

Abstract
Y‐box‐binding protein‐1 (YB‐1) is a multifunctional protein involved in the regulation of transcription, translation, and mRNA splicing. In recent years, several laboratories have demonstrated that YB‐1 is directly involved in the cellular response to genotoxic stress. Importantly, YB‐1 is increased in tumor cell lines resistant to cisplatin, and the level of nuclear expression of YB‐1 is predictive of drug resistance and patient outcome in breast tumors, ovarian cancers, and synovial sarcomas. YB‐1 binds to several DNA repair enzymes in vitro including human endonuclease III (hNTH1). Human NTH1 is a bifunctional DNA glycosylase/apurinic/apyrimidinic lyase involved in base excision repair. In this study, we show that YB‐1 binds specifically to the auto‐inhibitory domain of hNTH1, providing a mechanism by which YB‐1 stimulates hNTH1 activity. Indeed, YB‐1 strongly stimulates in vitro the activity of hNTH1 toward DNA duplex probes containing oxidized bases, lesions prone to be present in cisplatin treated cells. We also observed an increase in YB‐1/hNTH1 complex formation in the mammary adenocarcinoma MCF7 cell line treated with UV light and cisplatin. Such an increase was not observed with mitomycin C or the topoisomerase I inhibitor camptothecin. Accordingly, antisense RNAs against either YB‐1 or hNTH1 increased cellular sensitivity to UV and cisplatin but not to mitomycin C. An antisense RNA against YB‐1 increased camptothecin sensitivity. In contrast, an antisense against hNTH1 did not. Finally, siRNA against hNTH1 re‐established cytotoxicity in otherwise cisplatin‐resistant YB‐1 overexpressing MCF7 cells. These data indicate that hNTH1 is a relevant target to potentiate cisplatin cytotoxicity in YB‐1 overexpressing tumor cells. (Cancer Sci 2008; 99: 762–769)
YB‐1 (Y‐box‐binding protein‐1) was originally described as a transcription regulator that binds to inverted CCAAT box DNA sequences present in the control regions of several genes.( 1 ) Subsequent studies indicated that YB‐1 is a multifunctional protein that also affects the splicing and the translation of specific mRNAs.( 2 , 3 ) In recent years, several laboratories have demonstrated that YB‐1 is also directly involved in the cellular response to genotoxic stress. Upon UV, mitomycin C, or cisplatin treatments, YB‐1 translocates from the cytoplasm to the nucleus and is known to bind modified nucleic acids.( 4 , 5 ) Interestingly, nuclear YB‐1 is spontaneously increased in cultured cell lines resistant to cisplatin. In fact, the nuclear level of YB‐1 is predictive of drug resistance and patient outcome in breast tumors, ovarian cancers, and synovial sarcomas.( 6 , 7 , 8 , 9 , 10 ) Accordingly, it has been shown that depletion of YB‐1 protein expression with specific antisense RNA in such cells results in increased sensitivity to cisplatin.( 11 ) YB‐1 affinity chromatography experiments have indicated that it is associated directly or indirectly with several DNA repair proteins in vitro.( 12 ) One such DNA repair protein is the human endonuclease III or hNTH1.( 13 , 14 ) This enzyme is involved in base excision repair. It is a bifunctional DNA glycosylase/apurinic/apyrimidinic lyase which removes either hydrated, reduced, or oxidized bases from the DNA backbone as the initial step of base excision repair.( 13 ) YB‐1 stimulates the hNTH1 activity in vitro. ( 13 ) These results suggest that YB‐1 modulates hNTH1 activity during DNA damage. However, it is unknown whether hNTH1 is required for cisplatin resistance in YB‐1 overexpressing tumor cells. In this report, we present a mechanism by which YB‐1 stimulates hNTH1 activity, and we show for the first time that siRNA specific to hNTH1 mRNA increases cisplatin‐sensitivity of YB‐1 overexpressing cells, and indicate that hNTH1 enzymatic activity can be a potential target against cisplatin‐resistant tumors overexpressing nuclear YB‐1.
Materials and Methods
Cell lines and antibodies. The human MCF7 breast adenocarcinoma cell line was maintained in DMEM supplemented with either 10% fetal bovine serum at 37°C in atmosphere of 5% CO2. When indicated, cells were treated with increasing doses of either cisplatin, UV light, camptothecin, or mitomycin C. The antiproliferative effect of the treatments were measured by standard MTT cell survival assays. A polyclonal antibody against the YB‐1 peptide (residues 299–313; CDGKETKAADPPAENS) was generated in rabbits as described before.( 15 ) In addition, a rabbit polyclonal antibody against YB‐1 was purchased from AbCam. (Cambridge, MA, USA). Antibodies against hNTH1 were purchased from Oncogene Research Products (Boston, MA, USA). Finally, all horseradish peroxidase‐conjugated secondary antibodies were purchased from AmershamPharmacia (Piscataway, NJ, USA). The above antibodies were used as indicated by the manufacturers. Western blotting analyses were performed as described.( 12 )
Plasmid. The YB‐1 cDNA was cloned in the pNTAP‐B vector in‐frame with the TAP‐epitope consisting of both calmodulin and streptavidin epitopes (Stratagene, LaJolla, CA, USA). The YB‐1 cDNA was also cloned into pCMV‐SPORT6 vector for mammalian cell expression. Several GST‐YB‐1 fusion proteins were constructed for the in vitro binding assay as described before.( 12 ) The human NTH1 cDNA was cloned into the PCRII plasmid from Invitrogen (Carlsbad, CA, USA). Subsequently, it was cloned into the EcoRI site of the pGEX‐2TK vector. In addition, the hNTH1 cDNA was cut with EcoRI and PvuII (amino acid residues 1–94 of hNTH1) and cloned into the appropriate modified restriction sites in the pGEX‐2TK vector. The remaining PvuII‐EcoRI 3′ fragment (residues 95–312) was cloned into the pGEX‐3X vector. The hNTH1 cDNA was also cloned into the pET24d vector in‐frame with the Histidine tag and into the pNTAP‐B vector (Stratagene, LaJolla, CA, USA) in‐frame with the TAP‐epitope. Finally, to knock down the expression of YB‐1, a small hairpin sequence specific to YB‐1 (5′‐GGTCATCGCAACGAAGGTT‐3′) was cloned into the pSuper vector (GeneScript, Seattle, WA, USA) to generate the pSuperYB‐1 plasmid.
Protein purifications and chromatography experiments. Purifications of the different GST‐hNTH1 and GST‐YB‐1 constructs for affinity chromatography experiments with MCF7 cells were performed as described previously.( 12 ) The His‐hNTH1 protein was produced in the BL21 bacterial strain. Bacteria were lyzed in a denaturing buffer (50 mmol NaCl, 50 mmol phosphate pH 7.5, 4 mol urea) and the His‐hNTH1 protein was purified by chromatography on a nickel column (ProBond; Invitrogen, Carlsbad, CA, USA) followed by gel filtration on a Superdex‐200 column (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Purified His‐hNTH1 was present in fraction 29 (0,5 mL fractions). Protein concentration was determined using the Bradford assay. The purity of the proteins was analyzed on Coomassie‐stained SDS‐polyacrylamide gels. For the in vitro binding assays, beads containing either GST alone or GST‐YB‐1 were incubated overnight with the indicated amounts of purified hNTH1 in a binding buffer containing 50 mmol Tris (pH 8.0), 120 mmol NaCl, 0.5% NP‐40, and a cocktail of protease inhibitors (complete protease inhibitor cocktail tablet from Boehringer Mannheim, Ridgefield, CT, USA). The next day, beads were washed four times with binding buffer. Proteins bound to the affinity matrices were analyzed by Western blotting.
In vitro translation and protein binding. The IVTT/GST‐binding assay was done as described previously.( 16 ) Full length and fragments of hNTH1 open reading frame were cloned into pCR‐BluntII‐TOPO (Invitrogen, Carlsbad, CA, USA). 35S‐labeled hNTH1 protein was generated by in vitro transcription/translation (IVTT) using the TNT Coupled Reticulocyte Lysate System (Promega, Madison, WI, USA) as described by the manufacturer. YB‐1 open reading frame was cloned into pGEX vector (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA) and transformed into BLR(DE3)pLysS (Novagen, Madison, WI, USA) cells to facilitate the expression of a GST‐YB‐1 fusion protein. Cells were induced with isopropyl‐1‐thio‐β‐D‐galactopyranoside (IPTG) for 2 h to induce expression, lyzed, and treated with DNAseI for 30 min. Subsequently, the lysate was centrifuged, and the supernatant was incubated with glutathione‐agarose beads (Sigma, St. Louis, MO, USA) on a rocking platform at 4°C for 1 h. The beads were centrifuged, washed, and exposed to 35S‐labeled proteins generated by IVTT as described above for 1 h at 4°C. Subsequently, the beads were centrifuged, washed, resuspended in SDS loading buffer, and separated by SDS‐PAGE. The bands were visualized and quantitated using a Molecular Dynamics PhosphorImager system (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA) as described.( 16 ) GST without YB‐1 expressed and treated in the same way was used as a negative control in each experiment.
Nuclease assays. The DNA substrates used for the strand separation and nuclease assays are depicted in Table 1. One strand was labeled with T4 polynucleotide kinase and [γ‐32P] ATP and annealed to its complementary strand as described previously.( 12 ) DNA with cisplatin adducts were created by incubating oligonucleotides with cisplatin overnight as described previously.( 17 ) DNA substrate were incubated with the proteins as indicated in the figure legends for 30 min at 37°C in the reaction buffer (40 mmol Tris‐HCl, pH 7.5, 4 mmol MgCl2, 5 mmol DTT, 2 mmol ATP, and 0.1 µg/mL bovine serum albumin). Four µL of loading buffer was added to the reaction (40% glycerol, 50 mmol sodium EDTA, 2% SDS, and 1% bromophenol blue, pH 8.0) and the DNA was analyzed on a denaturing gel (14% polyacrylamide, 8 mol urea in TBE) by autoradiography. The TAP‐hNTH1 protein was purified from a stable MCF7 clone expressing this protein construct with a TAP purification kit (Stratagene, LaJolla, CA, USA) as described by the manufacturer. MCF7 cells containing an empty TAP vector were used as controls. When indicated, the TAP‐hNTH1 was incubated in the presence of purified YB‐1 construct in the nuclease assay. GST‐YB‐1 beads were treated with biotinylated thrombin (Novagen, Madison, WI, USA) for 2 h at room temperature in thrombin cleavage buffer (20 mmol Tris‐HCl pH 8.4, 150 mmol NaCl, 2.5 mmol CaCl2). Beads were spun down and the supernatant was kept for the next step. Thrombin was captured by incubation with streptavidine agarose (Novagen, Madison, WI, USA) for 2 h on a rocking platform at room temperature. Agarose beads were spun down and YB‐1 protein from the supernatant was concentrated onto Centricon‐30 filters (Amicon, Bedford, MA, USA). Protein concentration was determined using the Bradford assay.
Table 1.
DNA structures
| Name | Nucleotides |
|---|---|
| Control & Cisplatin treated | ATATACCGCGGCCGGCCGATCAAGCTTATT |
| TATATGGCGCCGGCCGGCTAGTTCGAATAA | |
| 8‐oxoG | ATATACCGCG † CCGGCCGATCAAGCTTATT |
| TATATGGCGCGGGCCGGCTAGTTCGAATAA |
8‐oxoG
Transfections of MCF7 cells. MCF7 cells were transfected with a siRNA specific to hNTH1 (NTHL1 Stealth Select RNAi; HSS107332) with the Lipofectamine 2000 transfection reagent as described by the manufacturer (Invitrogen, Carlsbad, CA, USA). Cells were lyzed in RIPA buffer after 48 h for Western blot analysis. The pCMV‐YB‐1 expression vector or the pSuperYB‐1 plasmid were transfected into MCF7 cells with the Amaxa nucleofector kit (Amaxa Biosystems, Gaithersburg, MD, USA) when indicated in the figure legends. Expression of YB‐1 was already detectable by Western analyses 5 h after transfection (data not shown). Knock down of YB‐1 with the shRNA (pSuperYB‐1) was achieved within 48 h.
FACS analysis. MCF7 cells were transfected with either an empty expression vector or a YB‐1 expression vector. After 48 h, cells were fixed in 50% ethanol overnight. Cells were then washed in phosphate‐buffered saline (PBS) and incubated for 30 min at 37°C in a buffer containing propidium iodide and RNAses. Cells were then analyzed on a Beckman‐Coulter Epics Elite ESP (Cambridge, MA, USA) flow activated cell sorter. Data were analyzed with the MultiCycle software (Phoenix Flow System, San Diego, CA, USA).
Results
Previous reports have indicated that YB‐1 stimulates hNTH1 activity in vitro.( 13 , 14 ) However, a detailed analysis of the YB‐1/hNTH1 interaction had not been performed. Using a panel of GST‐YB‐1 deletion mutants, we mapped the interaction region in YB‐1 responsible for the association with hNTH1. We first performed a series of affinity chromatography experiments on MCF7 cell lysates with different GST‐YB‐1 fusion constructs as described previously.( 12 ) Proteins bound to the different affinity matrices were eluted and analyzed by Western blotting with an antibody against hNTH1 protein (Fig. 1a). A summary of the data is depicted in Figure 1b. As indicated, a region of YB‐1 peptide containing amino acids 51–129 was a minimum required for its association with hNTH1 in our experiments. Residues 51–129 form the YB‐1 cold shock domain and are essential for binding strongly to nucleic acids.( 18 ) To determine whether nucleic acid was responsible in the total lysate for the YB‐1/hNTH1 association, purified hNTH1 (Fig. 1c,d) was incubated with the GST‐YB‐1 matrix. During the purification procedure, nucleases were added to all buffers to eliminate nucleic acid molecules. As shown in Figure 1e, purified hNTH1 bound the GST‐YB‐1 beads but not the GST beads alone. The cold shock domain (CSD = amino acids 51–129) also bound hNTH1 but with a much weaker intensity in the absence of DNA or RNA molecules in the binding buffer. These results indicate that purified hNTH1 binds directly to YB‐1 in the absence of nucleic acids.
Figure 1.
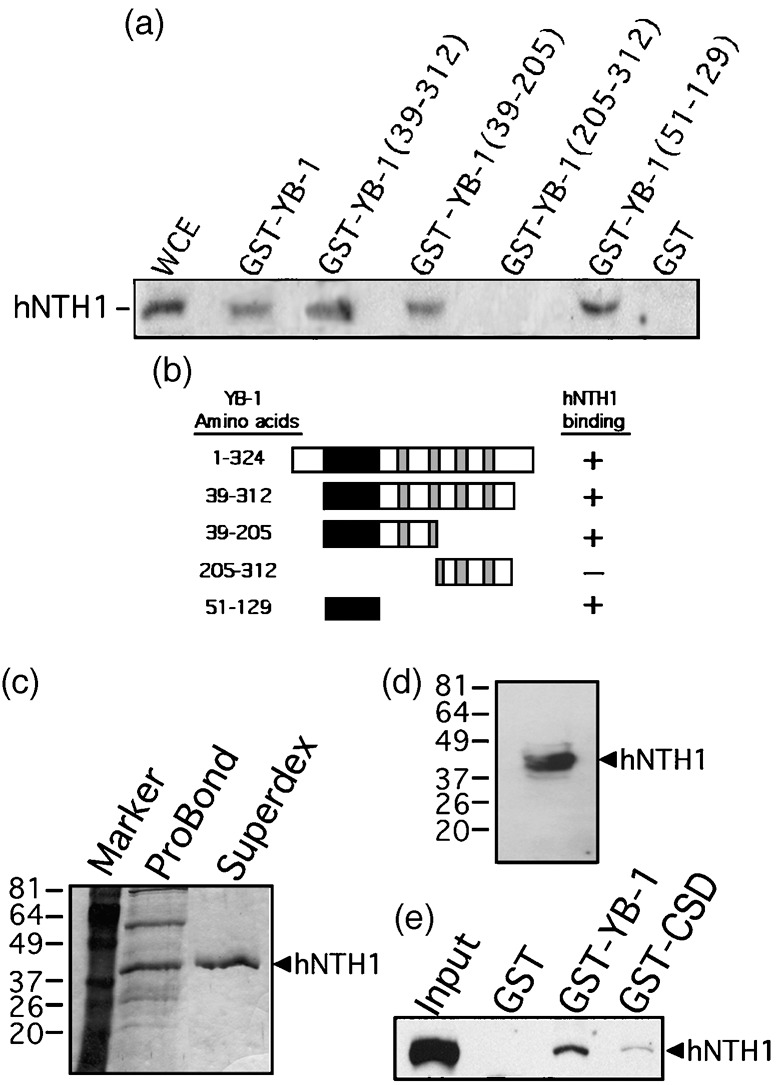
Interaction of different domains of Y‐box‐binding protein‐1 (YB‐1) with human endonuclease III (hNTH1) in total cell extract. (a) Immunoblot against hNTH1 proteins bound to different GST‐YB‐1 affinity Sepahrose beads. Human MCF7 whole cell extracts (WCE) were incubated with either 50 µg of GST‐YB‐1 or GST‐linked glutathione‐sepharose beads overnight. Proteins bound to the affinity beads were analyzed by SDS‐PAGE with antibodies against hNTH1. (b) Schematic representation of different YB‐1 polypeptides that were used in the YB‐1 affinity chromatography experiments. The black box is the cold shock domain and the gray boxes are the basic/acidic cluster domains. The amino acid residues of the YB‐1 fragments used in this study are indicated on the left. hNTH1 binding is indicated on the right by the ‘+’ sign. The ‘–’ sign indicates no binding detected. (c) Coomassie staining of a gel containing purified hNTH1 after the nickel column step (ProBond resin) and after fractionation on Superdex‐200 (fraction number 29). The molecular weight, in kDa, is indicated on the left. (d) Western blot of purified hNTH1 after the final Superdex‐200 step (fraction 29) with an antibody against hNTH1. (e) Interaction of purified hNTH1 (after the final Superdex‐200 step) with 50 µg of GST‐YB‐1, GST‐CSD (cold shock domain of YB‐1; amino acids 51–129), or GST‐linked glutathione‐sepharose beads.
The hNTH1 contains a well conserved catalytic domain and an auto‐inhibitory N‐terminal tail.( 19 ) To determine which part of the hNTH1 protein interacts with YB‐1, total cell lysate from MCF7 cells was incubated with different GST‐hNTH1 constructs. The affinity beads were washed extensively and the proteins bound to the GST‐hNTH1 constructs were analyzed on SDS‐PAGE with an antibody against YB‐1. As indicated in Figure 2a, YB‐1 bound the full length and to the first 94 amino acid residues of hNTH1. No signal was detected with GST alone or a GST chimeric protein containing residues 95–312 of hNTH1. These results were confirmed by incubating GST‐YB‐1 proteins with different hNTH1 peptide fragments that were 35S‐labeled (Fig. 2b). Full‐length hNTH1 protein selectively bound to GST‐YB‐1 beads (first lane) but not to the GST control (second lane). Similarly, fragments containing amino acids 1–176 and 1–250 bound GST‐YB‐1 (fifth and seventh lanes) but not GST alone. In contrast, a fragment missing the first 136 amino acids could not bind GST‐YB‐1 (third lane from the left). Figure 2c gives a summary of the data. We concluded that YB‐1 binds to the auto‐inhibitory N‐terminal tail of hNTH1.
Figure 2.
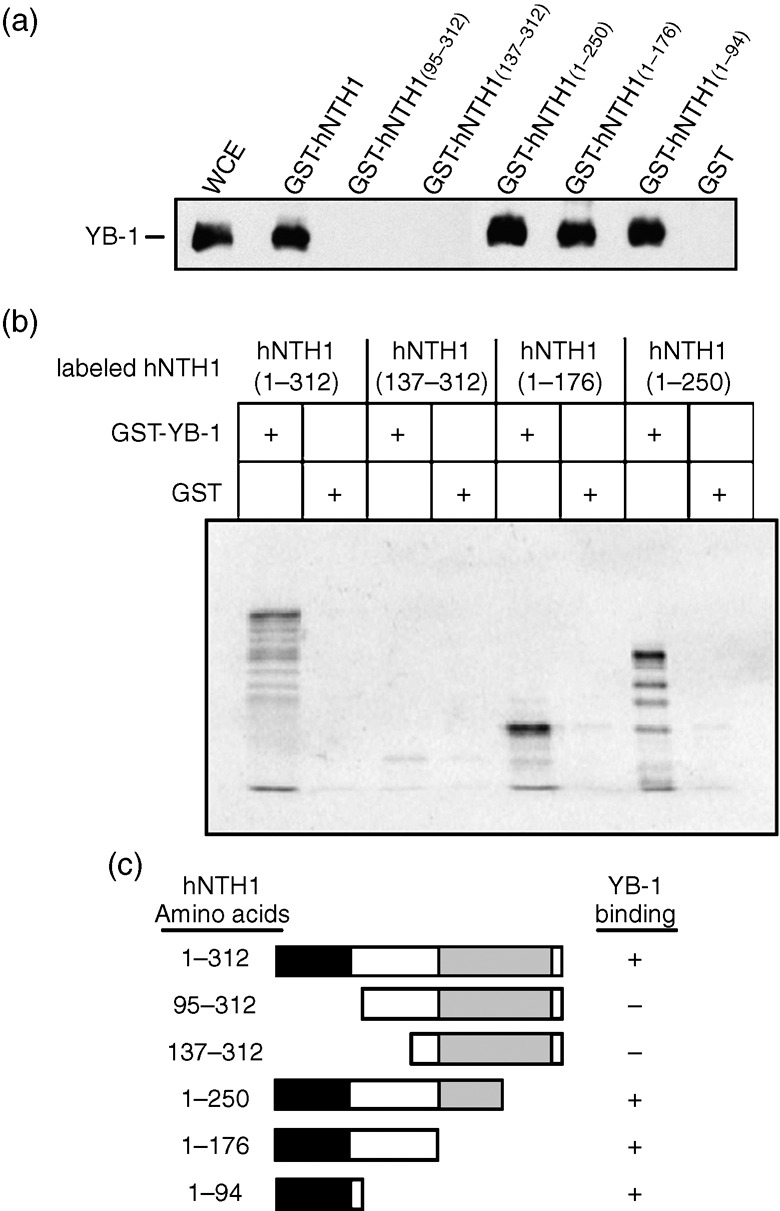
Interaction of the auto‐inhibitory domain of human endonuclease III (hNTH1) with the Y‐box‐binding protein‐1 (YB‐1) protein. (a) Immunoblot against YB‐1 proteins bound to different GST‐hNTH1 affinity Sepahrose beads. Human MCF7 cell extracts (WCE) were incubated with either 50 µg of GST‐hNTH1 constructs or GST‐linked glutathione‐sepharose beads overnight. Proteins bound to the affinity beads were analyzed by SDS‐PAGE with antibodies against YB‐1. (b) IVTT/GST interaction assay. In vitro‐transcribed/translated 35S‐labeled hNTH1 protein fragments bound to YB‐1‐GST affinity beads were separated by SDS‐PAGE and visualized using a PhosphoImager. (c) Schematic representation of the different hNTH1 polypeptides that were used in the hNTH1 affinity chromatography experiments. The black box is the auto‐inhibitory domain and the gray box is the catalytic domain. The amino acid residues of the hNTH1 fragments used in this study are indicated on the left. YB‐1 binding is indicated on the right by the ‘+’ sign. The ‘–’ sign indicates no binding detected.
To examine the impact of purified YB‐1 on the nuclease activity of cellular hNTH1, a stable cell line expressing a TAP‐hNTH1 construct was generated. The TAP‐hNTH1 can be purified from MCF7 cells with a streptavidin column followed by elution with a biotin solution. The eluted hNTH1 complex was then incubated with a radioactive cisplatin‐containing duplex DNA. No activity was detected with this TAP‐hNTH1 (data not shown). The TAP‐hNTH1 did not increase the nuclease activity of purified YB‐1( 12 , 18 ) toward the radioactive cisplatin‐containing duplex probe (data not shown). However, the MCF7‐eluted TAP‐hNTH1 proteins exhibited nuclease activities on a radioactive duplex containing 8‐oxoguanine residues (Fig. 3a). This is important information as cisplatin is known to induce oxidative stress in cells.( 20 , 21 ) No activity was detected using a similar probe without 8‐oxoguanine residue demonstrating the specificity of hNTH1 for oxidized DNA duplex (data not shown). To verify that the nuclease activity was from the TAP‐hNTH1 itself and not from a contaminating enzyme in the MCF7 eluate, the TAP‐hNTH1 expressing cells were transfected with a small interference RNA specific to hNTH1. The siRNA specific to hNTH1 decreased both protein levels (endogenous hNTH1 and TAP‐hNTH1) (Fig. 3b) and nuclease activities toward the radioactive duplex (Fig. 3a, panels on the left). A control siRNA (with scrambled sequence) had no impact on protein levels and hNTH1 enzymatic activity. The same purification method from MCF7 expressing an empty TAP vector exhibited no nuclease activity toward the same DNA probe (Fig. 3a, panel on the right). We then examined the impact of purified YB‐1 peptides on the TAP‐hNTH1 nuclease activity. We assayed a full length YB‐1 peptide and a YB‐1 peptide that does not bind to hNTH1 (amino acid 205–312 of YB‐1). The purified YB‐1 peptides were added to the nuclease reaction containing 0 or 0.2 µg of TAP‐hNTH1 eluate (Fig. 3c). This amount of TAP‐hNTH1 eluate was the minimum of protein required to see cleavage of the DNA duplex (second lane from the left). Upon addition of full length YB‐1, there was a huge increase (12‐fold) in cleavage activity (forth lane from the left). Purified YB‐1 alone exhibited a weak endonuclease activity by itself (fifth lane) as described before.( 12 ) The C‐terminus peptide of YB‐1 had no significant effect on the TAP‐hNTH1 cleavage activity (lanes 3 and 6). Finally, nuclease assays with eluate from MCF7 cells containing an empty TAP vector did not exhibit cleavage activity and did not increase YB‐1 associated nuclease activity. These results confirm previous findings demonstrating that YB‐1 increases hNTH1 activity by binding to it in vitro.( 13 )
Figure 3.
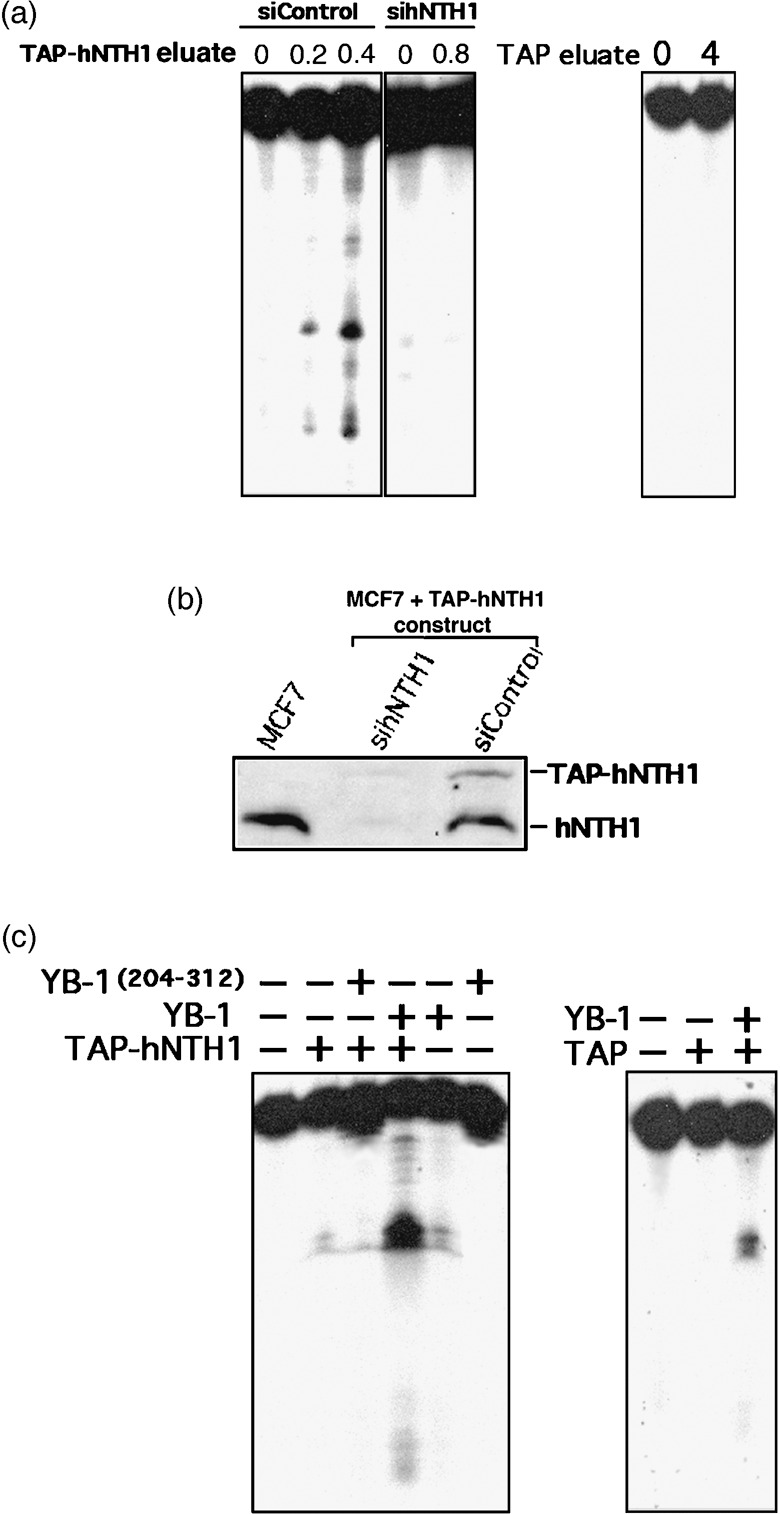
Nuclease activity of eluted TAP‐hNTH1 in the presence of purified Y‐box‐binding protein‐1 (YB‐1). (a) Tap‐hNTH1 MCF7 expressing cells were transfected with either siRNA control (siControl) or siRNA against human endonuclease III (hNTH1) (panels on the left). Two days later the TAP‐hNTH1 complex was purified on streptavidin column and eluted with biotin. Eluted TAP‐hNTH1 proteins (0, 0.2, or 0.4 µg) were incubated for 30 min at 37°C as described in ‘Materials and Methods’ with a radioactive DNA duplex containing 8‐oxoguanine residues. Reactions were stopped in the appropriate dye buffer and cleaved DNA products were analyzed on 14% denaturing polyacrylamide gels. The panel on the right represents nuclease assays with or without eluate from MCF7 cells containing an empty TAP vector (0 or 0.8 µg). (b) Western blot showing intracellular levels of endogenous hNTH1 and TAP‐hNTH1 after transfection with a siRNA specific to hNTH1 or with a scrambled control siRNA. The first lane contained whole lysate from untransfected parental MCF7 cells. Each lane contains 75 µg of whole cell lysate. (c) Nuclease activity with TAP‐hNTH1 in the absence or presence of either a full length YB‐1 peptide (2 µg) or a purified peptide containing amino acids 205–312 of YB‐1 (2 µg). (Panel on the left.) Note that YB‐1(205–312) does not bind to hNTH1. The panel on the right represents nuclease assays with eluate from MCF7 cells containing an empty TAP vector (0.8 µg) in the presence or absence of YB‐1 (2 µg).
To determine whether the YB‐1/hNTH1 association is physiologically relevant in cells, coimmunoprecipitation experiments were performed with the MCF7 mammary tumor cell line treated with different DNA‐damaging conditions. We also quantified the amount of YB‐1/hNTH1 complexes in such treated cells. As none of the polyclonal antibody immunoprecipitated YB‐1 well from total cell lysates, a stable MCF7 cell line expressing a TAP‐YB‐1 construct was generated. This tagged YB‐1 can also be precipitated with steptavidin beads. As indicated in Figure 4a, the TAP‐YB‐1 was expressed at levels similar to the endogenous YB‐1 on an immunoblot. Importantly, the Tap‐YB‐1 precipitate contained the hNTH1 protein (Fig. 4b). Transfected cells were treated with either increasing doses of cisplatin, UV light, camptothecin, or mitomycin C for 4 h, and the amount of hNTH1 associated with TAP‐YB‐1 was quantified from the Western blots. These experiments were repeated twice, and Figure 4c–f gives examples of such experiments. As indicated in Figure 4c, scanning analyses of the bands in the blots showed a significant two‐fold increase in YB‐1/hNTH1 complex formation after the highest concentration of cisplatin treatment. There was also a three‐fold increase in YB‐1/hNTH1 complex formation after the highest UV irradiation dose tested (Fig. 4d). Finally, there was no increase in the amount of YB‐1/hNTH1 complex after camptothecin or mitomycin C treatments compared to untreated MCF7 cells (Fig. 4e,f). Higher concentrations of these latter drugs were too toxic for these cells (data not shown). These results indicate that there is an increase in YB–1/hNTH1 association in the MCF7 mammary tumor cell line under specific DNA‐damaging conditions.
Figure 4.

Co‐precipitation of human endonuclease III (hNTH1) protein with TAP‐YB‐1 protein under different DNA‐damaging conditions. (a) Detection of endogenous hNTH1 and Y‐box‐binding protein‐1 (YB‐1) proteins in whole cell lystes after transfection of TAP‐YB‐1 construct into MCF7 cells. (b) Detection of TAP‐YB‐1 and hNTH1 proteins after streptavidin affinity precipitation of the TAP constructs. (c–f) Detection of TAP‐YB‐1 and hNTH1 proteins after streptavidin affinity precipitation of the TAP constructs in untreated cells and in MCF7 cells treated (4 h) with either increasing doses of cisplatin (c), UV light (d), mitomycin C (e), or camptothecin (f). Under each Western blot, scanning analyses of the Western blots (from two independent experiments) are presented. Data are expressed as the mean (±SD) ratio of hNTH1 signals over the affinity‐precipitated TAP‐YB‐1 signals.
We next investigated the effect of knocking down either YB‐1 or hNTH1 expression in MCF7 cells under different DNA‐damaging treatments. MCF7 cells were first transfected with a vector expressing a shRNA specific to YB‐1. Forty‐eight hours after transfection, cells were treated with either different doses of cisplatin, UV light, camptothecin, or mitomycin C for 18 h. The antiproliferative effect of the treatments was measured by the standard MTT assay. As indicated in Figure 5a, YB‐1 expression levels dropped by 90% compared to a control empty vector. Figure 5(b–d) shows that shRNA against YB‐1 significantly increased the sensitivity of cells to cisplatin, UV light, and camptothecin. Knock‐down of YB‐1 did not significantly sensitize cells to mitomycin C compared to control transfections in MCF7 cells (Fig. 5e). Even higher concentrations of the later drug did not increase sensitivity compared to control transfections (data not shown).
Figure 5.
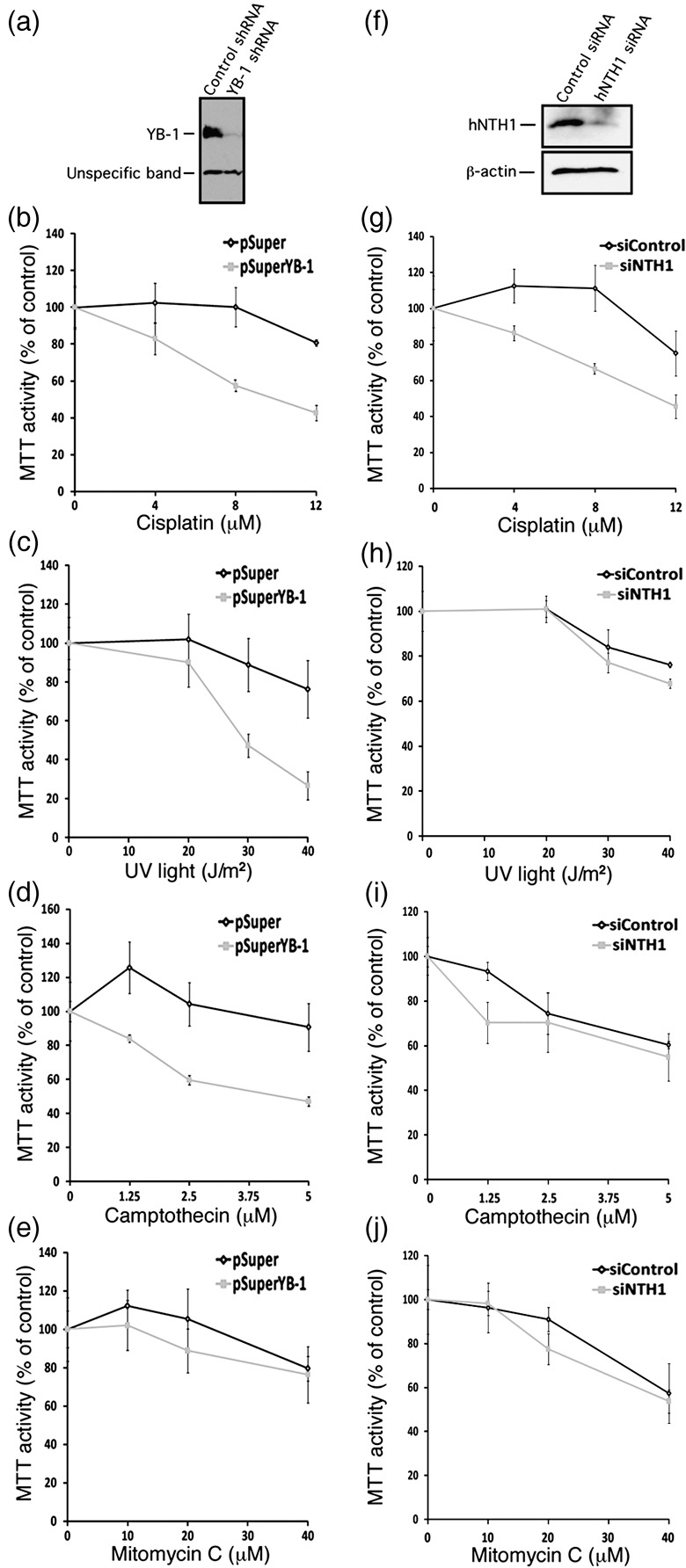
Impact of depleting either endogenous Y‐box‐binding protein‐1 (YB‐1) or human endonuclease III (hNTH1) proteins in MCF7 on survival after different DNA‐damaging conditions. (a) Western blots showing intracellular levels of YB‐1 48 h after transfection with a shRNA specific to YB‐1 or with a control construct. The lower molecular weight band is an unspecific protein recognized by the anti‐YB‐1 antibody. (b–e) MTT cell survival assays of MCF7 cells transfected with a shRNA specific to YB‐1 or with a control construct after of treatment with either cisplatin (b), UV light (c), camptothecin (d), or mitomycin C (e). Cells were first transfected with the appropriate constructs. Forty‐eight hours later, cells were plated in 96‐well plates. Cells were then treated with the drugs for 16 h in culture before the MTT assays. Results represent the mean ± SD of triplicate experiments. (f) Western blots showing intracellular levels of hNTH1 after 48 h after transfection with a siRNA specific to hNTH1 or with a scrambled control siRNA. The anti β‐actin was used as loading control for the blot. (g–j) MTT cell survival assays of MCF7 cells transfected with a siRNA specific to hNTH1 or with a scrambled control siRNA after treatment with either (g) cisplatin, (h) UV light, (i) camptothecin, or (j) mitomycin C. Cells were first transfected with the siRNAs. Forty‐eight hours later, cells were plated in 96‐well plates. Cells were then treated with the drugs for 16 h in culture before the MTT assays. Results represent the mean ± SD of triplicate experiments.
In a second series of experiments, siRNAs specific to hNTH1 were transfected into MCF7 cells. Forty‐eight hours later, the same treatments were applied. As shown in Figure 5f, the siRNA decreased hNTH1 protein levels by approximately 80% compared to control scrambled siRNA. Knock‐down of hNTH1 sensitized cells to both cisplatin and UV light (Fig. 5g,h). It did not sensitize MCF7 cells to camptothecin and mitomycin C (Fig. 5i,j). Thus, the effect of siRNA specific to hNTH1 was similar to a knock‐down of YB‐1 with regard to cisplatin and UV light treatments. These results suggest that hNTH1 and YB‐1 may be part of the same DNA repair pathway in response to cisplatin and UV treatments.
It has been reported by several groups that YB‐1 is spontaneously increased in cultured cell lines resistant to cisplatin, and the nuclear level YB‐1 expression is predictive of drug resistance and patient outcome in breast tumors, ovarian cancers, and synovial sarcomas.( 6 , 7 , 8 , 9 , 10 ) Since there is an increase in YB‐1/hNTH1 association during cisplatin treatment, we next examined the impact of knocking down hNTH1 expression in cisplatin resistant YB‐1 overexpressing mammary tumor MCF7 cells. We first transfected MCF7 cells with a siRNA specific to hNTH1 or scrambled control siRNA. Forty‐eight hours later, cells were re‐transfected with a YB‐1 expression vector or an empty expression control vector. (Forty‐eight hours was the minimum time to obtain efficient knock‐down of hNTH1 before YB‐1 transfection). As indicated in Figure 6, double controls transfected cells (scrambled siRNA plus empty expression vector) were more sensitive to cisplatin treatment than in single (siRNA) transfection experiments (Fig. 5). Thus, a double transfection protocol had an impact on cisplatin resistance to some extent. However, transfection of YB‐1 expression vector significantly increased cisplatin resistance in tranfected MCF7 cells. Importantly, knocking down the expression of hNTH1 in such YB‐1 overexpressing cells significantly sensitized them to cisplatin treatments (Fig. 6). The same conclusion was obtained by counting the transfected cells with a hemacytometer (trypan blue exclusion assay; Fig. 6a) or by the MTT assay (Fig. 6b).
Figure 6.
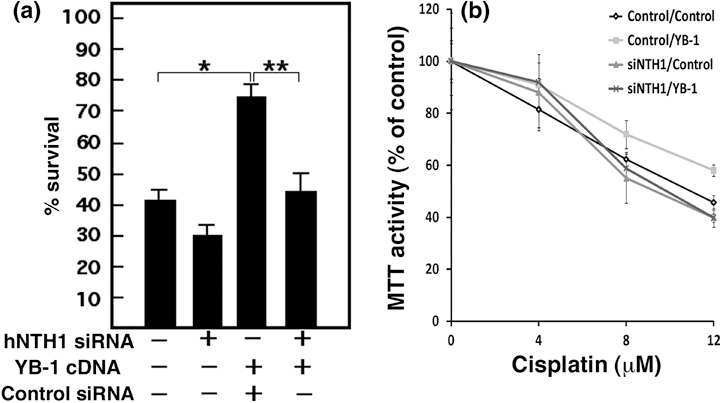
Impact of depleting endogenous human endonuclease III (hNTH1) proteins in Y‐box‐binding protein‐1 (YB‐1) overexpressing MCF7 cisplatin‐resistant cells. MCF7 cells were transfected with a siRNA specific to hNTH1 or scrambled control siRNA. Forty‐eight hours later, cells were transfected with YB‐1 expression vector or empty expression control vector. (a) Transfected cells were treated with 4 µM cisplatin for 16 h before counting with a hemacytometer (*P < 0.02; P < 0.05). The ‘–’ sign indicates scrambled siRNA or empty expression vector. The ‘+’ sign indicates sihNTH1 or YB‐1 expression vector. Experiments were done in triplicates. (b) Transfected cells were plated in 96‐well plates. Cells were then treated with the drugs for 16 h in culture before the MTT assays. Results represent the mean ± SD of the triplicate experiments.
It is well established that YB‐1 can activate cell cycle progression.( 22 ) However, a recent report has indicated that overexpresssion of YB‐1 can interfere with protein synthesis and transformation.( 23 ) To determine the impact of YB‐1 overexpression on MCF7 cell cycle, FACS analyses were performed on transfected cells. As indicated in Table 2, the percentage of cells in G1, S, and G2M phases were similar in MCF7 cells 48 h after transfection with either an empty expression vector or a YB‐1 expression plasmid. Thus, overexpression of YB‐1 did not significantly alter the cell‐cycle progression in the MCF7 tumor cell line. Furthermore, cell growth was unaltered in YB‐1 MCF7 overexpressing cells compared to control transfected cells (Fig. 7). We conclude that the cisplatin resistance conferred by YB‐1 overexpression in MCF7 was not due to simple increased cell proliferation in the MTT assay (Fig. 6).
Table 2.
Cell‐cycle progression in cells transfected with control or Y‐box‐binding protein‐1 (YB‐1) expression plasmids
| Cycle | Control | YB‐1 |
|---|---|---|
| G1 | 57% ± 3 | 59% ± 1 |
| S | 34% ± 2 | 31% ± 2 |
| G2/M | 9% ± 1 | 10% ± 2 |
FACS analyses were performed in duplicates.
Figure 7.
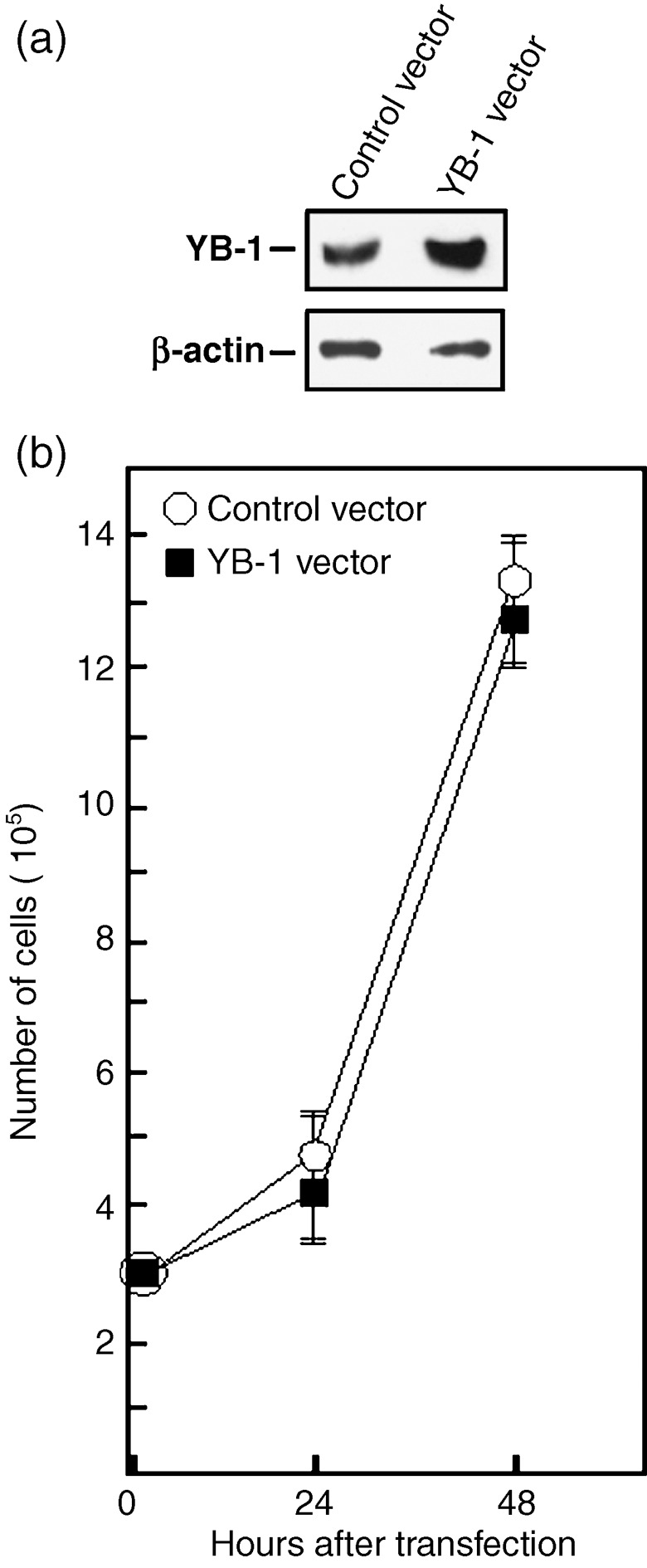
Cell growth of MCF7 cells overexpressing Y‐box‐binding protein‐1 (YB‐1). (a) Example of Western blots showing intracellular levels of YB‐1 48 h after transfection with either an empty expression vector or a YB‐1 expression vector. The anti β‐actin is used as loading control for the blot (bottom panel). On average, transfection of YB‐1 expression vector increased the levels of YB‐1 proteins by two‐fold. (b) Cell growth of transfected MCF7 cells. Cells were transfected with either an empty expression vector or a YB‐1 expression vector by electroporation with a nucleofector kit (see ‘Materials and Methods’ for details). Approximately, 3 × 105 transfected cells were plated on 90‐mm Petri dishes. Cells were counted with an hemocytometer 24 and 48 h after the transfections. Results represent the mean ± SD of four independent transfection experiments. All cultures reached confluence at 72 h (data not shown).
Discussion
Previous reports have indicated that YB‐1 stimulates hNTH1 activity in vitro and coimmunoprecipitates with hNTH1 in vivo. In this study, we found that YB‐1 interacts specifically with the N‐terminus tail of hNTH1 protein. This region of hNTH1 was recently shown to inhibit the catalytic activity of hNTH1.( 19 ) Thus, YB‐1 would stimulate hNTH1 activity by binding and masking the inhibitory effect of the hNTH1 N‐terminus tail. To confirm the impact of purified YB‐1 on hNTH1 nuclease activity, we generated a new system in which MCF7 cells were expressing a stable TAP‐hNTH1 construct. This TAP‐hNTH1 chimeric protein is fully functional as it can cleave a DNA duplex containing 8‐oxoguanine residues. Such residues are likely to be present in genomic DNA during cisplatin treatments as this drug is known to induce oxidative stress.( 20 , 21 ) This nuclease activity was specific to the TAP‐hNTH1 protein as transfection of siRNA specific to hNTH1 abolished TAP‐hNTH1 expression and nuclease activity in the eluate (Fig. 3). Added purified YB‐1, which also contains nuclease activities,( 12 , 18 ) increased the overall enzymatic activity in the reaction. Note that in these nuclease assays, we did not transfect the YB‐1 cDNA into TAP‐hNTH1 expressing MCF7 cells because we noticed that overexpression of YB‐1 also modulated the transcription of several DNA repair enzymes (manuscript in preparation), which would have influenced the interpretation of the results. We added the well‐characterized purified YB‐1 in the nuclease reaction instead.( 12 , 18 )
We observed an increase YB‐1/hNTH1 complex formation after UV light and cisplatin treatments in MCF7 cells. This was not observed with camptothecin and mitomycin C. Concomitant with these observations, we found that knocking down hNTH1 protein levels sensitized cells to both UV light and cisplatin actions similar to a YB‐1 knock‐down. This is the first report on the impact of siRNA specific to hNTH1 on cisplatin or UV light resistance. These results also suggest that YB‐1 acts in concert with hNTH1 in the response of MCF7 cells to cisplatin and UV light. Cisplatin is an anticancer drug often used in chemotherapy which creates both intra‐ and interstrand crosslinks as well as monofunctional adducts.( 24 ) It also induces an oxidative stress and hence oxidative DNA damage.( 20 , 21 ) UV treatment can also lead to oxidative DNA damage. Several oxidized bases are substrates for hNTH1 while DNA adducts created by mitomycin C or topoisomerase/DNA poison complexes created by camptothecin are not recognized by hNTH1. The results with mitomycin C are intriguing as a knock‐down of YB‐1 in a human epidermoid cancer KB cell line and in mouse embryonic stem cells confers sensitivity to this drug.( 15 , 25 ) However, mouse embryonic stem cells with reduced expression of YB‐1 did not exhibit higher sensitivity to UV light,( 25 ) unlike KB epidermoid cells,( 15 ) and MCF7 cells (Fig. 5a). This suggests that UV and mitomycin C toxicity also depend on the cell type.
The regulation of the cisplatin‐dependent association of YB‐1 with the autoinhibitory domain of hNTH1 is unknown at the present time. It has been reported, however, that YB‐1 is a substrate for Akt,( 26 ) a serine‐threonine kinase activated by either cisplatin or UV treatments.( 27 , 28 ) Phosphorylation of YB‐1 by Akt induces YB‐1 nuclear translocation.( 26 ) An increase in nuclear phospho‐YB‐1 would thus increase the pool of nuclear hNTH1/YB‐1 complex formation under such conditions. Mutagenesis of the serine (or threonine) residues of YB‐1 phosphorylated by Akt will be required in cells to explore this possibility.
A major problem with cisplatin regimens is the appearance of resistant tumor cells during the course of the treatment.( 22 ) Importantly, overexpression of YB‐1 correlates well with the appearance of cisplatin resistant tumor cells in several cancer patients.( 7 ) A principal conclusion of this study is that a knock down of hNTH1 protein levels in YB‐1 overexpressing MCF7 cells (thus more resistant to cisplatin) sensitized them to cisplatin treatments. Hence, these experiments indicate that inhibition of hNTH1 activity is a potential target to sensitize cells to cisplatin treatments. It will be possible in the future to perform large scale screens to isolate the small molecule inhibitor of DNA base excision repair targeted specifically to purified hNTH1 (or YB‐1) to potentiate the cytotoxicity of cisplatin, as it has been demonstrated for the endonuclease enzyme APE1.( 29 )
Acknowledgments
We thank N. Roberge (Hôpital Hôtel‐Dieu de Québec, Québec, Canada) for all FACS analyses. This study was supported in part by grants from the Canadian Institutes of Health Research (CIHR) and the Cancer Research Society awarded to Michel Lebel. David Guay is a scholar from the National Sciences and Engineering Research Council of Canada. Michel Lebel is a scholar from the Fonds de Recherche en Santé du Québec.
References
- 1. Swamynathan SK, Nambiar A, Guntaka RV. Role of single‐stranded DNA regions and Y‐box proteins in transcriptional regulation of viral and cellular genes. FASEB J 1998; 12: 515–22. [DOI] [PubMed] [Google Scholar]
- 2. Ashizuka M, Fukuda T, Nakamura T et al . Novel translational control through an iron–responsive element by interaction of multifunctional protein YB‐1 and IRP2. Mol Cell Biol 2002; 22: 6375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stickeler E, Fraser SD, Honig A, Chen AL, Berget SM, Cooper TA. The RNA binding protein YB‐1 binds A/C‐rich exon enhancers and stimulates splicing of the CD44 alternative exon v4. EMBO J 2001; 20: 3821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hayakawa H, Uchiumi T, Fukuda T et al . Binding capacity of human YB‐1 protein for RNA containing 8‐oxoguanine. Biochemistry 2002; 41: 12739–44. [DOI] [PubMed] [Google Scholar]
- 5. Koike K, Uchiumi T, Ohga T et al . Nuclear translocation of the Y‐box binding protein by ultraviolet irradiation. FEBS Lett 1997; 417: 390–4. [DOI] [PubMed] [Google Scholar]
- 6. Bargou RC, Jurchott K, Wagener C et al . Nuclear localization and increased levels of transcription factor YB‐1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat Med 1997; 3: 447–50. [DOI] [PubMed] [Google Scholar]
- 7. Janz M, Harbeck N, Dettmar P et al . Y‐box factor YB‐1 predicts drug resistance and patient outcome in breast cancer independent of clinically relevant tumor biologic factors HER2, uPA and PAI‐1. Int J Cancer 2002; 97: 278–82. [DOI] [PubMed] [Google Scholar]
- 8. Oda Y, Ohishi Y, Saito T et al . Nuclear expression of Y‐box‐binding protein‐1 correlates with P‐glycoprotein and topoisomerase II alpha expression, and with poor prognosis in synovial sarcoma. J Pathol 2003; 199: 251–8. [DOI] [PubMed] [Google Scholar]
- 9. Rubinstein DB, Stortchevoi A, Boosalis M, Ashfaq R, Guillaume T. Overexpression of DNA‐binding protein B gene product in breast cancer as detected by in vitro‐generated combinatorial human immunoglobulin libraries. Cancer Res 2002; 62: 4985–91. [PubMed] [Google Scholar]
- 10. Yahata H, Kobayashi H, Kamura T et al . Increased nuclear localization of transcription factor YB‐1 in acquired cisplatin‐resistant ovarian cancer. J Cancer Res Clin Oncol 2002; 128: 621–6. [DOI] [PubMed] [Google Scholar]
- 11. Ohga T, Uchiumi T, Makino Y et al . Direct involvement of the Y‐box binding protein YB‐1 in genotoxic stress‐induced activation of the human multidrug resistance 1 gene. J Biol Chem 1998; 273: 5997–6000. [DOI] [PubMed] [Google Scholar]
- 12. Gaudreault I, Guay D, Lebel M. YB‐1 promotes strand separation in vitro of duplex DNA containing either mispaired bases or cisplatin modifications, exhibits endonucleolytic activities and binds several DNA repair proteins. Nucl Acids Res 2004; 32: 316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marenstein DR, Ocampo MT, Chan MK et al . Stimulation of human endonuclease III by Y box‐binding protein 1 (DNA‐binding protein B). Interaction between a base excision repair enzyme and a transcription factor. J Biol Chem 2001; 276: 21242–9. [DOI] [PubMed] [Google Scholar]
- 14. Marenstein DR, Chan MK, Altamirano A et al . Substrate specificity of human endonuclease III (hNTH1). Effect of human APE1 on hNTH1 activity. J Biol Chem 2003; 278: 9005–12. [DOI] [PubMed] [Google Scholar]
- 15. Ohga T, Koike K, Ono M et al . Role of the human Y box‐binding protein YB‐1 in cellular sensitivity to the DNA‐damaging agents cisplatin, mitomycin C, and ultraviolet light. Cancer Res 1996; 56: 4224–8. [PubMed] [Google Scholar]
- 16. Schmutte C, Sadoff MM, Shim KS, Acharya S, Fishel R. Role of the human Y‐box binding protein YB‐1 in cellular sensitivity to DNA‐damaging agents cisplatin, mitomycin C, and ultraviolet light. J Biol Chem 2001; 276: 33011–8. 11427529 [Google Scholar]
- 17. Moggs JG, Yarema KJ, Essigmann JM, Wood RD. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3‐intrastrand d (GpTpG) ‐cisplatin adduct. J Biol Chem 1996; 271: 7177–86. [DOI] [PubMed] [Google Scholar]
- 18. Izumi H, Imamura T, Nagatani G et al . Y box‐binding protein‐1 binds preferentially to single‐stranded nucleic acids and exhibits 3′→5′ exonuclease activity. Nucl Acids Res 2001; 29: 1200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu X, Choudhury S, Roy R. In vitro and in vivo dimerization of human endonuclease III stimulates its activity. J Biol Chem 2003; 278: 50061–9. [DOI] [PubMed] [Google Scholar]
- 20. Martins NM, Santos NA, Curti C, Bianchi ML, Santos AC. Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J Appl Toxicol 2007. [Epub ahead of print]. [DOI] [PubMed]
- 21. Sasada T, Iwata S, Sato N et al . Redox control of resistance to cis‐diamminedichloroplatinum (II) (CDDP): protective effect of human thioredoxin against CDDP‐induced cytotoxicity. J Clin Invest 1996; 97: 2268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jurchott K, Bergmann S, Stein U et al . YB‐1 as a cell cycle‐regulated transcription factor facilitating cyclin A and cyclin B1 gene expression. J Biol Chem 2003; 278: 27988–96. [DOI] [PubMed] [Google Scholar]
- 23. Bader AG, Vogt PK. Inhibition of protein synthesis by Y box‐binding protein 1 blocks oncogenic cell transformation. Mol Cell Biol 2005; 25: 2095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res 2001; 478: 23–43. [DOI] [PubMed] [Google Scholar]
- 25. Shibahara K, Uchiumi T, Fukuda T et al . Targeted disruption of one allele of the Y‐box binding protein‐1 (YB‐1) gene in mouse embryonic stem cells and increased sensitivity to cisplatin and mitomycin C. Cancer Sci 2004; 95: 348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sutherland BW, Kucab J, Wu J et al . Akt phosphorylates the Y‐box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage‐independent growth of breast cancer cells. Oncogene 2005; 24: 4281–92. [DOI] [PubMed] [Google Scholar]
- 27. Winograd‐Katz SE, Levitzki A. Cisplatin induces PKB/Akt activation and p38 (MAPK) phosphorylation of the EGF receptor. Oncogene 2006; 25: 7381–90. [DOI] [PubMed] [Google Scholar]
- 28. Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultraviolet irradiation activates PI 3‐kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol 2001; 18: 461–6. [DOI] [PubMed] [Google Scholar]
- 29. Madhusudan S, Smart F, Shrimpton P et al . Isolation of a small molecule inhibitor of DNA base excision repair. Nucl Acids Res 2005; 33: 4711–24. [DOI] [PMC free article] [PubMed] [Google Scholar]