [Google Scholar]
- 36. Chen YJ, Kuo CD, Tsai YM, Yu CC, Wang GS, Liao HF. Norcantharidin induces anoikis through Jun‐N‐terminal kinase activation in CT26 colorectal cancer cells. Anticancer Drugs 2008; 19: 55–64. [DOI] [PubMed] [Google Scholar]