

Abstract
Centrosomes are important cytoplasmic organelles involved in chromosome segregation, defects in which can result in aneuploidy, and contribute to tumorigenesis. It is known that DNA damage causes the supernumerary centrosomes by a mechanism in which centrosomes continue to duplicate during cell cycle arrest at checkpoints. We show here that ionizing radiation induces the overduplication of centrosomes in a dose‐dependent manner, and that the level of overduplication is pronounced in BRCA1‐ and NBS1‐deficient cells, even though their checkpoint control is abrogated. Conversely, marginal increases in overduplication were observed in Ku70‐ and DNA‐PKcs‐deficient cells, which are intact in checkpoint control. The frequency of radiation‐induced overduplication of centrosomes might be associated with DNA repair, as it was decreased with reduced cell killing after protracted exposures to radiation. As a result, when the frequency of radiation‐induced centrosome overduplication was plotted against radiation‐induced cell killing, similar curves were seen for both protracted and acute exposures in wild‐type cells, Ku70‐deficient, and DNA‐PKcs‐deficient cells, indicating a common mechanism for centrosome overduplication. However, the absence of either BRCA1 or NBS1 enhanced radiation‐induced overduplication frequencies by 2–4‐fold on the basis of the same cell killing. These results suggest that radiation‐induced centrosome overduplication is regulated by at least two mechanisms: a checkpoint‐dependent pathway involved in wild‐type cells, Ku70‐deficient and DNA‐PKcs‐deficient cells; and a checkpoint‐independent pathway as observed in BRCA1‐deficient and NBS1‐deficient cells. (Cancer Sci 2010; 101: 2531–2537)
The centrosome, an organelle consisting of a pair of centrioles surrounded by pericentriolar material, is necessary for proper chromosome segregation during cell division.( 1 , 2 ) Centrosomes duplicate only once during each cell division cycle. When the proper number of centrosomes is not present, the formation of multiple mitotic spindles and aneuploidy can occur, leading to tumorigenesis.( 3 , 4 ) BRCA1, an ovarian and breast cancer‐specific tumor suppressor protein,( 5 , 6 , 7 ) localize at the centrosomes, and suppression of BRCA1 leads to centrosome overduplication.( 8 ) BRCA1 is an E3 ubiquitin ligase that ubiquitinates γ‐tubulin, a key protein for the formation of pericentriolar material.( 5 ) Defects in the ubiquitination activity of BRCA1 lead to centrosome overduplication and abnormal aster formation,( 9 , 10 ) suggesting that the ubiquitination is required to maintain the proper number of centrosomes in cells. Hypomorphic mutations in the NBS1 protein result in Nijmegen breakage syndrome (NBS), which is characterized by a predisposition towards malignancies.( 11 ) NBS1 localizes at the centrosomes by forming complexes with BRCA1; the knockdown of NBS1 in mammalian cells results in centrosome overduplication.( 12 ) Given that downregulation of the NBS1 protein reduces the ubiquitination of γ‐tubulin, NBS1 could be involved in the maintenance of the proper number of centrosomes through BRCA1‐mediated ubiquitination of γ‐tubulin.( 12 ) Other DNA repair proteins, such as the DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), also localize at the centrosome; however, their function at the centrosome is not clear.( 13 )
Ionizing radiation (IR) induces DNA double‐strand breaks (DSBs), a potentially lethal type of DNA damage, and generates chromosome aberrations and gene mutations, and leads to apoptosis.( 14 ) Two major pathways are involved in DSB repair: homologous recombination (HR) repair; and non‐homologous end‐joining (NHEJ) repair.( 15 , 16 , 17 ) Homologous recombination is an accurate repair system comprising a number of proteins, including the NBS1/MRE11/RAD50 (MRN) complex and BRCA1.( 15 ) The HR factors, BRCA1 and NBS1, are not only involved with the repair of DSBs but also regulate cell cycle checkpoints.( 16 , 18 ) In contrast, NHEJ enzymes are not associated with cell cycle checkpoints, but they constitute the dominant repair mechanism in mammalian cells. DNA‐PKcs forms a complex with Ku70 and Ku80, which then recruits the XRCC4/ligase 4 complex to directly rejoin the two ends of a DSB.( 17 ) The time required for rejoining DSBs depends on which repair process is used; NHEJ takes approximately 1–2 h,( 19 ) but HR requires more time.( 20 ) If not properly rejoined, DSBs could be mis‐repaired through interactions with other DSBs, which can convert it into a lethal lesion. Exposure of cells to protracted IR spacio‐temporally promotes the correct rejoining of DSBs, and consequently results in fewer DSBs being mis‐repaired. This will ultimately result in a lower degree of cell death after radiation exposure.( 21 , 22 )
It has been reported that IR induces the overduplication of centrosomes.( 23 , 24 ) Although the underlying mechanism is not well understood, this effect could be due to an uncoupling between cell cycle checkpoints and centrosome duplication. Specifically, centrosome duplication is triggered by the activation of cyclin‐dependent kinase (CDK)/cyclin‐A/E and continues when the cell cycle is arrested in response to DNA damage. This model is supported by evidence showing that centrosome overduplication is strongly reduced by the presence of a CDK/cyclin‐A/E inhibitor such as roscovitine,( 12 ) or by the abrogation of cell cycle checkpoints in Chk1‐deficient cells.( 25 , 26 ) Thus, a cell cycle arrest should increase the chances for overduplication to occur,( 23 ) while also providing more time for DNA repair by NHEJ and HR.( 27 , 28 ) Accumulating evidence regarding the associations of DNA repair, NHEJ and HR, with IR‐induced cell killing has been reported, but the contributions of NHEJ‐ and HR‐repair proteins to the overduplication of centrosomes following exposures to IR remains to be examined. The aim of this study was to investigate the underlying mechanisms involved in centrosome overduplication after IR exposures and the role of specific DNA repair proteins, which localize at the centrosomes.
Materials and Methods
Cell culture and transfection. The human cell line U2OS and the mouse cell line NIH3T3 were used as control cell lines. A31‐1 cells (Nbs1−/−) were established from mouse embryonic fibroblasts from Nbs1 knockout mice using SV40 infection. Brca1−/− and Brca1+/− mouse embryonic stem (ES) cells were gifts from M.E. Moynahan, Developmental Biology, Memorial Sloan‐Kettering Cancer Center, New York, NY, USA. Ku70−/−, SCID‐IM (DNA‐PKcs−/−), and CB‐17‐IM (DNA‐PKcs+/+) cells were established from mouse embryonic fibroblasts obtained from the appropriate knockout mice (a gift from F.W. Alt, Department of Genetics and Pediatrics, Harvard University, Boston, MA, USA) and the parental mice (Charles River, Wilmington, MA, USA) through SV40 infection. A31‐1 cells complemented with NBS1 cDNA (A31‐1 + NBS1 cDNA) and Ku70−/− cells complemented with Ku70 cDNA (Ku70−/−+ Ku70 cDNA) were used as wild‐type cell controls. Preparation of (A31‐1 + NBS1 cDNA) cells and (Ku70−/−+Ku70 cDNA) cells was carried out as previously reported.( 12 ) Cells were cultured in DMEM (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (HyClone, Waltham, MA, USA) at 37°C in a humidified atmosphere containing 5% CO2.
Immunostaining. The immunostaining was carried out according to the methods of Saladino et al. ( 29 ) Briefly, cells on coverslips were washed with PBS, fixed in cold methanol (−20°C) for 10 min, and incubated with a detergent solution (0.1% Triton X‐100, 20 mM HEPES [pH 7.4], 50 mM NaCl, 3 mM MgCl2, and 300 mM sucrose) at 4°C for 5 min. The cells were then incubated with 3% non‐fat milk for 30 min, followed by incubation with primary antibodies against γ‐tubulin (Sigma‐Aldrich) and centrin‐2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The cells were subsequently incubated with the following secondary antibodies: Alexa‐488‐conjugated anti‐rabbit IgG for γ‐tubulin; and Alexa‐546‐conjugated anti‐rabbit IgG for centrin‐2 (both Molecular Probes, Eugene, OR, USA). Fluorescence was visualized using a confocal laser‐scanning microscope (Olympus, Tokyo, Japan). At least 200 cells in each sample were examined for centrosome number and nuclear morphology.
Irradiation and cell survival assays. The fraction of surviving cells after IR exposures was determined by clonogenic survival assays. Cells were irradiated with 137Cs γ‐rays at a dose rate of 1 Gy/min using a Gammacell 40 system (MDS Nordion, Ottawa, Canada) or at 0.5 mGy/min using a low dose rate irradiation system (Sangyo Kagaku, Tokyo, Japan). During exposures (for a maximum period of 7 day), cells were maintained in a 5% CO2 atmosphere at 37°C. Following irradiation, cells were trypsinized and an appropriate number of cells were plated on 100 mm dishes. After a 10‐day incubation period, the surviving fraction of the cell population was determined by counting the number of colonies that had formed. An additional survival experiment was carried out in which NIH3T3 cells were treated with 25 μM roscovitine for 72 h after irradiation with 137Cs γ‐rays at a dose rate of 1 Gy/min.
Results
Excess centrosomes accumulate after exposure of cells to acute and protracted IR. To determine the number of centrosomes in each cell, the expression of γ‐tubulin, a centrosome‐specific protein, was quantified with immunostaining at various time points after exposure to IR. In U2OS cells, excess centrosomes in irradiated cells, defined as more than two centrosomes per cell, reached a peak at 96 h after an acute 5 Gy exposure at a dose rate of 1 Gy/min. The centrosome number then gradually declined over the next 48 h (Fig. 1a, left panel). To determine the dose rate effects of centrosome overduplication, we compared their frequencies at a dose rate of 1 Gy/min with that of 0.5 m Gy/min, in which we observed the repair from DNA damage after IR exposure.( 21 , 22 , 30 ) After a protracted exposure (5 Gy at a dose rate of 0.5 mGy/min), the frequency of cells containing excess centrosomes was relatively constant for at least 96 h (Fig. 1a, right panel). When human U2OS cells were compared at 96 h post‐exposure, it was found that IR increased the frequency of centrosomes in a dose‐dependent fashion (Figs 1b, left panel,S1). Moreover, as observed for cell death frequencies (Fig. 1c, left panel), the frequency of cells containing excess centrosomes after a protracted exposure to 5 Gy was reduced to 70–80% of that seen after an acute exposure to 5 Gy (Fig. 1b, left panel). Because cell death frequencies appeared to correlate with a reduced frequency of cells containing excess centrosomes, cells were next treated with the CDK inhibitor roscovitine to examine the causal relationship between the frequency of cells containing excess centrosomes and cell death. Consistent with a previous report,( 12 ) roscovitine strongly suppressed the accumulation of excess centrosomes in irradiated cells, whereas the levels of cell death remained unchanged (Fig. 1d). This observation indicates that there is no causal relationship between the frequency of cells containing excess centrosomes and cell death. A reduced frequency of cells containing excess centrosomes after protracted exposures was also observed in mouse NIH3T3 cells (Fig. 1b,c, right panel).
Figure 1.
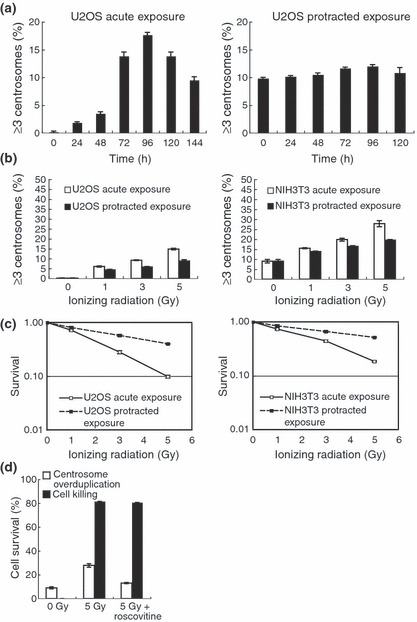
Production of excess centrosomes in human U2OS cells and mouse NIH3T3 cells after acute and protracted exposures to γ‐irradiation. (a) U2OS cells were irradiated with 5 Gy at a dose rate of 1 Gy/min (left panel), and with a total dose of 5 Gy at a dose rate of 0.5 mGy/min (right panel). Cells were examined for excess centrosomes at the indicated time points using γ‐tubulin antibodies. (b) Dose–response curves for centrosome overduplication. U2OS (left panel) and NIH3T3 (right panel) cells were irradiated with the indicated doses at a dose rate of 1 Gy/min and 0.5 mGy/min, and centrosome numbers were counted 96 h after irradiation. (c) Cell survival after protracted exposures to ionizing radiation. U2OS (left panel) and NIH3T3 (right panel) cells were irradiated with the indicated doses at a dose rate of 1 Gy/min and 0.5 mGy/min. (d) Roscovitine, a cyclin‐dependent kinase inhibitor, had no effect on the survival of NIH3T3 cells after exposure to 5 Gy of ionizing radition, but strongly inhibited the induction of centrosome overduplication. Bars represent the standard errors (n = 3).
Excess centrosomes result from overduplication, not frag‐mentation, following IR exposures. The production of an excess number of centrosomes can potentially occur through two mechanisms, the overduplication of centrosomes, or centrosome fragmentation. If centrosomes are produced through the first mechanism, they will contain a normal number of centriole(s), that is, we observed two centrioles in a centrosome when centrioles were immunostained with antibodies against centrin‐2 (Fig. 2a,b). The second mechanism (fragmentation) results in centrosomes containing no centrioles or centrosomes containing a reduced number of centriole(s). After acute exposure to 5 Gy, it was found that cells containing supernumerary centrioles, defined as more than four centrioles per cell, comprised 30% of the population (Fig. 2c, right panel). This result is consistent with the existence of excess centrosomes, defined as more than two centrosomes per cell (Fig. 2c, left panels). Moreover, centrosomes lacking centrioles were not observed. Similar results were obtained after protracted exposures (Fig. 2d), although the frequency of cells containing excess centrosomes was lower than that observed after acute exposures. These results indicate that the presence of excess centrosomes after an IR exposure is due to the overduplication of centrosomes, and that the frequency of fragmentation, if any occurs, is very low.
Figure 2.
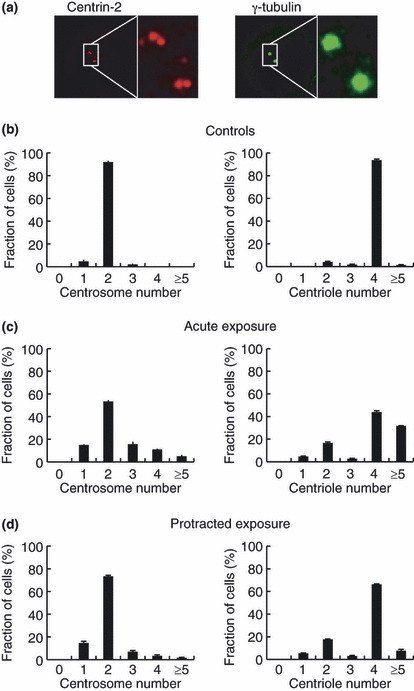
Quantification of the number of centrosomes and centrioles in U2OS cells after acute and protracted exposures to γ‐irradiation. Centrosomes and centrioles were immunostained with anti‐γ‐tubulin antibodies and anti‐centrin‐2 antibodies, respectively (a), and their numbers were quantified in untreated U2OS cells (b). U2OS cells were irradiated with 5 Gy of ionizing radiation at a dose rate of 1 Gy/min and 72 h later, at least 200 cells were counted for quantitative analysis of centrosomes and centrioles (c). Similarly, centrosomes and centrioles were counted after irradiation with 5 Gy delivered at a rate of 0.5 mGy/min (d).
Homologous recombination repair proteins are associated with centrosome overduplication after IR exposures. It was previously reported that HR repair proteins, including NBS1 and BRCA1, are localized at the centrosomes, and that the depletion of these proteins leads to centrosome overduplication.( 12 ) Consistent with this observation, after acute exposures to 5 Gy, the frequency of cells containing excess centrosomes increased threefold in Brca1‐deficient ES cells when compared to irradiated wild‐type ES cells (Brca1+/−) (Figs 3a,S1a,S2a). Protracted exposures to IR resulted in a high frequency of cells containing excess centrosomes. However, this was 80% and 79% of the frequency of cells containing excess centrosomes observed after acute IR exposures of Brca1‐deficient and wild‐type ES cells, respectively (Figs 3a,S2a). Similar results were obtained when using Nbs1‐deficient A31‐1 cells. Nbs1‐deficient cells showed a twofold frequency of cells containing excess centrosomes after acute and protracted exposures to 5 Gy when compared to cells complemented with wild‐type NBS1 cDNA (Figs 3b,S1b,c). As observed in Brca1−/− cells, the frequency of cells containing excess centrosomes decreased to 78% in wild‐type NBS1 cells and to 65% in A31‐1 cells after protracted exposures to IR (Figs 3b,S2b). Thus, the overduplication of centrosomes observed after both types of exposure was considerably enhanced in the absence of the BRCA1 and NBS1 proteins, although the protracted exposures similarly reduced these frequencies in either the presence or absence of HR proteins.
Figure 3.
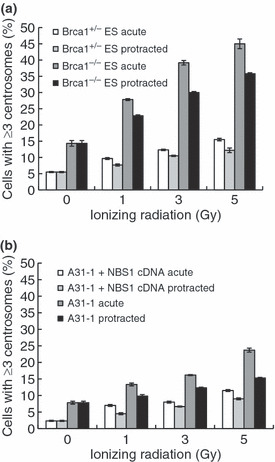
Centrosome overduplication in homologous recombination repair protein‐deficient cells. (a) Brca1−/− and Brca1+/− embryonic stem (ES) cells were stained with anti‐γ‐tubulin and anti‐centrin‐2 antibodies after irradiation with 5 Gy at dose rates of 1 Gy/min and 0.5 mGy/min. Cells were irradiated with the indicated doses and centrosomes were quantified 72 h after irradiation. (b) As in (a), Nbs1−/− fibroblast cells (A31‐1) and complemented A31‐1 + NBS1 cDNA cells were stained, and centrosomes were quantified 72 h after irradiation. Bars represent the standard errors (n = 3).
Non‐homologous end‐joining deficiency slightly potentiates IR‐induced centrosome overduplication. It was previously reported that DNA‐PKcs is localized at the centrosomes,( 13 ) so we examined the overduplication of centrosomes after IR exposures in DNA‐PKcs‐deficient, Ku70‐deficient, and parental wild‐type cells. In contrast to Brca1‐ and Nbs1‐deficient cells, centrosome overduplication was detected only at low levels in non‐irradiated Ku70‐ and DNA‐PKcs‐deficient cells. The frequency of cells containing excess centrosomes was only 10–30% higher in Ku70‐deficient cells than in wild‐type cells after exposure to 5 Gy (Figs 4a,S1d,S3a), even though cell death was enhanced markedly by Ku70 deficiency (Figs 5b, left panel,S1d). Similarly, DNA‐PKcs deficiency provided only marginal effect and the increase in their frequencies was insignificant (Figs 4b,S1e,S3b). In Ku70−/− cells irradiated at a low dose rate, the frequency of cells containing excess centrosomes was 77% of that observed when these cells were irradiated acutely. In addition, all of the excess centrosomes observed in both the irradiated Ku70‐deficient and wild‐type cells contained a normal number of centrioles (Fig. 4a). Therefore, in contrast to Brca1‐ and Nbs1‐deficient cells, the disruption of NHEJ repair proteins slightly enhanced centrosome overduplication after IR exposures.
Figure 4.
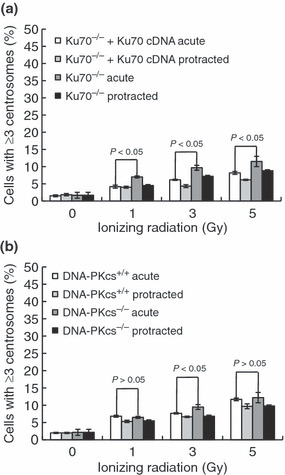
Centrosome overduplication in non‐homologous end‐joining repair protein‐deficient cells. (a) Ku70−/− fibroblast cells and complemented Ku70−/−+Ku70 cDNA cells were stained with anti‐γ‐tubulin antibodies and anti‐centrin‐2 antibodies after irradiation with 5 Gy at dose rates of 1 Gy/min and 0.5 mGy/min. Cells were irradiated with the indicated doses and centrosomes were quantified 72 h after irradiation. *Student’s t‐test indicated a statistically significant increase (P > 0.05). (b) As in (a), DNA‐PKcs−/− fibroblast cells (SCID) and parental CB17 mouse fibroblast cells (DNA‐PKcs+/+) were stained, and centrosomes were quantified 72 h after irradiation. Bars represent the standard errors (n = 3). *Student’s t‐test indicated no statistically significant increase (P < 0.05).
Figure 5.
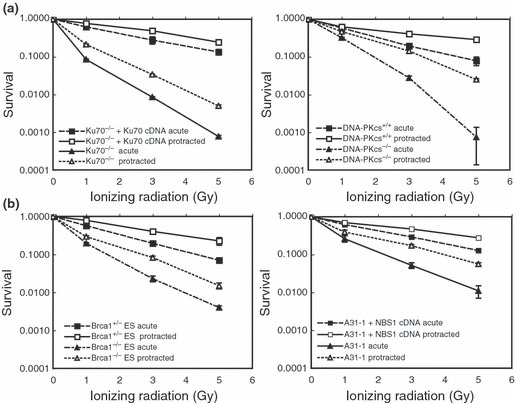
Survival curves for homologous recom‐bination‐deficient and non‐homologous end‐joining‐deficient cells after exposures to acute and protracted irradiations. (a) Survival curves for Ku70‐ and DNA‐PKcs‐deficient cells and the corresponding wild‐type cells irradiated with dose rates of 1 Gy/min (acute) and 0.5 mGy/min (protracted). Bars represent standard errors (n = 3). (b) Survival curves for Brca1‐ and Nbs1‐deficient cells and the corresponding wild‐type cells irradiated at dose rates of 1 Gy/min and 0.5 mGy/min. ES, embryonic stem (cells).
Association between radiosensitivity and centrosome over‐duplication. Because the extent of DNA damage remaining after exposure to IR can vary depending on the cell’s ability to repair such damage, it was of interest to examine relationships between centrosome overduplication and DNA damage, or resulting cell death, in HR‐ and NHEJ‐deficient cells. As expected, cell death after acute exposure to 5 Gy was 100–1000 times higher in Ku70‐ and DNA‐PKcs‐deficient cells than in the corresponding wild‐type cells, and their cell death levels were significantly lower after exposures to protracted IR (Fig. 5a). Similarly, Brca1‐ and Nbs1‐deficient cells were more sensitive than their wild‐type counterparts after an acute exposure to IR, and they were less sensitive to protracted exposures (Fig. 5b). As a result, both NHEJ‐ and HR‐deficient cells showed increased survival after a protracted exposure to IR. This result is consistent with the fact that, although NHEJ is the dominant repair mode in mammalian cells, both NHEJ and HR repair mechanisms are involved in the repair of DSBs.( 20 , 31 )
We next evaluated the frequencies of cells containing centrosome overduplication in radiation sensitive cells on the basis of cell killing, as we described previously.( 32 ) When the frequency of cells containing overduplicated centrosomes was plotted against cell death, which was derived from 3, 4, 5 (1–5 Gy) and from Fig. S1 (8–15 Gy), the curves of all wild‐type cells were similar, regardless of the radiation dose rate (Fig. 6a). This trend was also observed in the curves of NHEJ‐deficient cells, although the frequency of cells containing centrosome overduplication is slightly lower in NHEJ‐deficient cells than that in wild‐type cells (Fig. 6b). However, Brca1 and Nbs1 deficiencies produced differently shaped curves, in which the frequency of centrosome overduplication increased in Brca1‐ and Nbs1‐deficient cells, even in the presence of high levels of cell death (Fig. 6b). These results indicate that NBS1 and BRCA1 are required for centrosome maintenance after exposure to IR, although Ku70 and DNA‐PKcs appears to have marginal roles in centrosome duplication.
Figure 6.
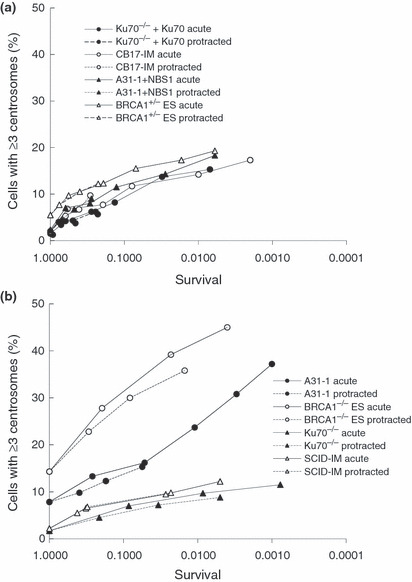
Frequency of cells containing overduplicated centrosomes with respect to cell survival. (a) The frequency of cells containing excess centrosomes in all wild‐type cells was plotted as shown. (b) The frequency of cells containing excess centrosomes in Brca1‐deficient, Nbs1‐deficient, and non‐homologous end‐joining‐deficient cells was plotted as shown. Acute, 1 Gy/min; ES, embryonic stem (cells); Protracted, 0.5 mGy/min.
Discussion
In this report, it was shown that centrosome overduplication after IR exposure is dose‐dependent and is significantly reduced after protracted exposures to IR when compared to acute exposures. Centrosome duplication is triggered by the activation of CDK/cyclin‐A/E, and the production of excess centrosomes is caused by continued centrosome duplication during cell cycle arrest in the G2 phase with the introduction of DNA damage.( 33 ) This model is supported by the observation that the number of cells containing excess centrosomes increased during the post‐irradiation period (Fig. 1a, left panel)( 23 ) and the number of excess centrosomes observed after IR exposures decreased in response to treatments with the CDK inhibitor, roscovitine (Fig. 1d).
Ionizing radiation delivers the same number of initial DSBs during both protracted and acute exposures, therefore, the lower rates of cell death observed after protracted exposures could be due to a reduction in the number of DSBs after DNA repair. To determine the relationship between centrosome overduplication and amount of DSBs, or cell killing, the frequency of cells containing excess centrosomes was plotted against cell death levels (Fig. 6). Surprisingly, with different cell lines and different dose rates, these plots showed similar curves (Fig. 6a), and centrosome overduplication frequencies closely correlate with cell death frequencies, although there is no direct causal relationship (Fig. 1b). These results indicate that centrosome overduplication in irradiated cells, under different conditions of repair time (acute or protracted exposures), is associated with the number of DSBs which lead to cell killing. This association is consistent with a mechanistic model: uncoupling between cell cycle arrest and centrosome duplication, as the period of cell cycle arrest depends on the amount of DNA damage.( 34 ) This trend was also observed in DNA‐PKcs‐ and Ku70‐deficient cells, although the frequency of cells containing centrosome overduplication was slightly reduced (Fig. 6b). This might be due to their defects in the rejoining of chromosomal aberration,( 20 , 31 ) which is generated in cells containing centrosome overduplication.( 35 )
Recently, it has been reported that many HR proteins, including NBS1, BRCA1, and BRCA2 can localize at the centrosomes.( 36 , 37 ) It was shown here that the frequency of cells containing excess centrosomes after an IR exposure is considerably enhanced by deficiencies in the HR proteins BRCA1 and NBS1 on the same basis of cell killing, although their frequencies is higher in BRCA1‐deficient cells than in NBS1‐deficient cells (Fig. 6). This different frequency could be due to partial function of NBS1 in NBS cells as a result of the hypomorphic mutation.( 38 ) The elevated frequencies of both cells is an unexpected result, because NBS1 and BRCA1 are checkpoint proteins( 39 ) and thus should abrogate G2 arrest,( 40 ) reducing centrosome overduplication. Consequently, these results suggest that BRCA1‐ and NBS1‐mediated centrosome aberrations are induced by a pathway independent of checkpoint activation. It was previously reported that centrosome overduplication is significantly increased by disruptions in the BRCA1 and NBS1 proteins in the absence of DNA damage.( 12 ) Both proteins are involved in the ubiquitination of γ‐tubulin for the maintenance of centrosomes, a process in which the number of centrosomes present in a cell is likely monitored by BRCA1 and NBS1.( 9 ) Therefore, an absence of their monitoring functions could enhance centrosome overduplication during growth arrest, even though such an arrest may be shorter than in wild‐type cells.
In summary, these results describing DNA repair on centrosome overduplication in both wild‐type and, most likely, NHEJ‐deficient cells, are consistent with a model in which excess centrosomes appearing after exposure to IR are generated by continued centrosome duplication during an induced cell cycle arrest.( 33 ) In addition, a deficiency of either BRCA1 or NBS1 greatly increases centrosome overduplication in irradiated cells, possibly through an alternative pathway, because the absence of either of these two proteins shortens or abolishes the cell cycle arrest after an exposure to IR.( 39 , 40 ) Therefore, overduplication of centrosomes after IR exposure is generated either through the cell cycle arrest or through direct control of centrosome duplication by centrosome‐maintaining protein, such as BRCA1/NBS1. BRCA1 is inactivated in familial breast cancer, and this is observed in approximately 5% of breast cancer patients.( 41 ) Alternatively, in the absence of a mutation, BRCA1 is transcriptionally downregulated under hypoxic conditions.( 42 ) When a patient with breast cancer receives a fractionated dose from a total dose of 40–60 Gy in radiation therapy or post‐mastectomy adjuvant radiation therapy, IR is delivered at 2 Gy/day for 4–6 weeks.( 43 ) Given that centrosome overduplication is persistent over, at least, 144 h after IR exposure (Fig. 1a), the average dose rate for 4–6 weeks was calculated as 1 mGy/min (=10 Gy/weeks), which is comparable with the condition of protracted exposure used in this experiment. Based on these results, it is suggested that an additional risk of malignant progression may exist due to the occurrence of aneuploidy through centrosome overduplication, particularly in BRCA1‐deficient patients.
Supporting information
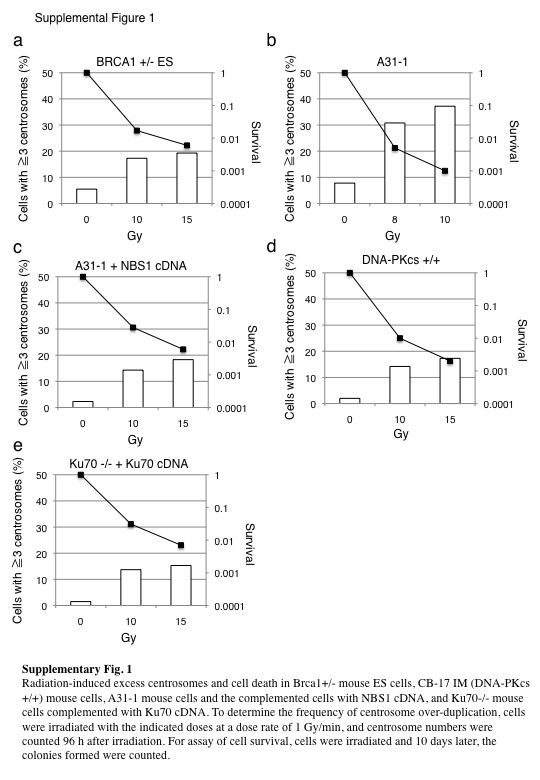
Fig. S1. Radiation‐induced excess centrosomes and cell death.
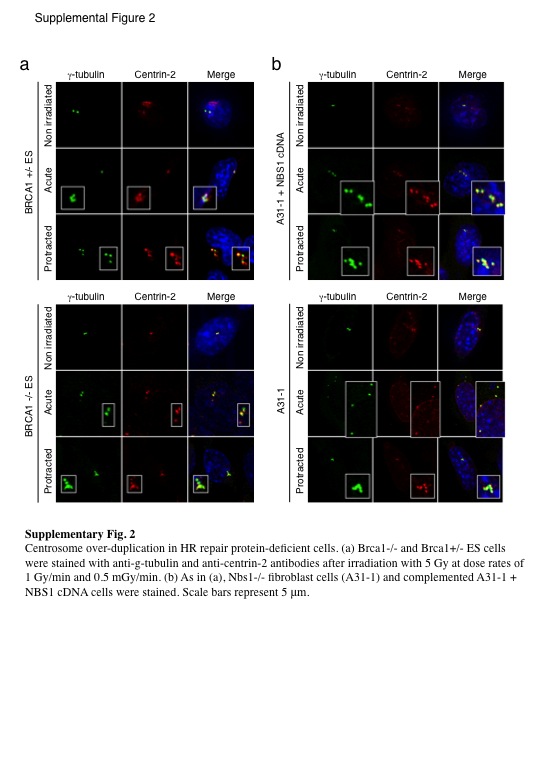
Fig. S2. Centrosome overduplication in homologous recombination repair protein‐deficient cells.
Fig. S3. Centrosome overduplication in non‐homologous end‐joining repair protein‐deficient cells.
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors wish to thank Dr. T. Habu and Mr. D. Oliveira for critical discussions, as well as our colleagues, particularly Ms. R. Sagae and Ms. Y. Hayuka, for excellent technical assistance.
References
- 1. Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer 2007; 7: 911–24. [DOI] [PubMed] [Google Scholar]
- 2. Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2002; 2: 815–25. [DOI] [PubMed] [Google Scholar]
- 3. Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol 2005; 15: 303–11. [DOI] [PubMed] [Google Scholar]
- 4. Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer 2006; 119: 2717–23. [DOI] [PubMed] [Google Scholar]
- 5. Sankaran S, Starita LM, Groen AC, Ko MJ, Parvin JD. Centrosomal microtubule nucleation activity is inhibited by BRCA1‐dependent ubiquitination. Mol Cell Biol 2005; 25: 8656–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology‐directed DNA repair. Mol Cell 1999; 4: 511–8. [DOI] [PubMed] [Google Scholar]
- 7. Starita LM, Machida Y, Sankaran S et al. BRCA1‐dependent ubiquitination of gamma‐tubulin regulates centrosome number. Mol Cell Biol 2004; 24: 8457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sankaran S, Starita LM, Simons AM, Parvin JD. Identification of domains of BRCA1 critical for the ubiquitin‐dependent inhibition of centrosome function. Cancer Res 2006; 66: 4100–7. [DOI] [PubMed] [Google Scholar]
- 9. Parvin JD, Sankaran S. The BRCA1 E3 ubiquitin ligase controls centrosome dynamics. Cell Cycle 2006; 5: 1946–50. [DOI] [PubMed] [Google Scholar]
- 10. Joukov V, Groen AC, Prokhorova T et al. The BRCA1/BARD1 heterodimer modulates ran‐dependent mitotic spindle assembly. Cell 2006; 127: 539–52. [DOI] [PubMed] [Google Scholar]
- 11. Matsuura S, Tauchi H, Nakamura A et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 1998; 19: 179–81. [DOI] [PubMed] [Google Scholar]
- 12. Shimada M, Sagae R, Kobayashi J, Habu T, Komatsu K. Inactivation of the Nijmegen breakage syndrome gene leads to excess centrosome duplication via the ATR/BRCA1 pathway. Cancer Res 2009; 69: 1768–75. [DOI] [PubMed] [Google Scholar]
- 13. Zhang S, Hemmerich P, Grosse F. Centrosomal localization of DNA damage checkpoint proteins. J Cell Biochem 2007; 101: 451–65. [DOI] [PubMed] [Google Scholar]
- 14. Jeggo PA. Risks from low dose/dose rate radiation: what an understanding of DNA damage response mechanisms can tell us. Health Phys 2009; 97: 416–25. [DOI] [PubMed] [Google Scholar]
- 15. Tauchi H, Kobayashi J, Morishima K et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002; 420: 93–8. [DOI] [PubMed] [Google Scholar]
- 16. Stiff T, Reis C, Alderton GK, Woodbine L, O’Driscoll M, Jeggo PA. Nbs1 is required for ATR‐dependent phosphorylation events. EMBO J 2005; 24: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burma S, Chen BP, Chen DJ. Role of non‐homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006; 5: 1042–8. [DOI] [PubMed] [Google Scholar]
- 18. Kobayashi J, Tauchi H, Sakamoto S et al. NBS1 localizes to gamma‐H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 2002; 12: 1846–51. [DOI] [PubMed] [Google Scholar]
- 19. Komatsu K, Kubota N, Gallo M, Okumura Y, Lieber MR. The scid factor on human chromosome 8 restores V(D)J recombination in addition to double‐strand break repair. Cancer Res 1995; 55: 1774–9. [PubMed] [Google Scholar]
- 20. Takata M, Sasaki MS, Sonoda E et al. Homologous recombination and non‐homologous end‐joining pathways of DNA double‐strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 1998; 17: 5497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura H, Yasui Y, Saito N, Tachibana A, Komatsu K, Ishizaki K. DNA repair defect in AT cells and their hypersensitivity to low‐dose‐rate radiation. Radiat Res 2006; 165: 277–82. [DOI] [PubMed] [Google Scholar]
- 22. Ishizaki K, Hayashi Y, Nakamura H, Yasui Y, Komatsu K, Tachibana A. No induction of p53 phosphorylation and few focus formation of phosphorylated H2AX suggest efficient repair of DNA damage during chronic low‐dose‐rate irradiation in human cells. J Radiat Res (Tokyo) 2004; 45: 521–5. [DOI] [PubMed] [Google Scholar]
- 23. Sato N, Mizumoto K, Nakamura M et al. A possible role for centrosome overduplication in radiation‐induced cell death. Oncogene 2000; 19: 5281–90. [DOI] [PubMed] [Google Scholar]
- 24. Sato N, Mizumoto K, Nakamura M, Tanaka M. Radiation‐induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp Cell Res 2000; 255: 321–6. [DOI] [PubMed] [Google Scholar]
- 25. Duensing A, Liu Y, Tseng M, Malumbres M, Barbacid M, Duensing S. Cyclin‐dependent kinase 2 is dispensable for normal centrosome duplication but required for oncogene‐induced centrosome overduplication. Oncogene 2006; 25: 2943–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bourke E, Dodson H, Merdes A et al. DNA damage induces Chk1‐dependent centrosome amplification. EMBO Rep 2007; 8: 603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mendiola‐Cruz MT, Morales‐Ramirez P. Repair kinetics of gamma‐ray induced DNA damage determined by the single cell gel electrophoresis assay in murine leukocytes in vivo. Mutat Res 1999; 433: 45–52. [DOI] [PubMed] [Google Scholar]
- 28. Riballo E, Kuhne M, Rief N et al. A pathway of double‐strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma‐H2AX foci. Mol Cell 2004; 16: 715–24. [DOI] [PubMed] [Google Scholar]
- 29. Saladino C, Bourke E, Conroy PC, Morrison CG. Centriole separation in DNA damage‐induced centrosomes amplification. Environ Mol Mutagen 2009; 50: 725–32. [DOI] [PubMed] [Google Scholar]
- 30. Bedford JS, Mitchell JB. Dose‐rate effects in synchronous mammalian cells in culture. Radiat Res 1973; 54: 316–27. [PubMed] [Google Scholar]
- 31. Essers J, Hendriks RW, Swagemakers SM et al. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell 1997; 89: 195–204. [DOI] [PubMed] [Google Scholar]
- 32. Kawamura K, Fujikawa‐Yamamoto K, Ozaki M et al. Centrosome hyperamplification and chromosomal damage after exposure to radiation. Oncology 2004; 67: 460–70. [DOI] [PubMed] [Google Scholar]
- 33. Shoji S, Watanabe H, Katoh O et al. Developmental malformations and intrauterine deaths in gamma‐ray‐irradiated scid mouse embryos. Int J Radiat Biol 1998; 73: 705–9. [DOI] [PubMed] [Google Scholar]
- 34. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9: 297–308. [DOI] [PubMed] [Google Scholar]
- 35. Rosario CO, Ko MA, Haffani YZ et al. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc Natl Acad Sci USA 2010; 107: 6888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Date O, Katsura M, Ishida M et al. Haploinsufficiency of RAD51B causes centrosome fragmentation and aneuploidy in human cells. Cancer Res 2006; 66: 6018–24. [DOI] [PubMed] [Google Scholar]
- 37. Nakanishi A, Han X, Saito H et al. Interference with BRCA2, which localizes to the centrosome during S and early M phase, leads to abnormal nuclear division. Biochem Biophys Res Commun 2007; 355: 34–40. [DOI] [PubMed] [Google Scholar]
- 38. Matsuura S, Kobayashi J, Tauchi H, Komatsu K. Nijmegen breakage syndrome and DNA double strand break repair by NBS1 complex. Adv Biophys 2004; 38: 65–80. [PubMed] [Google Scholar]
- 39. Van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 2009; 19: 207–17. [DOI] [PubMed] [Google Scholar]
- 40. Buscemi G, Savio C, Zannini L et al. Chk2 activation dependence on Nbs1 after DNA damage. Mol Cell Biol 2001; 21: 5214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baselga J, Norton L. Focus on breast cancer. Cancer Cell 2002; 1: 319–22. [DOI] [PubMed] [Google Scholar]
- 42. Bindra RS, Gibson SL, Meng A et al. Hypoxia‐induced down‐regulation of BRCA1 expression by E2Fs. Cancer Res 2005; 65: 11597–604. [DOI] [PubMed] [Google Scholar]
- 43. Gebski V, Lagieva M, Keech A, Simes J, Langiands AO. Survival effects of postmastectomy adjuvant radiation therapy using biologically equivalent doses: a clinical perspective. J Natl Cancer Inst 2006; 98: 26–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Radiation‐induced excess centrosomes and cell death.
Fig. S2. Centrosome overduplication in homologous recombination repair protein‐deficient cells.
Fig. S3. Centrosome overduplication in non‐homologous end‐joining repair protein‐deficient cells.
Supporting info item
Supporting info item
Supporting info item