

Abstract
Hepatocellular carcinoma (HCC) is widely known to develop more frequently in cirrhotic patients with a high expression of Hepatitis B virus X protein (HBx), which is controlled by the enhancer 1 (Enh1)/X‐promoter. To examine the effect of the mutations in the Enh1/X‐promoter region in hepatitis B virus (HBV) genomes on the development of HCC, we investigated the differences in HBV isolated from cirrhotic patients with or without HCC along with the promoter activities of certain specific mutations within the Enh1/X‐promoter. We examined 160 hepatitis B surface antigen (HBsAg)‐positive cirrhotic patients (80 HCC patients, 80 non‐HCC patients) by evaluating the biochemical, virological, and molecular characteristics. We evaluated the functional differences in certain specific mutations within the Enh1/X‐promoter. The isolated sequences included all of the subgenotypes C2. The sites that showed higher mutation rates in the HCC group were G1053A and G1229A, which were found to be independent risk factors through multiple logistic analysis (P < 0.05). Their promoter activities were elevated 2.38‐ and 4.68‐fold, respectively, over that of the wild type in the HepG2 cells. Similarly, both the mRNA and protein levels of HBx in these two mutants were much higher than that in wild type‐transfected HepG2 cells. Mutated nucleotides of the Enh1/X‐promoter, especially G1053A and G1229A mutations in the HBV subgenotype C2 of patients with cirrhosis, can be risk factors for hepatocarcinogenesis, and this might be due to an increase in the HBx levels through the transactivation of the Enh1/X‐promoter. (Cancer Sci 2010)
The hepatitis B virus (HBV) genome is a 3.2‐kb circular, partially double‐stranded molecule with four overlapping open reading frames (ORF; P, S, C, and X). Hepatitis B virus (HBV) produces several viral proteins that are influenced by promoters and enhancers located upstream.( 1 ) Hepatic injury due to HBV is mediated by immune‐related mechanisms; these can induce chronic liver disease (CLD) and eventually lead to hepatocellular carcinoma (HCC).( 2 , 3 , 4 ) However, the exact mechanism underlying hepatocarcinogenesis in chronic HBV infection remains elusive.
The development of HCC by HBV is enhanced by several risk factors such as cirrhosis, carcinogen exposure, alcohol abuse, genetic factors, a higher viral load, viral genotype, male gender, and advanced age.( 5 , 6 , 7 , 8 ) Among them, cirrhosis is the strongest risk factor.( 9 , 10 , 11 ) Therefore, the ability of HBV to induce inflammation that affects the host immune response might be a primary contributing factor for hepatocarcinogenesis.( 5 , 7 , 12 )
Additional explanations of hepatocarcinogenesis include the genomic integration of HBV in the host( 13 , 14 ) and the multiple regulatory activities caused by viral proteins.( 15 , 16 ) Among them, Hepatitis B virus X protein (HBx) has been most commonly implicated in hepatocarcinogenesis, as HCC does not occur in avian hosts that lack X‐ORF.( 17 ) Hepatocellular carcinoma (HCC) development occurs more frequently in humans with certain viral genotypes; in transgenic mice, it depends upon the inserted HBV‐DNA genotypes and portions of viral DNA.( 18 , 19 ) Hepatocellular carcinoma (HCC) also develops more frequently in transgenic mice with higher levels of HBx expression. Hepatocellular carcinoma (HCC) patients have higher levels of HBx expression than cirrhotic patients.( 20 , 21 ) Furthermore, specific nucleotide (nt.) mutations that are located in functional areas develop HCC more frequently.( 22 , 23 ) The locations of these specific nucleotide mutations correlated with HCC occurrence differ for each genotype. It is possible for nucleotide mutations to occur at the same site, but their functions differ for each genotype.( 24 ) These results suggest that the viral genotype, HBx expression levels, and underlying conditions of the host are important for hepatocarcinogenesis. Thus, further study is required to elucidate the correlation between hepatocarcinogenesis by HBV and the expression of HBx in similar conditions.
HBx expression is regulated by Enhancer 1 located just upstream of the X‐gene. In addition to regulating HBx expression through modulation of the X‐promoter, Enhancer 1 also regulates all the other viral promoters.( 25 , 26 ) Enhancer 1 consists of three domains (modulator element, core domain, and 3′ end overlaps with the x‐promoter) and eight known functional sites.( 27 ) Of these, three functional sites (Retinoid X receptor α, hepatocyte nuclear factor 3 and 4) in the central core domain are crucial to the functioning of Enhancer 1.( 27 , 28 ) Nuclear factor 1 in the 3′ end overlaps with the X‐promoter and works as an essential X‐promoter element.( 29 ) In addition, the p53‐like binding sequence and the androgen response element in the modulator element have been reported to play a role in HBV replication and the development of HCC.( 30 , 31 , 32 )
In this study, nucleotide mutations in the enhancer 1(Enh1)/X‐promoter region that influences HBx expression in cirrhosis patients with or without HCC was investigated. We also examined the Enh1/X‐promoter activities of specific mutations within the Enh1/X‐promoter, which might alter due to these specific mutations.
Materials and Methods
Patients and clinical samples. We explained the protocol of our study and enrolled the subjects who accepted our protocol and voluntarily provided written informed consent. We chose 172 naïve chronic hepatitis B surface antigen (HBsAg) carriers who showed progression of their liver disease (liver cirrhosis [LC] with HCC, 80; LC without HCC, 92) between January 1999 and August 2006. Subjects were selected with matching factors of selection criteria known to be largely associated with HCC development such as age, sex, HBV‐DNA value, and LC severity (platelet count, Child‐Turcotte‐Pugh score). Liver cirrhosis (LC) without HCC patients were followed up for 2 years after recruitment in this study. Further, we checked the ultrasonography or abdominal computed tomography (CT) and alfa‐fetoprotein (AFP) levels at an interval of 3–6 months during the follow‐up period for surveillance of developing HCC. Of these 92 LC without HCC patients, 12 subjects developed HCC during the follow‐up period. Therefore, only the remaining 80 LC without HCC patients were recruited.
Inclusion criteria for subjects in this study were as follows: (i) seropositivity for HBsAg for 6 months or longer; (ii) regular follow‐ups for at least 36 months; (iii) no special antiviral treatment administered before being admitted to our hospital and during follow up; and (iv) cases where direct sequencing was possible after HBV‐PCR.
This study excluded (i) patients who developed HCC (detected by ultrasound, computerized tomography, or AFP performed every 3–6 months) during the follow‐up period of 2 years after recruitment; and (ii) patients who had other viral infections including hepatitis viruses A, C, D, or E; consumed alcohol in excess of 40 gm/day for 1 year; and/or had other debilitating systemic diseases.
Liver cirrhosis (LC) was diagnosed by histology or imaging studies (CT or ultrasonography) accompanied by biochemical and endoscopic findings (esophageal varix, congestive gastropathy, etc.) and clinical features of portal hypertension such as thrombocytopenia, high‐albumin gradient ascites, and hepatic encephalopathy.
Hepatocellular carcinoma (HCC) was diagnosed by histology or a combination of ultrasonography, CT, or magnetic resonance imaging and/or hepatic angiography as well as AFP levels >200 ng/mL, according to the guidelines for the diagnosis of HCC from the European Association for the Study of the Liver (EASL).
Serum samples from each subject were maintained at −80°C until use. This study protocol was approved by the ethics committees of the concerned institutions.
Laboratory assays. The chemical laboratory tests for transaminase were performed according to recommendations protocol from the International Federation of Clinical Chemistry. Serological testing for HBV was assayed with commercial kits (HBsAg, anti‐HBc, anti‐HBc IgM, hepatitis B e antigen (HBeAg), and anti‐HBe; Abbott, Chicago, IL, USA). The viral load of HBV was assayed by RealArt‐PCR (RealArt HBV LC PCR Reagents; Artus, Hamburg, Germany). However, before February 2003, the viral load of HBV was measured with a hybridization assay (Digene, Gaithersburg, MD, USA); therefore, these patients were reevaluated by performing RealArt‐PCR on their frozen sera, which were stored at −20°C. We used the modified Child–Pugh index (CPI) score as a measure of the severity of LC, as based on the clinical evidence and laboratory data.
Amplification, sequencing, genotyping, and subgenotyping of HBV‐DNA. For sequencing, HBV‐DNA was extracted from serum using a commercial kit (QIAmp DNA Blood Mini Kit; Qiagen, Alameda, CA, USA) according to the manufacturer’s instructions. The positions and sequences of the PCR primers are given in Table 1. To sequence the PCR products, agarose gel electrophoresis was performed, and the band corresponding to the expected size was excised from the gel. The products were purified using a commercial kit (QIAquick; Qiagen) to remove the residual primers and dNTPs. The purified PCR products were directly sequenced by the dideoxy method using the BigDye Terminator Cycle Sequencing Ready Reaction Kit V2 and analyzed on an ABI Prism 377 analyzer (Applied Biosystems, Foster City, CA, USA). The nucleotide sequences of the Enh1/X‐promoter region were aligned using DNASTAR SeqMan software. These sequences were used for genotyping the HBV genome as previously described.( 33 ) The 160 isolates from our subjects were compared with the isolates of HBV genotype C (24 strains) and non‐genotype C HBV (52 stains) retrieved from the NCBI Genebank (http://www.ncbi.nlm.nih.gov/Genbank). The HBV genomic sequences were multiple‐aligned using ClustalW software. Genetic distances were estimated by Kimura’s two‐parameter method, and phylogenetic trees were constructed by the neighbor‐joining method.( 33 , 34 )
Table 1.
Sequences and positions of primers used for amplification and sequencing of the Enh1/X‐promoter regions, cloning, and measurement of X‐gene mRNA
| Position | Nucleotide sequence (5′–3′) | Polarity |
|---|---|---|
| Primer for PCR and sequencing | ||
| For 1st round of PCR | ||
| 953→968 | AAC TKC CTG TAA AYC AG | Sense |
| 1433→1416 | GGG ACG TAA RAC AAA GGA C | Antisense |
| For 2nd round of PCR | ||
| 970→988 | CCT ATT GAT TGG AAA GTW TG | Sense |
| 1430→1413 | ACG TAR ACA AAG GAC GTC | Antisense |
| Primer for cloning (pGL3 basic) | ||
| 950→966 | CTA GCT AGC GGA AAC TGC CTG TAA AT (including the restriction enzyme NheI site) | Sense |
| 1373→1354 | GGT GCA AGC TTG GGA AGG AGG TGT ATT TCC G (including the restriction enzyme HindIII site) | Antisense |
| Primer for mRNA of X‐gene | ||
| 1374→1400 | ATG GCT GCT CGG GTG TGC TGC CAA CTG | Sense |
| 1602→1579 | GTG CAG AGG TGA AGC GAA GTG CAC | Antisense |
Nucleotide variation was defined according to the difference in consensus sequences within the same subgenotype. A hotspot was defined as any nucleotide with more than 15% variability for the corresponding sequence.
Constructions and site‐directed mutagenesis. We selected a sequence that corresponded to the consensus sequence (wild type, pGL3–WT) and then inserted the Enh1/X‐promoter regions (nt. 950–1373, genotype C2) into the multicloning sites of a pGL3 basic vector (Promega, Madison, WI, USA). Among the multicloning sites, NheI and HindIII were selected as sites for cleavage and ligation, respectively. The details of the primers used are given in Table 1. Using the wild‐type constructs (pGL3–WT), three types of mutant constructs were generated: the first carried a point mutation at nt. 1053 (pGL3–G1053A, G→A at nt. 1053; G1053A), the second carried mutations at nt. 1229 (pGL3–G1229A, G1229A), and the third carried mutations at both sites (nt. 1053 and nt. 1229; pGL3–G1053/G1229A, G1053A combined with G1229A). The three types of mutant constructs (pGL3–G1053A, pGL3–G1229A, pGL3–G1053A/G1229A) were confirmed by direct sequencing.
In order to confirm the synthesis of HBx mRNA and the X‐protein concentration, it was necessary to construct another vector that included the X‐gene sequence. The pHBV1.2X construct was a kind gift from Professor Jung (Seoul National University).( 35 ) Three mutant constructs were generated from pHBV1.2X using the Quick Change Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s instructions. The first carried a point mutation at nt. 1053 (pHBV1.2X–G1053A, G1053A), the second carried mutations at nt. 1229 (pHBV1.2X–G1229A, G1229A), and the third carried mutations at both sites (nt. 1053 and nt. 1229; pHBV1.2X–G1053A/G1229A, G1053A combined with G1229A). The three mutant constructs (pHBV1.2X–G1053A, pHBV1.2X–G1229A, and pHBV1.2X–G1053A/G1229A) were confirmed by direct sequencing.
Cells, transfection, and luciferase assay. We selected wild‐type p53‐positive HepG2 cells (American Type Culture Collection, Manassas, VA, USA) for functional assay. HepG2 cells were cultured in DMEM (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum. Approximately 2 × 105 cells were plated on six‐well plates 24 h before transfection. The cells were transfected with 2 μg of the pGL3–HBV constructs and 0.5 μg of pcDNA1‐galactosidase (Stratagene) by using the FuGENE 6 transfection reagent (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s instructions. After 48 h of culture, the total cell extract was obtained by lysis of the cells. Luciferase activity was measured with a luciferase assay system (Promega) according to the manufacturer’s instructions. Luciferase activity was measured in triplicate, averaged, and then normalized with β‐galactosidase activity using the galactosidase assay system (Galacto‐Light; Tropix, Bedford, MA, USA) according to the manufacturer’s instructions.
Reverse transcriptase‐PCR (RT‐PCT) and quantitative real‐time PCR amplification. The total RNA was extracted with the use of TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The single‐stranded cDNA was synthesized from the total RNA. Polymerase chain reaction (PCR) was performed with Taq DNA polymerase (Takara Shuzo, Shiga, Japan) for 30 cycles and with the following protocol: 94°C for 30 s, 61°C for 30 s, and 72°C for 50 s. Then, 10 μL of each PCR product was separated on 1.2% agarose gels and the products were visualized under ultraviolet light. Real‐time PCR analysis using the TaqMan fluorescence methods was performed for quantitatively analyzing the mRNA. The products were amplified using a LightCycler (Roche Diagnostics, Basel, Switzerland) in a reaction mixture (20 μL) containing 2 μL of LightCycler‐FastStart DNA Master Taqman (Roche), 0.5 μmol/L of each primer, and 3 mmol/L MgCl2. The details of the primers used are given in Table 1. The copy numbers of mRNA were measured in triplicate, averaged, and then standardized according to those of β‐actin.
Western blotting. Western blot analysis was performed as follows. Cells were harvested and washed twice with ice‐cold phosphate‐buffered saline (PBS). Whole‐cell lysates were prepared by resuspending the cell pellet in RIPA buffer (50 mm Tris Hcl [pH 8.0], 150 mm NaCl, 1% [v/v] NP‐40, 0.5% [w/v] deoxycholic acid, 0.1% [w/v] sodium dodecyl sulfate [SDS]) that contained a protease inhibitor cocktail (Roche, Mannheim, Germany). The extracted proteins were loaded onto SDS‐polyacrylamide gels for electrophoresis and then transferred to a nitrocellulose membrane. The membrane was incubated in 5% (w/v) dried milk protein in PBS containing 0.05% (v/v) Tween‐20 (PBS‐T) for 1 h, and washed with PBS‐T and then further reacted with primary antibodies against HBX (Chemicon, Temecula, CA, USA) and β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h. The membrane was extensively washed with PBS‐T, and then incubated with the secondary antibody against either mouse or goat IgG conjugated to horseradish peroxidase for 1 h. After extensive washing, the protein bands on the membrane were visualized with chemiluminescent reagents according to the manufacturer’s instructions (Supersignal Substrate; Pierce, Rockford, IL, USA).
Statistical analysis. The data was analyzed using SPSS version 11.5 (SPSS, Chicago, IL, USA). For comparing continuous data, statistical analyses were performed with the Kruskal–Wallis test. For comparing the categorical data, statistical analyses were performed with the χ2‐test, and Fisher’s exact test was used when a cell value <5 in two‐by‐two tables. In order to confirm the factors that encouraged the development of HCC, we analyzed the data by multiple logistic regression tests. Variables with P‐values ≤0.05 on single logistic analysis were candidates for multiple logistic analyses. All P‐values were two‐sided, and P < 0.05 was considered statistically significant.
Results
The baseline demographic characteristics of the patients are summarized in Table 2. There were no differences in the variables examined between the groups, including age, gender, laboratory findings, and severity of LC (Table 2). The time at which HCC was newly diagnosed in the 12 subjects leading to their exclusion from the LC groups was as follows: eight subjects in 1 year after recruitment (1 at 6 months, 2 at 8 months, 2 at 9 months, and 3 at 12 months) and four subjects at 2 years after recruitment. Therefore, the yearly HCC incidence in this study was 4–8%. Of the 80 patients included in the LC‐HCC (LC with HCC) group, 29 were confirmed to have HCC by histology, while 51 were verified by CT, ultrasonography, magnetic resonance imaging, and AFP rise.
Table 2.
Demographic data of the 160 patients with cirrhosis, with and without HCC
| LC (80) | LC‐HCC (80) | P‐value | |
|---|---|---|---|
| Age (years) | 54.26 ± 8.05 | 55.50 ± 9.27 | 0.369 |
| Gender (male:female) | 46:34 | 56:24 | 0.100 |
| HBV‐DNA (Log10 copies/mL) | 6.80 ± 1.08 | 6.86 ± 1.22 | 0.741 |
| AST (IU/L) | 98.36 ± 124.62 | 110.60 ± 111.99 | 0.515 |
| PLT (×103/mm3) | 102.38 ± 48.79 | 101.51 ± 65.86 | 0.924 |
| CTP A (5–6) | 33 | 22 | 0.186 |
| B (7–9) | 27 | 34 | |
| C (10–15) | 20 | 24 | |
| HBeAg (+:−) | 46:34 | 41:39 | 0.427 |
−, HBeAg negative; +, HBeAg positive; AST, aspartate aminotransferase; CTP, Child‐Turcotte‐Pugh score; HBeAg, hepatitis B e antigen; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; LC, liver cirrhosis; PLT, platelet.
Subgenotyping of HBV according to nucleotide sequences and divergence from the consensus sequence. The isolated sequences were included in the HBV genotype C and subgenotype C2 (data not shown). Consensus sequence in the Enh1/X‐promoter regions (nt. 951–1373) is shown in Figure 1. The rate of divergence of nucleotides from the consensus sequences was 1.69% in 119 sites with 945 divergent nucleotides. The mean divergence in one site was 7.94 ± 14.83 nucleotides (range, 1–81). The mean number of divergent nucleotides in one isolate was 5.91 ± 4.83 (range, 0–14; not shown in the Figure).
Figure 1.

Consensus sequences in the Enh1/X‐promoter regions (nt. 951–1373, genotype C2). Enh1/X‐promoter regions were inserted into the multi‐cloning sites of a pGL3 basic vector and a pUC18 vector. Nt. 1053 and nt. 1229 locations with red color indicated. Functional sites, including nt.1053 and nt.1229, indicated by italic type. NF 1, nuclear factor 1.
Nucleotide variability, hotspots, and the relationship among hotspots from isolated sequences. The number of divergent nucleotides was 5.62 ± 2.82 in the LC without HCC group and 6.20 ± 2.81 in the LC with HCC group; thus, the rate of divergence was 1.60% and 1.76%, respectively. There was no difference in the divergences according to the disease state. There were 12 hotspots with >15% divergence in the examined region; they were located at nt. 1050, 1053, 1078, 1126, 1134, 1167, 1206, 1229, 1317, 1323, and 1342 (Table 3). There were certain specific relationships among them; for example, the T1126C nucleotide mutation was strongly associated with the T1134C mutation (R = 0.892, P < 0.001) (data not shown).
Table 3.
Nucleotide substitutions in several sites of the HBV enhancer 1 and X‐promoter region (1021–1373) compared with the consensus sequence and the significance of these mutations
| No. of nt.* (wild→mutant) | LC (80) | LC‐HCC (80) | P‐value |
|---|---|---|---|
| 1050 (C→T) (C→A) | 14 (17.50%) 2 (2.50%) | 13 (16.25%) 0 (0.00%) | 0.833 0.155 |
| 1053 (G→A) | 7 (8.75%) | 18 (22.50%) | 0.017 |
| 1078 (T→G) | 31 (38.75%) | 30 (37.50%) | 0.871 |
| 1126 (A→C) | 22 (27.5%) | 24 (30.00%) | 0.727 |
| 1134 (T→C) | 19 (23.75%) | 18 (22.50%) | 0.851 |
| 1167 (A→C) (A→T) | 8 (10.00%) 5 (6.25%) | 5 (6.25%) 14 (17.50%) | 0.385 0.028 |
| 1206 (A→C) (A→T) (A→W) | 12 (15.00%) 1 (1.25%) 1 (1.25%) | 10 (12.50%) 3 (3.75%) 0 (0.00%) | 0.646 0.311 0.316 |
| 1229 (G→A) | 23 (28.75%) | 41 (51.25%) | 0.004 |
| 1317 (G→A) (G→T) (G→R) | 34 (42.50%) 2 (2.50%) 2 (2.50%) | 41 (51.25%) 1 (1.25%) 1 (1.25%) | 0.267 0.560 0.560 |
| 1320 (A→C) (A→G) | 14 (17.50%) 0 (0.00%) | 10 (12.50%) 1 (1.25%) | 0.376 0.316 |
| 1323 (T→C) | 13 (16.25%) | 8 (10.00%) | 0.242 |
| 1341 (T→C) (T→G) (T→A) | 4 (5.00%) 6 (7.50%) 2 (2.50%) | 3 (3.75%) 3 (3.75%) 0 (0.00%) | 0.699 0.303 0.155 |
| 1053 (G→A)/1229 (G→A) | 1 (1.25%) | 7 (8.75%) | 0.002 |
The significance of differences (P‐value) in the indicated group is shown. P < 0.05 is indicated by italic type. IUPAC (Table 3) codes for nucleotides and ambiguous nucleotides: A, adenine; C, cytosine; G, guanine; T, thymine; D, W, A+T; R, A+G. *No. of nt., number of nucleotides; 1053 (G→A)/1229 (G→A), combined mutation of G1053A and G1229A. HBV, hepatitis B virus. IUPAC, International Union of Pure and Applied Chemistry.
Differences in mutations according to disease groups. There were several mutation sites with different trends according to the disease state: nt. 1053, 1167, and 1229. Table 3 shows the mutation sites with the frequencies and P values. The comparison of the mutation frequencies in the HCC group and the non‐HCC group with cirrhosis showed statistical significance for G1053A, A1167T, and G1229A (P = 0.017, 0.028, and 0.004, respectively; Table 3).
Univariate and multivariate analyses between the disease groups according to the examined items and the mutations of nucleotides. In this study, the G1053A, A1167T, and G1229A mutations were identified as the risk factors for HCC from the single logistic analysis in this study (odds ratio [OR] [95% confidence interval, CI] = 3.028 [1.187–7.723], 3.075 [1.046–9.042], and 2.605 [1.356–5.006], respectively; P = 0.020, 0.041, and 0.004, respectively). However, A1167T as a risk factor was eliminated after multiple logistic regression analysis (OR [95% CI] = 2.642 [0.830–8.412], P = 0.100, Table 4).
Table 4.
Factors associated with the development of HCC by multiple logistic analysis
| OR | 95% CI | P‐value* | |
|---|---|---|---|
| G1053A | 3.484 | 1.146–10.593 | 0.028 |
| G1229A | 2.627 | 1.283–5.382 | 0.008 |
| G1053A/G1229A | 12.461 | 1.399–111.019 | 0.024 |
| A1167T | 2.642 | 0.830–8.412 | 0.100 |
The significance of differences (P‐value) in the indicated group is shown. * P < 0.05 (compared to wild type) is indicated by italic type. CI, confidence interval; HCC, hepatocellular carcinoma; OR, odds ratio. G1053A/G1229A, combined mutation of G1053A and G1229A.
The developing mutation patterns of G1053A and G1229A, which showed different mutation rates depending on the disease status from multiple logistic regression analysis, showed the rate of 1:2 (LC:LC‐HCC) for the single mutation and 1:7 (LC:LC‐HCC) for the combined mutation. Accordingly, the combined mutation rate of the LC‐HCC group was significantly higher than that of the LC group. These mutations – single and combined – differed significantly between the LC and LC‐HCC groups (P = 0.002, Table 3). Interestingly, an increased risk of developing HCC was observed for the combined mutation. (G1053A, G1229A, both mutations; OR = 3.484, 2.627, and 12.461, respectively; P = 0.028, 0.008, and 0.024, respectively, Table 4).
Comparison of the luciferase activity, the mRNA, and protein expression of HBx between the wild types and mutant types of the Enh1/X‐promoter. We constructed the wild‐type vector (pGL3–WT) by inserting the Enh1/X‐promoter into the pGL3 basic luciferase reporter vector. Based on multiple logistic regression analysis, we constructed pGL3–G1053A, pGL3–G1229A, and pGL3–G1053A/G1229A by using site‐directed mutagenesis, and then measured the luciferase activity from pGL3–G1053A, pGL3–G1229A, pGL3–G1053A/G1229A, and pGL3–WT. As a result, pGL3–G1053A, pGL3–G1229A, and pGL3–G1053A/G1229A showed 2.38‐, 4.68‐, and 7.52‐fold increases in luciferase activity in the HepG2 cells compared to pGL3–WT, respectively (all, P < 0.001) (Fig. 2).
Figure 2.
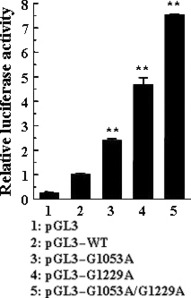
Luciferase activity of wild and mutant vectors, pGL3 (pGL3 basic vector), pGL3–WT (wild type), pGL3–G1053A, pGL3–G1229A, and pGL3–G1053A/G1229A in HepG2 Cells. Luciferase activity was measured in triplicate, averaged, and then normalized with β‐galactosidase activity using the galactosidase assay system. * P < 0.05 vs pGL3–WT, ** P < 0.01 vs pGL3–WT.
This was a retrospective study, using samples which were collected and stored at −80°C during the period between January 1999 to August 2006. Consequently, we would expect different extents of mRNA degradation in the samples, making the quantification and comparison of HBx expression in these clinical samples extremely challenging. We therefore created a complete HBV genome replicon (pHBV1.2X) with the wild‐type or mutated sequence(s), and showed differences in HBx mRNA and protein expression levels as a result of point mutations including G1053A and G1229A.
We measured the level of HBx mRNA in cells transfected with pHBV1.2X–G1053A, pHBV1.2X–G1229A, pHBV1.2X–G1053A/G1229A, or pHBV1.2X (wild type). Compared with pHBV1.2X, cells transfected with pHBV1.2X–G1053A, pHBV1.2X–G1229A, and pHBV1.2X–G1053A/G1229A exhibited significantly higher levels of HBx mRNA (Fig. 3). Next, to investigate whether the significant mutation on the Enh1/X‐promoter affected the translation of HBx, the amount of the expressed X‐protein was analyzed by western blotting. pHBV1.2X, pHBV1.2X–G1053A, pHBV1.2X–G1229A, and pHBV1.2X–G1053A /G1229A transfected HepG2 cells. And then we tested each cell extract. This fold difference indicated the results are due to the extract of each vector transfected cell (Fig. 4).
Figure 3.
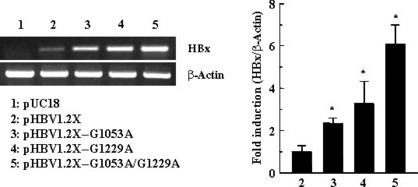
mRNA amount of wild and mutant vectors, pHBV1.2X (wild type), pHBV1.2X–G1053A, pHBV1.2X–G1229A, and pHBV1.2X–G1053A/G1229A in HepG2 cells. Cells were transfected with wild and mutant vectors as described in the Material and Methods. Total RNA was then isolated by TRIzol and cDNA was synthesized by reverse transcription. pHBV1.2X, pHBV1.2X–G1053A, pHBV1.2X–G1229A, and pHBV1.2X–G1053A/G1229A cDNAs were amplified by RT‐PCR or quantitative real‐time PCR using specific primer sets, respectively. * P < 0.05 vs pHBV1.2X, ** P < 0.01 vs pHBV1.2X.
Figure 4.
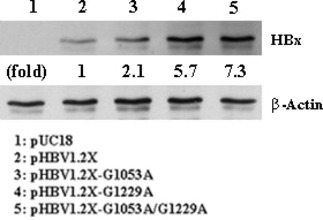
Western blotting results for wild and mutant vectors, pHBV1.2X (wild type), pHBV1.2X–G1053A, pHBV1.2X–G1229A, and pHBV1.2X–G1053A/G1229A in HepG2 cells. Upper panel shows the expression of Hepatitis B virus X protein (HBx), whereas the bottom panel gives β‐actin, which was used as a loading control.
Discussion
There are many studies concerning the risk factors for HCC development by HBV on the epidemiological, experimental, and biological aspects.( 5 , 6 , 7 , 8 , 9 , 10 , 11 ) Cirrhosis and HBx protein is reported to have a significant contribution to HCC development. When the cirrhosis appears, the rate of developing HCC is 5–20%.( 5 , 36 ) Previous studies regarding differences between HCC and non‐HCC cirrhosis patients have examined various clinical factors. One study showed differences between HCC and non‐HCC only for cirrhosis patients; the significant factors were age (>55 years), prothrombin time (PT) (<75%), platelet count (<75000/mm3), AFP level (>20 ng/mL, i.e. higher than normal), the existence of LC complications, and the severity of LC,( 11 ) most of which are related to the severity of LC. The differences in the viral aspect in the LC groups with HCC and without HCC remain to be established. In our study, there were no significant differences in age and LC severity between the LC with HCC group and the LC without HCC group at the time of enrollment. The influence of these factors was therefore excluded and the differences between the two groups was analyzed only in terms of the viral aspect.
HBx protein is found in most HCC tissues, and among the HBV genes that are inserted in the DNA of the host during the development of HCC, the HBx gene is known to survive for relatively long time periods in the host’s chromosomes.( 37 ) The oncogenic role of HBx is strongly suggested by the development of hepatocellular carcinoma in a number of transgenic mouse models.( 18 ) However, since HCC did not occur in HBx transgenic mouse models in other studies, the direct oncogenic role of HBx is still controversial.( 38 ) Since it takes over 20 years for HCC to develop in a HBV carrier, it is believed that expression of HBx is not the sole causative factor in HCC, and it is helped by the presence of other carcinogenic reagents (such as oncogene and genotoxic stress).( 39 ) The goal of our study was to find nucleotide mutations that frequently occur in HCC patients and to examine the effects of such mutations on the expression of HBx. This information would also be valuable in exploring the relationship of HBx expression with the occurrence of HCC.
The extent of HBx protein expression is controlled by the Enh1/X‐promoter located at the front of the X‐gene. Several studies have investigated the relationship between the occurrence of HCC and nucleotide mutations in the X‐gene; however, nucleotide mutations of the Enh1/X‐promoter region, which controls the X‐gene, have rarely been studied. Thus, our study aimed to find nucleotide mutations in the Enh1/X‐promoter region correlated with the occurrence of HCC and their underlying mechanisms, albeit with a limited sample of LC patients only. Also, this study was designed to exclude any disparity in other known risk factors in the development of HCC, such as genotypes, age, disease status (all LC), and the severity of LC, between the control and the study groups; therefore, our study group was well suited for examining the role of nucleotide mutations in HCC development in LC patients.
In this study, all isolated sequences were included within a single cluster among the HBV subgenotype C2. The nucleotide variability (1.69%) in the examined area was greater than the previously reported diversities from the whole genome and X‐gene studies.( 40 , 41 ) Generally, nucleotide diversities of the sequences is higher in the areas of singly coding regions (SCR) than in doubly coding regions (DCR).( 40 ) Such higher nucleotide diversities in nucleotide sequences have occurred in potent antigenic epitope areas, which were reflected by immune surveillance, such as that of the C‐gene and pre‐S gene.( 12 , 23 , 41 , 43 , 44 ) Considering the above‐mentioned characteristics of HBV‐induced CLD, the nucleotide sequences in our examined area might be less influenced by immune response. The sequence identified in this study appears to be a single coding region that is known to be one of the highly variable portions in the whole HBV genome. Therefore, the results of our study are consistent with expected findings as follows: the nucleotide variability remained more or less identical regardless of the disease severity, there were no potent antigenic epitopes present in the examined area, and the highly variable portion showed somewhat high nucleotide diversities.
In our study, G1053A and G1229A mutations were identified as independent risk factors in the development of HCC. The frequency of G1053A and G1229A mutations were significantly different according to the underlying diseases in our study. The G1053A mutation is located in the reverse transcriptase (RT) region and G1229A is located in the RNase H region in the P gene. However, this may not be important for enzymatic polymerase function, as G1053A is a non‐synonymous substitution; and G1229A, a synonymous substitution from R to Q, is not the catalytic and substrate‐binding residue.( 45 ) However, they are located in the functional area of the Enh1/X‐promoter region of HBV. Nt. 1053 is located in the middle of the p53 binding sequence‐like element, and this element is located in the modulatory domain of the Enh1.( 32 , 46 ) Nt. 1229 is located in an important transactivating portion in the X‐promoter region, where the domain sequences (nt. 1223 to nt. 1242) are known to bind with X‐PBP.( 47 ) Recently, nuclear respiratory factor 1 (NRF1) was found to be a binding protein in this domain and to contribute to the transcription of the X‐gene.( 48 ) Therefore, we hypothesize that these nucleotide mutations, G1053A and G1229A, have certain effects on the transcription and translation of HBx. There are only a few related studies with HCC patients regarding nucleotide mutations in the Enh1/X‐promoter region. Considering that there is evidence that HBx levels affect hepatocarcinogenesis and that the Enh1/X‐promoter region directly controls the X‐gene, which produces the HBx protein, this is slightly surprising. In addition, there is evidence that the HBV Enh1 region gets integrated into the HCC cell line and plays a role in viral replication.( 26 , 49 , 50 ) A recent study reported that the occurrence of mutations at other sites (nt. 1317 and nt. 1341) on the Enh1/X promoter increased the risk of developing HCC in chronic viral hepatitis B patients.( 51 ) Although these are not located in the transactivating region, the authors of the study suggest that the two sites of mutation have an effect on X‐gene transcription. However, these sites were not significant for the disease status in our study. This difference between our study and theirs might arise from the differences in the study designs concerning the underlying disease state and the follow‐up procedures, and the difference in the examined sequence and size, etc. Another study reported a nucleotide mutation related to HCC at nt.1165 in genotype B.( 24 ) Although our study also found a greater number of nucleotide mutations in the HCC group (LC vs LC‐HCC, 2 vs 6), the difference was statistically insignificant. The difference in the genotype is probably responsible for the different results, but a further study with more subjects is needed. Further, the areas in which the relationships between the progression of CLD and the HCC development with nucleotide mutations have been studied, the most frequent are the pre‐core/core‐promoter (including T1762/A1764 and T1766 and/or A1768), pre‐S, X‐gene, Pre‐S, and X‐gene.( 22 , 23 , 52 , 53 , 54 , 55 ) The region of study most researched in mutations associated with HCC development is the core promoter mutation (T1762/A1764), which is the most important mutation for disease progression according to previous studies. Of the 160 subjects included in this study, 88 subjects underwent nucleotide sequencing of the core promoter region. Most (85.23%) had a core promoter mutation; this is more frequent than that observed in previous studies.( 22 , 23 ) Because these subjects had advanced liver disease (LC) and high viral load, they had a more frequent mutation rate of the basal core promoter region compared with that in previous studies. The mutation rate did not differ from that of the disease group (LC vs LC‐HCC, 85.71%vs 84.61%, P = 0.885). The core promoter mutation was not related to the development of G1053A and G1229A mutations (McNemar, P = 0.000) (data not shown). Therefore, the basal core promoter mutation did not affect the G1053A and G1229A mutations.
Based on our functional study of the Enh1/X‐promoter, it was confirmed that G1053A, G1229A, and combined mutations increased the luciferase activity, the amount of mRNA, and HBx protein expression in HepG2 cells. Interestingly, the combined mutation (G1053A with G1229A) showed a higher expression level of HBx than did the single mutation (G1053A, G1229A). Therefore, we suggest that the combined mutation has an additive effect in HCC development. We found a higher G1229A mutation rate in the HCC group of genotype C in whole genomic HBV sequences reported to NCBI; however, this has not been observed in previous reports.( 56 , 57 ) Our study is the first research on the relationship between nucleotide mutations in the Enh1/X‐promoter regions and HCC in cirrhotic patients.
The limitations of this study were as follows: first, an unavoidable selection bias since the subjects comprised patients admitted to a single center. Second, the result was attained from targets with one genotype; it would be necessary to verify our findings to apply them to other genotypes. Third, this study selected individuals in whom direct sequencing was possible. Since the DNA value would be higher than that of other studies, it would be necessary to verify the significance of having these mutations even among the greater proportion of patients with LC having low HBV‐DNA. Nevertheless, these mutations were attained from 160 LC patients with risk factors that were similar to other conditions of HCC development. We objectively verified the fact that all the mutations had increased HBX expression.
In summary, we suggest that G1053A and G1229A mutations in the cirrhotic patient with the subgenotype C2 are independent risk factors for HCC development and that the mutations might be related to increased levels of HBx protein. However, further studies are needed to confirm the significance of the mutations in other subgenotypes, races, ethnicities, and disease states.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- AST
aspartate aminotransferase
- CTP
Child‐Turcotte‐Pugh score
- HBeAg
hepatitis B e antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- LC
liver cirrhosis
- PLT
platelet
- CI
confidence interval
- HCC
hepatocellular carcinoma
- OR
odds ratio
Acknowledgments
This study was supported with a grant from The Korean Association of Study of Liver disease (Fund of KASL 2006‐04) and Wonkwang University (2008).
References
- 1. Ganem D, Varmus HE. The molecular biology of the hepatitis B viruses. Annu Rev Biochem 1987; 56: 651–93. [DOI] [PubMed] [Google Scholar]
- 2. Eddleston AL, Mondelli M. Immunopathological mechanisms of liver cell injury in chronic hepatitis B virus infection. J Hepatol 1986; 3(Suppl 2): S17–23. [DOI] [PubMed] [Google Scholar]
- 3. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet 1981; 2: 1129–33. [DOI] [PubMed] [Google Scholar]
- 4. Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer 1988; 61: 1942–56. [DOI] [PubMed] [Google Scholar]
- 5. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004; 127: S35–50. [DOI] [PubMed] [Google Scholar]
- 6. Yu MC, Yuan JM. Environmental factors and risk for hepatocellular carcinoma. Gastroenterology 2004; 127: S72–8. [DOI] [PubMed] [Google Scholar]
- 7. Ikeda K, Arase Y, Kobayashi M et al. Hepatitis B virus‐related hepatocellular carcinogenesis and its prevention. Intervirology 2005; 48: 29–38. [DOI] [PubMed] [Google Scholar]
- 8. Kao JH, Chen PJ, Lai MY, Chen DS. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 2000; 118: 554–9. [DOI] [PubMed] [Google Scholar]
- 9. Thompson Coon J, Rogers G, Hewson P et al. Surveillance of cirrhosis for hepatocellular carcinoma: systematic review and economic analysis. Health Technol Assess 2007; 11: 1–206. [DOI] [PubMed] [Google Scholar]
- 10. Benvegnù L, Noventa F, Bernardinello E, Pontisso P, Gatta A, Alberti A. Evidence for an association between the aetiology of cirrhosis and pattern of hepatocellular carcinoma development. Gut 2001; 48: 110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Velázquez RF, Rodríguez M, Navascués CA et al. Prospective analysis of risk factors for hepatocellular carcinoma in patients with liver cirrhosis. Hepatology 2003; 37: 520–7. [DOI] [PubMed] [Google Scholar]
- 12. Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol 1995; 13: 29–60. [DOI] [PubMed] [Google Scholar]
- 13. Dejean A, Bougueleret L, Grzeschik KH, Tiollais P. Hepatitis B virus DNA integration in a sequence homologous to v‐erb‐A and steroid receptor genes in a hepatocellular carcinoma. Nature 1986; 322: 70–2. [DOI] [PubMed] [Google Scholar]
- 14. Hino O, Shows TB, Rogler CE. Hepatitis B virus integration site in hepatocellular carcinoma at chromosome 17;18 translocation. Proc Natl Acad Sci USA 1986; 83: 8338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol 2004; 78: 12725–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haviv I, Vaizel D, Shaul Y. pX, the HBV‐encoded cofactor, interacts with components of the transcription machinery and stimulates transcription in a TAF independent manner. EMBO J 1996; 15: 3413–20. [PMC free article] [PubMed] [Google Scholar]
- 17. Sprengel R, Kuhn C, Will H, Schaller H. Comparative sequence analysis of duck and human hepatitis B virus genomes. J Med Virol 1985; 15: 323–33. [DOI] [PubMed] [Google Scholar]
- 18. Yu DY, Moon HB, Son JK et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitisB virus X‐protein. J Hepatol 1999; 31: 123–32. [DOI] [PubMed] [Google Scholar]
- 19. Reifenberg K, Lhler J, Pudollek HP et al. Long‐term expression of the hepatitis B virus core‐e‐ and X‐proteins does not cause pathologic changes in transgenic mice. J Hepatol 1997; 26: 119–30. [DOI] [PubMed] [Google Scholar]
- 20. Koike K, Moriya K, Iino S et al. High‐level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994; 19: 810–9. [PubMed] [Google Scholar]
- 21. Su Q, Schroder CH, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P. Expression of hepatitis B virus X protein in HBV‐infected human livers and hepatocellular carcinomas. Hepatology 1998; 27: 1109–20. [DOI] [PubMed] [Google Scholar]
- 22. Tanaka Y, Mukaide M, Orito E et al. Specific mutations in enhancer II/core promoter of hepatitis B virus subgenotypes C1/C2 increase the risk of hepatocellular carcinoma. J Hepatol 2006; 45: 646–53. [DOI] [PubMed] [Google Scholar]
- 23. Chen CH, Changchien CS, Lee CM et al. Combined mutations in pre‐s/surface and core promoter/precore regions of hepatitis B virus increase the risk of hepatocellular carcinoma: a case‐control study. J Infect Dis 2008; 198: 1634–42. [DOI] [PubMed] [Google Scholar]
- 24. Sung JJ, Tsui SK, Tse CH et al. Genotype‐specific genomic markers associated with primary hepatomas, based on complete genomic sequencing of hepatitis B virus. J Virol 2008; 82: 3604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kosovsky MJ, Huan B, Siddiqui A. Purification and properties of rat liver nuclear proteins that interact with the hepatitis B virus enhancer 1. J Biol Chem 1996; 271: 21859–69. [DOI] [PubMed] [Google Scholar]
- 26. Doitsh G, Shaul Y. Enhancer I predominance in hepatitis B virus gene expression. Mol Cell Biol 2004; 24: 1799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bock CT, Malek NP, Tillmann HL, Manns MP, Trautwein C. The enhancer I core region contributes to the replication level of hepatitis B virus in vivo and in vitro. J Virol 2000; 74: 2193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huan B, Siddiqui A. Retinoid X receptor RXR alpha binds to and trans‐activates the hepatitis B virus enhancer. Proc Natl Acad Sci USA 1992; 89: 9059–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gustin K, Shapiro M, Lee W, Burk RD. Characterization of the role of individual protein binding motifs within the hepatitis B virus enhancer I on X promoter activity using linker scanning mutagenesis. Virology 1993; 193: 653–60. [DOI] [PubMed] [Google Scholar]
- 30. Wang SH, Yeh SH, Lin WH et al. Identification of androgen response elements in the enhancer I of hepatitis B virus: a mechanism for sex disparity in chronic hepatitis B. Hepatology 2009; 50: 1392–402. [DOI] [PubMed] [Google Scholar]
- 31. Ori A, Zauberman A, Doitsh G et al. p53 binds and represses the HBV enhancer: an adjacent enhancer element can reverse the transcription effect of p53. EMBO J 1998; 17: 544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qu JH, Zhu MH, Lin J et al. Interaction of p53 with p53 response element like binding sequence at upstream of hepatitis B virus enhancer I. Ai Zheng 2004; 23: 502–7. [PubMed] [Google Scholar]
- 33. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL‐X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997; 25: 4876–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 1980; 16: 111–20. [DOI] [PubMed] [Google Scholar]
- 35. Park SG, Lee SM, Jung G. Antisense oligodeoxynucleotides targeted against molecular chaperonin Hsp60 block human hepatitis B virus replication. J Biol Chem 2003; 278: 39851–7. [DOI] [PubMed] [Google Scholar]
- 36. Bruix J, Sherman M, Llovet JM et al. Clinical management of hepatocellular carcinoma: conclusions of the Barcelona‐2000 EASL conference. J Hepatol 2001; 35: 421–30. [DOI] [PubMed] [Google Scholar]
- 37. Hsu T, Möröy T, Etiemble J et al. Activation of c‐myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma. Cell 1988; 55: 627–35. [DOI] [PubMed] [Google Scholar]
- 38. Lee TH, Finegold MJ, Shen RF et al. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J Virol 1990; 64: 5939–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Terradillos O, Billet O, Renard CA et al. The hepatitis B virus X gene potentiates c‐myc‐induced liver oncogenesis in transgenic mice. Oncogene 1997; 14: 395–404. [DOI] [PubMed] [Google Scholar]
- 40. Norder H, Courouc AM, Magnius LO. Complete genomes, phylogenetic relatedness, and structural proteins of sixstrains of the hepatitis B virus, four of which represent two new genotypes. Virology 1994; 198: 489–503. [DOI] [PubMed] [Google Scholar]
- 41. Kim HC, Seo GS, Kim YS, Song WG, Moon HB, Cho JH. Hepatitis B virus (HBV) genotype in korean chronic HBV carriers: Whole HBV genome and it's nucleotide sequence by single polymerization chain reaction (PCR) method. Korean J Med 2001; 61: 479–488. [Google Scholar]
- 42. Akarca US, Lok AS. Naturally occurring hepatitis B virus core gene mutations. Hepatology 1995; 22: 50–60. [PubMed] [Google Scholar]
- 43. Chen CH, Hung CH, Lee CM et al. Pre‐S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg‐negative patients. Gastroenterology 2007; 133: 1466–74. [DOI] [PubMed] [Google Scholar]
- 44. Choi MS, Kim DY, Lee DH et al. Clinical significance of pre‐S mutations in patients with genotype C hepatitis B virus infection. J Viral Hepat 2007; 14: 161–8. [DOI] [PubMed] [Google Scholar]
- 45. Potenza N, Salvatore V, Raimondo D et al. Optimized expression from a synthetic gene of an untagged RNase H domain of human hepatitis B virus polymerase which is enzymatically active. Protein Expr Purif 2007; 55: 93–9. [DOI] [PubMed] [Google Scholar]
- 46. Takada S, Kaneniwa N, Tsuchida N, Koike K. Hepatitis B virus X gene expression is activated by X protein but repressed by p53 tumor suppressor gene product in the transient expression system. Virology 1996; 216: 80–9. [DOI] [PubMed] [Google Scholar]
- 47. Nakamura I, Koike K. Identification of a binding protein to the X gene promoter region of hepatitis B virus. Virology 1992; 191: 533–40. [DOI] [PubMed] [Google Scholar]
- 48. Tokusumi Y, Zhou S, Takada S. Nuclear respiratory factor 1 plays an essential role in transcriptional initiation from the hepatitis B virus x gene promoter. J Virol 2004; 78: 10856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shamay M, Agami R, Shaul Y. HBV integrants of hepatocellular carcinoma cell lines contain an active enhancer. Oncogene 2001; 20: 6811–9. [DOI] [PubMed] [Google Scholar]
- 50. Keasler VV, Hodgson AJ, Madden CR, Slagle BL. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J Virol 2007; 81: 2656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang KY, Imazeki F, Fukai K et al. Analysis of the complete hepatitis B virus genome in patients with genotype C chronic hepatitis and hepatocellular carcinoma. Cancer Sci 2007; 98: 1921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yotsuyanagi H, Hino K, Tomita E, Toyoda J, Yasuda K, Iino S. Precore and core promoter mutations, hepatitis B virus DNA levels and progressive liver injury in chronic hepatitis B. J Hepatol 2002; 37: 355–63. [DOI] [PubMed] [Google Scholar]
- 53. Buckwold VE, Xu Z, Chen M, Yen TS, Ou JH. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J Virol 1996; 70: 5845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li J, Buckwold VE, Hon MW, Ou JH. Mechanism of suppression of hepatitis B virus precore RNA transcription by a frequent double mutation. J Virol 1999; 73: 1239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang HI, Lu SN, Liaw YF et al. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N Engl J Med 2002; 347: 168–74. [DOI] [PubMed] [Google Scholar]
- 56. Takahashi K, Akahane Y, Hino K, Ohta Y, Mishiro S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full‐length isolates. Arch Virol 1998; 143: 2313–26. [DOI] [PubMed] [Google Scholar]
- 57. Song BC, Kim H, Kim SH, Cha CY, Kook YH, Kim BJ. Comparison of full length sequences of hepatitis B virus isolates in hepatocellular carcinoma patients and asymptomatic carriers of Korea. J Med Virol 2005; 75: 13–9. [DOI] [PubMed] [Google Scholar]