

Abstract
Mutations of DNA double‐strand breaks (DSB) repair genes, ATM, MRE11, RAD50, NBS1 and ATR, are postulated to play a role in the development of gastrointestinal malignancies with an impaired mismatch repair (MMR) function. In the present study, mutations of these genes together with the presence of microsatellite instability (MSI) were examined in 50 leukemia–lymphoma cell lines. MSI was detected in 13 (26%) lines. Mutations of intronic mononucleotide repeats in ATM and MRE11 were found in nine and six lines, respectively, whereas mutations of mononucleotide repeats of RAD50 were found in only one line, and none were found in either NBS1 or ATR. Frequencies of ATM and MRE11 mutations were significantly higher in MSI‐positive than MSI‐negative lines. These mutations generated aberrant splicing in both genes. The intensity of the aberrant transcript of ATM (497del22) was stronger in five lines harboring mononucleotide mutations of 2 bp or more than in the lines without or with a 1‐bp mutation. The intensity of the aberrant transcript of MRE11 (315del88) was stronger in four lines with mononucleotide mutations than in lines without. The expression levels of ATM and MRE11 transcripts in MSI‐positive lines were significantly higher than those in MSI‐negative lines. MSI‐positive cell lines showed delay or abrogation of DSB repair. The present study suggests that impairment of the MMR system causes aberrant transcripts in the DSB repair genes ATM and MRE11. This might result in inactivation of the DSB repair system, thus inducing an acceleration of genome instability and accumulation of genetic damage. (Cancer Sci 2006; 97: 226–234)
Abbreviation used:
- ATM
Ataxia telangiectasia mutated
- ATR
ATM‐ and Rad3‐related
- DSB
double‐strand break
- HR
homologous recombination
- IR
ionizing radiation
- IVS
intervening sequence
- MSI
microsatellite instability
- MMR
mismatch repair
- MRN
MRE11‐RAD50‐NBS1
- ORF
open reading frame
- PCR
polymerase chain reaction
- PFGE
pulsed‐field gel electrophoresis
- RI
ratio of intensity
- TAE
Tris‐acetate/EDTA
Exposure to hazardous agents in the environment may induce DNA damage that could be harmful to the maintenance of cell homeostasis and transmission of high‐fidelity genetic information. Thus, cells have evolved several mechanisms for DNA repair to ensure genomic integrity and prevent mutations. The DNA MMR system corrects errors that might occur during DNA replication, whereas HR and non‐homologous end joining are involved in the repair of DNA DSB.( 1 , 2 )
Double‐strand breaks, which are extremely cytotoxic DNA lesions, activate an extensive array of responses that lead to repair of the damage and allow continuation of cellular life.( 1 , 3 ) The nuclear protein kinase ATM is regarded as the primary activator of this network, phosphorylating key proteins in numerous signaling pathways.( 4 ) The highly conserved MRN complex plays a role in DSB repair, particularly in the HR pathways.( 5 ) While NBS1 is a target of ATM, recent observation has shown that the MRN complex itself contributes to the direct activation of ATM.( 6 ) ATR, another protein kinase, has been reported to regulate responses to a broad range of damage, including DSB.( 7 , 8 ) Whereas ATM is engaged primarily in DSB repair, several observations suggest that ATR has a critical role in virtually all cellular responses to the arrest of DNA replication forks, which are the DNA structures formed during replication.( 8 , 9 , 10 )
The increased rate of uncorrected replication errors at simple repeat sequences is known as MSI. Disruption of the MMR system, as revealed by MSI, is characterized by the accelerated accumulation of single nucleotide mutations and resultant alterations in the microsatellite DNA sequences, which affect the genome ubiquitously.( 11 ) MSI is correlated with hereditary non‐polyposis colorectal cancer, as well as a variety of sporadic cancers, including gastric, endometrial and colorectal cancers, through inactivation of several MMR target genes.( 12 , 13 ) Although MSI was previously regarded to be uncommon in hematolymphoid malignancies,( 14 , 15 ) recent studies have revealed it to be involved in the development of these malignancies, especially diseases involving the T‐cell lineage.( 16 , 17 )
Recent studies have suggested a link between impaired MMR and mutations of DSB repair genes in cancers. In colorectal tumors with an impaired MMR system, mutations of intronic mononucleotide repeats in ATM and MRE11 were found to result in aberrant splicing, followed by reduced expression of the wild‐type protein.( 18 , 19 , 20 ) Ottini et al. reported impaired MRE11 expression in gastric cancer with impaired MMR.( 21 ) Meanwhile, mutations in coding mononucleotide repeats in RAD50, NBS1 and ATR were reported in a subset of gastrointestinal cancers with MSI.( 22 , 23 ) In the present study, we provide evidence that impaired MMR function may result in aberrant splicing of ATM and MRE11 genes, which correlates with impairment of the DSB repair system in leukemia–lymphoma cell lines.
Materials and Methods
Cell lines
A total of 50 leukemia–lymphoma cell lines derived from Burkitt lymphoma (BJAB, BL36, BL60, BL137, Daudi, Katata, NAB‐2, Namalwa, Raji and Ramos), diffuse large B‐cell lymphoma (OPL‐1, OPL‐2, OPL‐3, OPL‐4, OPL‐5, OPL‐7 and Pal‐1),( 24 , 25 , 26 ) precursor B‐cell acute lymphoblastic leukemia (NALM‐6 and REH), T‐cell acute lymphoblastic leukemia (CCRF‐CEM, HPB‐ALL, MOLT‐4, MOLT‐14, Jurkat and Peer), adult T‐cell leukemia (MT‐1, MT‐2, MT‐4 and TL‐Om1), NK/NKT‐cell lymphoma (KHYG‐1, MOTN‐1, MTA, NK‐92, SNK‐6, SNT‐8 and YT),( 27 ) Hodgkin lymphoma (HDLM‐2, HD‐My‐Z, KM‐H2, L‐428, L‐540 and L‐1236), non‐B non‐T acute lymphoblastic leukemia (P30/Ohkubo), pyothorax‐associated lymphoma (PAL) with dual B/T‐cell phenotype (Deglis),( 28 ) acute myeloid leukemia (CTV‐1, P31/Fujioka, HEL, HL‐60 and KG‐1), and chronic myeloid leukemia (K‐562) were used in this study. OPL and Pal‐1 were derived from tumor cells in PAL, a B‐cell lymphoma of mostly large cell morphology that develops in long‐standing inflammation of the pleura, usually consequent to lung tuberculosis. Two colorectal carcinoma cell lines (HCT 116 and LS 174T) known to harbor MSI were used as positive controls, whereas two lymphoblastoid cell lines (IB‐4 and OS) were used as negative controls. Five normal peripheral blood samples as normal tissue controls were obtained from healthy volunteers.
BJAB, Daudi, Namalwa, Raji, Ramos, CCRF‐CEM, MOLT‐4, MOLT‐14, Jurkat, Peer, MTA, P30/Ohkubo, P31/Fujioka, HL‐60, KG‐1 and K‐562 were obtained from the Japanese Collection of Research Bioresources (Tokyo, Japan). HDLM‐2, HD‐My‐Z, KM‐H2, L‐428, L‐540, L‐1236, NALM‐6, REH, HPB‐ALL, YT, NK‐92 and CTV‐1 were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). HCT 116 and LS 174T were purchased from American Type Culture Collection (Manassas, VA, USA). OPL‐1, OPL‐2, OPL‐3, OPL‐4, OPL‐5, OPL‐7 and OS were established in our laboratory.( 24 , 25 ) Other cell lines were gifts from investigators listed in the Acknowledgments. All cell lines were cultured in RPMI 1640 medium (Sigma, St Louis, MO, USA) supplemented with 10% fetal bovine serum and antibiotics at 37°C in 5% CO2 in air.
Detection of MSI and mutations of ATM, MRE11, RAD50, NBS1 and ATR
Genomic DNA was extracted from the cell lines by the sodium dodecylsulfate‐proteinase K and phenol‐chloroform extraction method. MSI was determined as present when mutations in both BAT‐25 and BAT‐26 were observed. Polymorphisms of both of these microsatellites have been described as being rare. Deletions of both of these markers have been shown to establish MSI status with an accuracy of > 99.5% without the requirement of matching normal DNA.( 29 , 30 , 31 )
Fragments containing poly(T)15 nucleotide repeats in IVS 7 of ATM, poly(T)11 nucleotide repeats in IVS 4 of MRE11, poly(A)9 nucleotide repeats in the ORF of RAD50, poly(A)7 nucleotide repeats in the ORF of NBS1, and poly(A)10 nucleotide repeats in the ORF of ATR were amplified by PCR. Forward and reverse primer sequences were: ATM, 5′‐AAATCCTTTTTCTGTATGGGAT‐3′ and 5′‐(Hex)‐GTTACTGAGTCTAAAACATGG‐3′; MRE11, 5′‐GGAGGAGAATTTTAGGGA‐3′ and 5′‐(Hex)‐TGCTTTCCACAGACAAAC‐3′; RAD50, 5′‐GAGGCTGAGTTACAAGAAGTC‐3′ and 5′‐(Fam)‐CATTTCATCACGCCGCTTTTC‐3′; NBS, 5′‐CTACTTTCAGCCGTCTACC‐3′ and 5′‐(Hex)‐GCCACATCATCCATTTCC‐3′; ATR, 5′‐(Hex)‐CCTTTCTCTGAACACGGAC‐3′ and 5′‐CAAGTTTTACTGGACTAGGTA‐3′; BAT‐25, 5′‐(XRITC)‐TCGCCTCCAAGAATGTAAGT‐3′ and 5′‐CCACACTTCAAAATACATTCTGC‐3′; and BAT‐26, 5′‐(FITC)‐TGACTACTTTTGACTTCAGCC‐3′ and 5′‐AACCATTCAACATTTTTAACCC‐3′. Briefly, 0.5 µL of each PCR product was mixed with 12.0 µL of deionized formamide and 0.5 µL of dye‐labeled Internal Lane Size Standard (GeneScan 400 HD; Applied Biosystems, Foster City, CA, USA). The samples were run on an ABI PRISM 310 Genetic Analyzer, and the sizes of the PCR products were analyzed with GeneScan analysis software version 3.1 (Applied Biosystems). The experiment was repeated at least twice.
Detection of aberrant splicing in ATM (497del22) and MRE11 (315del88)
RNA was extracted from the cell lines in the presence of TRIzol reagent (Life Technologies, Rockville, MD, USA), and was reverse transcribed by random hexamer priming. PCR amplification of the indicated fragments was carried out in a total volume of 15 µL consisting of 1 µL of cDNA template and 0.5 units of AmpliTaq Gold (Applied Biosystems), using the primer pairs as follows: ATM exon 8, 5′‐CTCAGCAACAGTGGTTAG‐3′ and 5′‐GATATGATTTAGACCTGAAGAG‐3′; and MRE11 exon 5, 5′‐CCAGGGGTTCTTGGAGAAG‐3′ and 5′‐CCAGCACAACTTAAAATGTC‐3′. PCR was carried out at least twice for all samples. The PCR products (5 µL) were electrophoresed on a 1.5–2.5% agarose gel and visualized by ethidium bromide staining. The photographs taken under a UV transilluminator were scanned with an EpsonTM ES‐6000 HS scanner (Suwa, Japan). By using Adobe PhotoshopTM version 3.0 J (San Jose, CA, USA), the intensity of the wild‐type and aberrant bands on the scanned digital images were measured as follows: the degrees of average brightness for the same area of each band were quantified on a gray scale (the level ranging from 0 to 256) through the subtraction of the background brightness, then the ratio of average brightness of the aberrant band to that of the wild‐type band was calculated. An intensity of less than 0.10 was regarded as negative.
Sequencing analysis
Sequencing was carried out bythe dideoxy chain termination method using a DNA sequencing kit (Applied Biosystems) and the ABI PRISM 310 Genetic Analyzer as described previously.( 32 )
Real‐time PCR for assessment of expression levels of DSB repair genes
Expression levels of ATM, MRE11, RAD50, NBS1 and ATR were analyzed using the TaqMan Gene Expression AssaysTM according to the protocol of the manufacturer (Applied Biosystems): Expression of combined wild‐type and aberrant‐type genes was measured using the following primers: ATM, ID Hs00175892_m1, MRE11; Hs00271551_m1, RAD50; Hs00194871_m1, NBS1; Hs00159537_m1, ATR; Hs00169878_ m1, β‐actin; Hs99999903_m1 (Applied Biosystems). Standard curves for quantification of the molecules were constructed from the results of simultaneous amplifications of serial dilutions of cDNA from a lymphoblastoid cell line, IB‐4. Real‐time PCR and subsequent calculations were carried out with an ABI Prism 7700 Sequence Detector System (Applied Biosystems). To normalize the differences in RNA degradation and RNA loading for reverse transcription‐PCR in individual samples, the expression level of each molecule was divided by that of β‐actin in the same samples. All experiments were performed at least in duplicate. The expression level of each gene in IB‐4 was defined as 100, and the relative gene expression levels in each cell line was compared.
DNA DSB repair assays
Ability of repair for DNA DSB induced by IR in each cell line was analyzed as described elsewhere.( 33 , 34 ) Briefly, more than 1 × 105 cells from each cell line were embedded in agarose plugs as described previously.( 33 , 34 ) The plugs, on ice, were exposed to 20 Gy of IR, covered with RPMI 1640 medium, and kept at 37°C for up to 6 h to allow the cells to repair damaged DNA. Untreated plugs containing the same concentration of cells were used as controls. The plugs were embedded into wells of a 0.8% agarose gel in 1 × TAE and subjected to PFGE in a CHEF apparatus (Bio‐Rad, Hercules, CA, USA) at 3 V/cm, 14°C for 48 h with a pulse time of 45 s. Under these PFGE conditions, DNA fragments between 2 and 6 Mb in size migrate as a single band and form a compression zone under each well. Much smaller DNA from apoptotic fragmentations could be detected as thin smears, but not bands. Rejoined DNA fragments were too large to enter the gel. After electrophoresis, DNA was stained with ethidium bromide and visualized with UV light. The signals from the wells and compression zones were measured using FMBIO Analysis V8.0 (TaKaRa, Kusatsu, Japan). DSB were quantified as the fraction of DNA in the compression zone relative to that in the wells.
Statistical analysis
The frequency of ATM and MRE11 gene mutations between MSI‐positive and MSI‐negative lines was calculated using Fisher's exact test. P‐values of less than 0.01 were considered to be statistically significant. The expression levels of ATM, MRE11, RAD50, NBS1 and ATR and DSB repair ability were calculated using the Mann–Whitney test. P‐values of less than 0.05 were considered to be statistically significant.
Results
MSI in leukemia–lymphoma cell lines
Microsatellite instability, as determined by the presence of mutations in both BAT‐25 and BAT‐26, was observed in 13 of 50 leukemia–lymphoma cell lines, including 11 lymphoid and two myeloid lines (Table 1). Among the B‐cell lineage, MSI was found in four lines: one of 10 Burkitt lymphomas (Daudi), one of seven diffuse large B‐cell lymphomas (OPL‐3), and two of two precursor B‐cell acute lymphoblastic leukemia lines (NALM‐6 and REH). Among the T‐cell lineage, MSI was found in four lines derived from T‐cell acute lymphoblastic leukemia (HPB‐ALL, MOLT‐4, MOLT‐14 and Jurkat). MSI was found in one of seven NK/NKT‐cell (SNK‐6), one of six Hodgkin lymphoma (KM‐H2), and one of one non‐B non‐T acute lymphoblastic lymphoma (P30/Ohkubo) cell lines. These results showed the frequency of MSI to be highest in acute lymphoblastic leukemia lines irrespective of immunophenotype. Among the myeloid cell lines, MSI was found in two acute myeloid leukemia lines (CTV‐1 and P31/Fujioka). No MSI was found in five normal blood samples.
Table 1.
Summary of microsatellite instability and mutation of intronic mononucleotide repeats of ATM and MRE11 in leukemia–lymphoma cell lines
| Cell line | Microsatellite instability | Shortening | Mutation of intronic mononucleotide repeat | Aberrant transcript | Real‐time expression value | ||||
|---|---|---|---|---|---|---|---|---|---|
| BAT‐25 | BAT‐26 | ATM | MRE11 | ATM | MRE11 | ATM | MRE11 | ||
| Leukemia–lymphoma | |||||||||
| OPL−3 | MSI | −11 | −12 | −2 | −1 | + | + | 12.62 | 18.32 |
| Jurkat † | MSI | −4 | −9 | −4 | −1 | + | + | 23.9 | 78.5 |
| P31/Fujioka | MSI | −4 | −3 | −1 | −1 | + | + | 185.75 | 487.37 |
| MOLT‐4 | MSI | −2 | −4 | −1 | 0 | + | + | 349.65 | 513.83 |
| NALM‐6 | MSI | −2 | −4 | 0 | 0 | + | + | 7.5 | 13.75 |
| CTV‐1 | MSI | −3 | Deleted | −2 | 0 | + | + | 6.17 | 16.15 |
| HPB‐ALL | MSI | −1 | −4 | 0 | −1 | + | + | 42.4 | 101.4 |
| REH | MSI | −1 | −4 | 0 | 0 | + | + | 116.08 | 191.55 |
| P30/Ohkubo | MSI | −4 | −5 | −2 | 0 | + | − | 290.36 | 406.28 |
| Daudi | MSI | −3 | −2 | 0 | Het + 1 | + | − | 3.17 | 9.7 |
| MOLT‐14 | MSI | −1 | −1 | −3 | 0 | + | − | 2.93 | 5.2 |
| KM‐H2 | MSI | −1 | 1 | 0 | 0 | + | − | 1.2 | 1.66 |
| SNK‐6 | MSI | 1 | −1 | −1 | 0 | + | − | 2.2 | 5.87 |
| OPL‐4 | MSS | −6 | 0 | 0 | −1 | + | − | 4.5 | 4.9 |
| Katata | MSS | 0 | 0 | Het + 1 | 0 | + | − | 30.5 | 17.3 |
| Colorectal cancer (control) | |||||||||
| HCT 116 † | MSI | −8 | −13 | −3 | −2 | + | + | 3.3 | 1.62 |
| LoVo † | MSI | −7 | −1 | −1 | −1 | + | + | ND | ND |
| LS 174T † | MSI | −6 | −12 | −3 | −2 | + | + | 25.4 | 26.7 |
Shortening/lengthening of mononucleotide repeat in RAD50 was observed. Het, heterogenous; MSI, microsatellite unstable; MSS, microsatellite stable.
Mutations of mononucleotide repeats in ATM, MRE11, RAD50, NBS1 and ATR
Mutations of the intronic poly(T)15 nucleotide repeats in IVS 7 of ATM were found in nine lines (Table 2): homozygous shortening in eight lines with MSI (OPL‐3, MOLT‐4, MOLT‐14, Jurkat, SNK‐6, P30/Ohkubo, CTV‐1 and P31/Fujioka) and 1‐bp heterozygous lengthening in an MSI‐negative cell line (Katata). Shortening by 4 bp was found in Jurkat, 3 bp in MOLT‐14, 2 bp in OPL‐3, P30/Ohkubo and CTV‐1, and 1 bp in MOLT‐4, SNK‐6 and P31/Fujioka. No mutations were observed in five normal blood samples. The frequency of ATM mutation was significantly higher in MSI‐positive than MSI‐negative lines (P < 0.01).
Table 2.
Mutations of mononucleotide repeats and microsatellite instability (MSI) in ATM, MRE11, RAD50, NBS1 and ATR
P < 0.01 Fisher's exact test.
Mutations in the intronic poly(T)11 nucleotide repeats in IVS 4 of MRE11 were found in six lines (Table 2): five mutations in MSI‐positive lines (OPL‐3, Daudi, HPB‐ALL, Jurkat and P31/Fujioka) and one mutation in an MSI‐negative line (OPL‐4). All but one (Daudi) of these lines showed shortening by only 1 bp. Except for the 1‐bp heterozygous lengthening found in the Daudi cell line, all mutations were homozygous. No mutations were observed in five normal blood samples. The frequency of MRE11 mutation was significantly higher in MSI‐positive than MSI‐negative cell lines (P < 0.01).
As for the ORF of RAD50, 1‐bp heterozygous lengthening was found in the poly(A)9 nucleotide repeats in the Jurkat cell line. No mutation was found in the poly(A)7 nucleotide repeats in the ORF of NBS1 or poly(A)10 nucleotide repeats in the ORF of ATR. It is noteworthy that the MSI‐positive Jurkat line showed mutations in ATM, MRE11 and RAD50.
Aberrant splicing in ATM (497del22) and MRE11 (315del88)
The intronic mononucleotide repeat mutations were expected to generate aberrant splicing in the next exon of ATM (497del22) and MRE11 (315del88), as previously reported in colorectal cancer cell lines (Fig. 1A).( 18 , 19 ) PCR amplification of the cDNAs for fragments containing exon 8 of ATM and exon 5 of MRE11 was carried out using primer pairs as mentioned before. The electrophoresis revealed that aberrant bands in ATM were observed in two of five normal blood samples, suggesting the presence of transcript at a lesser intensity in normal controls (Fig. 1B). Aberrant bands were observed in 13 of 13 (100%) MSI‐positive lines and in 21 of 37 (56.8%) MSI‐negative lines. Among the 13 MSI‐positive lines, eight (61.5%) showed intronic mononucleotide mutations, whereas only one of the 21 MSI‐negative lines with aberrant bands (4.8%) showed an intronic mutation. The RI was stronger in five MSI‐positive lines (P30/Ohkubo, Jurkat, OPL‐3, CTV‐1 and MOLT‐14) harboring intronic mononucleotide mutations of 2 bp or more (RI, mean 1.03 ± 0.35) compared to MSI‐positives lines without mutations or with only 1‐bp mutations (RI, mean 0.32 ± 0.15) (Fig. 1B). The RI in one MSI‐negative line (Katata) and two normal blood samples in which aberrant bands were observed was 0.13 ± 0.07, 0.21 ± 0.04 and 0.29 ± 0.03, respectively.
Figure 1.
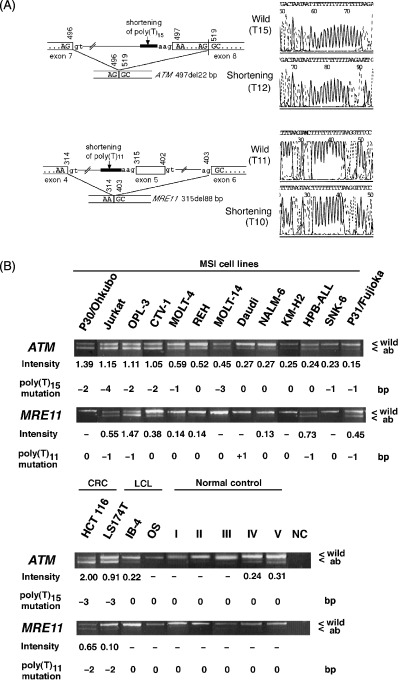
(A) Schematic representation of a 22‐bp deletion of exon 8 of ATM (497del22) and the skipping of exon 5 of MRE11 (315del88). Length variation of the poly(T)15 nucleotide repeats in intervening sequence (IVS) 7 of ATM was expected to cause aberrant splicing, which results in a 22‐bp deletion in exon 8. Length variation of the intronic poly(T)11 nucleotide in IVS 4 of MRE11 was expected to cause aberrant splicing, which results in the skipping of exon 5. Nucleotide numbering starts with the adenine residue in the start codon. (B) Aberrant transcripts of ATM and MRE11. Polymerase chain reaction amplification of the cDNA of 13 microsatellite instability (MSI)‐positive leukemia–lymphoma cell lines for fragments containing exon 8 of ATM and exon 5 of MRE11 were carried out. Aberrant bands for ATM and MRE11 were observed in 13 (100%) and eight (61.5%) lines, respectively. Two lymphoblastoid cell lines (IB‐4 and OS) and five peripheral lymphocytes from healthy volunteer were used as controls, whereas two colorectal carcinoma cell lines (HCT 116 and LS 174T) known to harbor MSI were used as positive controls. The ratios of intensity (RI) of the aberrant to wild‐type band was measured using Adobe PhotoshopTM. A ratio of less than 0.10 was regarded as negative. The intensity of the ATM aberrant transcript was stronger in five lines (P30/Ohkubo, Jurkat, OPL‐3, CTV‐1 and MOLT‐14) harboring a mutation of 2 bp or more in intronic poly(T)15 nucleotide repeats in IVS 7 of ATM (RI, mean 1.03 ± 0.35) than in those lines without a mutation or with only a 1‐bp mutation (RI, mean 0.32 ± 0.15). Of the lines with a detectable MRE11 aberrant transcript, strong intensity was observed in four lines (Jurkat, OPL‐3, HPB‐ALL and P31/Fujioka) with mutations in the intronic poly(T)11 nucleotide repeats in IVS 4 of MRE11 (RI, 0.80 ± 0.46) compared to those without mutation (RI, 0.20 ± 0.12). ab, aberrant band; CRC, colorectal cancer cell line; NC, negative control; wild, wild‐type band. *Length variation was detected as heterozygous.
The aberrant bands for MRE11 were found in eight of 13 (61.5%) MSI‐positive and two of 37 (5.4%) MSI‐negative lines. Aberrant bands were not observed in normal blood samples. Among the eight MSI‐positive lines with aberrant bands, 1‐bp intronic mononucleotide mutations were found in four lines (Jurkat, OPL‐3, HPB‐ALL and P31/Fujioka), which also showed a stronger RI of the aberrant to wild‐type bands (RI, mean 0.80 ± 0.46) as compared to lines without intronic mononucleotide mutations (RI, mean 0.20 ± 0.12). The RI of two MSI‐negative lines with aberrant bands was 0.11 ± 0.01 for CCRF‐CEM and 0.19 ± 0.09 for Namalwa.
As representatives, the aberrant bands found in the OPL‐3 line were cloned and sequenced, revealing a 22‐bp deletion in exon 8 of ATM (497del22, GenBank accession no. U33841) and the exclusion of the whole exon 5 (which is normally included in mRNA) of MRE11 (315del88, GenBank accession no. NM_005591) (data not shown). Adenine within the start codon was defined as nucleotide number 1.
Expression levels of ATM, MRE11, RAD50, NBS1 and ATR
In the real‐time PCR, the expression levels of ATM and MRE11 in 13 MSI‐positive lines (mean 80.30 ± 120.19 and 142.28 ± 195.27, respectively) were significantly higher than those in 37 MSI‐negative lines (mean 30.07 ± 41.44 and 57.86 ± 91.81, respectively; P < 0.05) (Fig. 2A). In contrast, no significant differences were found in the expression levels of RAD50, NBS1 and ATR between MSI‐positive and MSI‐negative lines (mean 33.03 ± 50.52 and 26.54 ± 35.18, 106.83 ± 183.28 and 51.79 ± 77.82, 64.63 ± 85.03 and 37.82 ± 49.02, respectively).
Figure 2.
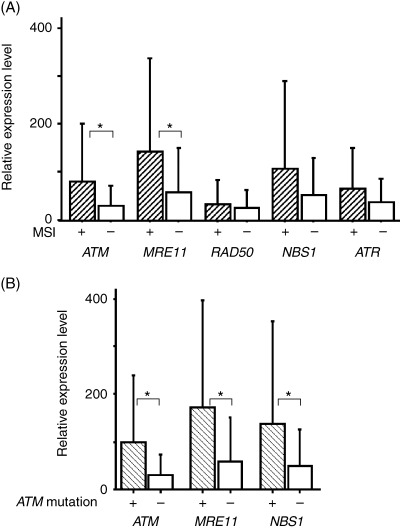
(A) Gene expression levels of ATM and MRE11 were significantly higher in microsatellite instability (MSI)‐positive than MSI‐negative leukemia–lymphoma cell lines, whereas there was no significant difference in the expression of RAD50, NBS1 or ATR. (B) In cell lines with a mutation in the intronic poly(T)15 nucleotide repeats in intervening sequence (IVS) 7 of ATM, the expression levels of MRE11 and NBS1 were also significantly different from those in lines without the mutation. The expression level of each gene in IB‐4 was defined as 100, and the relative gene expression levels in each cell line was calculated. *P < 0.05.
The expression levels of ATM, MRE11 and NBS1 were higher in cell lines with ATM mutations (mean 100.45 ± 137.82, 172.09 ± 225.61 and 137.08 ± 215.73, respectively) than in lines without mutations (mean 30.57 ± 42.30, 59.64 ± 90.61 and 50.46 ± 74.43, respectively; P < 0.05) (Fig. 2B). Mutation of MRE11 upregulated the expression of MRE11 itself, whereas the expression of other genes was not affected (data not shown).
DSB repair assays
To evaluate whether defects in MMR function may correlate with impairment of the DSB repair system in leukemia–lymphoma cell lines, kinetics of DNA DSB repair were examined using the PFGE. More than 80% of the DSB in IB‐4, a lymphoblastoid cell line derived from healthy individuals, were repaired within 2 h of IR exposure (Fig. 3A). Kinetics of DNA DSB repair was examined in 22 cell line selected randomly, comprising eight MSI‐positive and 14 MSI‐negative cell lines (Fig. 3B). DSB that were unrepaired after 2 h of incubation were then compared. Degrees of delay or abrogation of DSB repair, as shown by unrepaired DSB in eight MSI‐positive lines (mean 0.42 ± 0.25), were significantly higher than those in 14 MSI‐negative lines (mean 0.16 ± 0.10; P < 0.05) (Fig. 3C).
Figure 3.
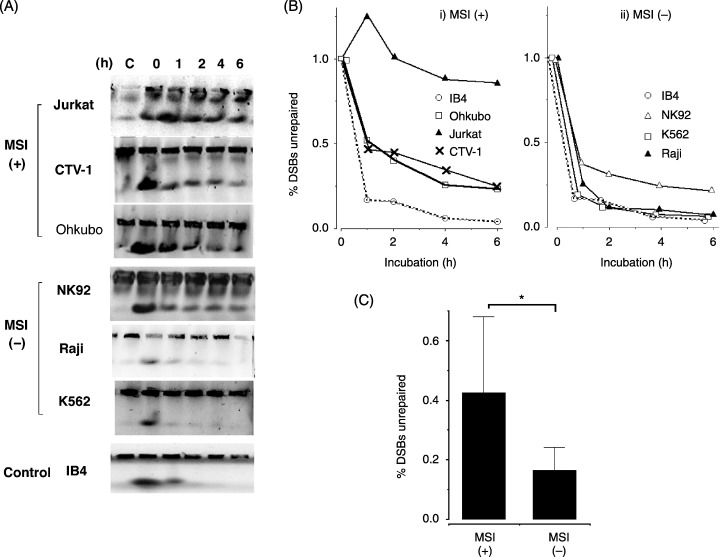
Induction and repair of DNA double‐strand breaks (DSB) after ionizing radiation (IR) exposure determined by pulsed‐field gel electrophoresis (PFGE). (A) Ethidium bromide‐stained gel after PFGE. Equal numbers of cells were embedded in 0.8% agarose plugs and exposed to 20 Gy IR. Cells were allowed to repair DSB for 0–6 h at 37°C, they were then lysed to extract DNA and subjected to PFGE. Untreated control is shown in the first lane. The upper bands are derived from undamaged DNA or repaired DNA in wells. The lower bands show the compression zones (CZ) consisting of DSB DNA. (B) Quantification of DSB repair using the results of representative PFGE. The percentage of unrepaired DSB was calculated by the ratio of signal from DSB DNA in the CZ to DNA in the corresponding well. The percentage of unrepaired DSB at 0 h of incubation was set at 100%. Symbols represent cell lines: (i) IB‐4 (○), Ohkubo (□), Jurkat (▴), CTV‐1 (×) (ii) IB‐4 (○), NK‐92 (▵), K‐562 (□), Raji (▴). (C) Repair of DSB after 2 h incubation were compared between MSI‐positive and MSI‐negative cell lines. Delay or abrogation of DSB repair as shown by unrepaired DSB in eight MSI‐positive lines (mean 0.42 ± 0.25) was significantly higher than that in 14 MSI‐negative lines (mean 0.16 ± 0.10) (P < 0.05).
Discussion
Thirteen (26.0%) of 50 leukemia–lymphoma cell lines showed MSI, as determined by mutations in both BAT‐25 and BAT‐26. Among these 13 lines, mutations of intronic mononucleotide repeats in ATM and MRE11 were found in eight (61.5%) and five (38.5%) lines, respectively. MSI status in leukemia–lymphoma cell lines were also reported by Takeuchi et al.( 35 ) (12 of 49 lines; 24.5%), Inoue et al.( 17 ) (11 of 91 lines; 12.1%) and Matheson and Hall( 36 ) (five of nine lines; 55.6%). Several cell lines showed different MSI status among these studies. The REH cell line was MSI‐positive in the present study as well as in those of Matheson and Hall( 36 ) and Takeuchi et al.,( 35 ) but was MSI‐negative in the study by Inoue et al.( 17 ) The Daudi cell line, which was MSI‐positive in the present study, was previously reported as MSI‐negative by Inoue et al.( 17 ) and by Matheson and Hall.( 36 ) These discrepancies might be caused by differences in the methods and definitions used in each study. The criteria in hematolymphoid malignancies has not been established as yet. In the present study, the presence of MSI was determined when mutations of both BAT‐25 and BAT‐26 were observed at GeneScan analysis. This criteria has been validated in defining high‐frequency of MSI in colorectal neoplasms,( 11 ) and was used in hematolymphoid malignancies.( 31 )
The same mutation of MRE11 has been reported in MSI‐positive colorectal, endometrial, ovarian and stomach tumors,( 19 , 20 , 21 ) whereas the information for mutation of ATM is limited to colorectal cancer cell lines.( 18 ) The mutations in mononucleotide repeats result in splicing abnormalities, which generate aberrant transcripts of ATM and MRE11. The RI of the aberrant ATM transcripts was higher in lines harboring a mutation of 2 bp or more in the intronic mononucleotide repeats than in those without the mutations. The mononucleotide repeats, especially polypyrimidine tracts together with GU and AG dinucleotides at the exon–intron and intron–exon junctions, respectively, function as splicing regulatory elements to ensure high accuracy of pre‐mRNA splicing site selection.( 37 ) The present data provided evidence that a defective MMR system affects the polypyrimidine tracts, thus resulting in a disorder of the splicing mechanism.
Despite the fact that both ATM and MRE11 were frequently mutated and aberrantly spliced in the MSI‐positive cell lines, the present data also showed differences. Eight of 13 MSI‐positive lines showed aberrant MRE11 transcripts, whereas only two MSI‐negative lines and no normal controls showed aberrant MRE11 transcripts. Furthermore, the DNA repair function was much more impaired in those lines harboring MRE11 mutations than those without the mutations. These findings confirmed that MRE11 is a target gene for MMR inactivation in leukemia–lymphoma cell lines as well as in other malignancies reported previously.( 19 , 20 , 21 )
As for ATM, strong expression of aberrant ATM transcripts were detected in lines with the MSI phenotype. Ejima et al. described the same aberrant splicing (497del22) exclusively in four colorectal cell lines with polypyrimidine tract mutations within the preceeding intron.( 18 ) Aberrantly spliced transcripts with lesser intensities were also observed in half of the MSI‐negative lines (56.8%) and in normal controls (40.0%). Recent studies have demonstrated that ATM has multiple functions other than DSB repair, such as cell‐cycle checkpoint, telomere maintenance and induction of apoptosis.( 1 , 3 , 4 , 6 , 7 ) The significance of the aberrant ATM transcripts is not clear as yet, because of the limited number of studies pertaining to its function.
Gene expression levels of ATM and MRE11 were significantly higher in MSI‐positive lines than MSI‐negative lines in the present study, which is in contrast to the studies of Ejima et al. and Giannini et al. using colorectal cancer cell lines.( 18 , 19 ) The present experiment included the same two cell lines (HCT 116 and LS 174T) used in their studies. Expression levels of all genes examined in these lines were apparently lower than the mean expression levels of MSI‐positive leukemia–lymphoma cell lines. Therefore the difference in gene expression levels found between our study and that of Giannini et al. might be due to the origin of the tumors. NBS1 expression was increased in the cell lines with ATM mutations, although there was no mutation in the NBS1 gene. ATM phosphorylates NBS1 on serine‐343, which may change the kinetics of the NBS1 protein as well as NBS1 mRNA. This might be a reason why NBS1 expression was increased in cell lines with ATM mutations even though there was no mutation in the NBS1 gene.( 3 , 4 )
Other genes engaged in DNA repair, RAD50, NBS1 and ATR, did not show mutations in any mononucleotide repeats except in one cell line. Expression levels of these genes did not differ between MSI‐positive and MSI‐negative leukemia–lymphoma cell lines. The sensitivity of detecting the mutations in the present study would be adequate because the mutations detected in the two cell lines (HCT116 and LS174T) of this study were identical to those reported in a previous study (data not shown). ATR and RAD50 were reported to be necessary not only for DNA damage repair but also for normal growth of the individual cells.( 8 , 9 , 10 , 38 ) The absence of mutations in mononucleotide repeats in ATR and RAD50 might be due to their essential role for cell survival.
The present study suggests that impairment of the MMR system generates aberrant transcripts in DSB repair genes, especially ATM and MRE11 but not RAD50, NBS1 or ATR, in hematolymphoid malignancies. This might result in inactivation of the DSB repair system, thus inducing a harmful chain reaction such as an acceleration of genome instability and accumulation of genetic damage.
Acknowledgments
We thank Dr T. Al Saati (Laboratoire d’Anatomie Pathologique CHU Purpan, Toulouse, France) for providing the Deglis cell line, Dr G. W. Bornkamm (GSF‐Research Center for Environment and Health, Munich, Germany) for BL36, BL60 and BL137, Dr E. Kieff (Brigham and Women's Hospital, Boston, MA, USA) for IB‐4, Dr P. W. Tucker (University of Texas, Austin, TX, USA) for NAB‐2, Drs M. Daibata and S. Imai (Kochi Medical School, Nangoku, Japan) for Pal‐1 and Katata, Drs M. Matsuoka and Y. Taniguchi (Kyoto University, Kyoto, Japan) for MT‐1, MT‐2, MT‐4 and TL‐Om1, Dr Y. Matsuo (Hayashibara Biochemical Laboratories, Okayama, Japan) for MOTN‐1 and HEL, Dr N. Shimizu (Tokyo Medical and Dental University, Tokyo, Japan) for SNT‐8 and SNK‐6, and Dr M. Yagita (Tazuke‐Kofukai Medical Research Institute, Osaka, Japan) for the KHYG‐1 cell line. This work was supported by grants from the Ministry of Education, Science, Culture and Sports, Japan (15406013, 16390105, 40244933).
References
- 1. Khanna KK, Jackson SP. DNA double‐strand breaks: signaling, repair and the cancer connection. Nat Genet 2001; 27: 247–54. [DOI] [PubMed] [Google Scholar]
- 2. Kolodner R. Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends Biochem Sci 1995; 20: 397–401. [DOI] [PubMed] [Google Scholar]
- 3. Hopfner KP, Putnam CD, Tainer JA. DNA double‐strand break repair from head to tail. Curr Opin Struct Biol 2002; 12: 115–22. [DOI] [PubMed] [Google Scholar]
- 4. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3: 155–68. [DOI] [PubMed] [Google Scholar]
- 5. D’Amours D, Jackson SP. The MRE11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 5: 317–27. [DOI] [PubMed] [Google Scholar]
- 6. Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004; 304: 93–6. [DOI] [PubMed] [Google Scholar]
- 7. Kastan MB, Bartek J. Cell‐cycle checkpoints and cancer. Nature 2004; 432: 316–23. [DOI] [PubMed] [Google Scholar]
- 8. Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17: 615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- 10. de Klein A, Muijtjens M, van Os R et al. Targeted disruption of the cell‐cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10: 479–82. [DOI] [PubMed] [Google Scholar]
- 11. Boland CR, Thibodeau SN, Hamilton SR et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58: 5248–57. [PubMed] [Google Scholar]
- 12. Muller A, Fishel R. Mismatch repair and the hereditary non‐polyposis colorectal cancer syndrome (HNPCC). Cancer Invest 2002; 20: 102–9. [DOI] [PubMed] [Google Scholar]
- 13. Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol 1995; 7: 83–9. [PubMed] [Google Scholar]
- 14. Gamberi B, Gaidano G, Parsa N et al. Microsatellite instability is rare in B‐cell non‐Hodgkin's lymphomas. Blood 1997; 89: 975–9. [PubMed] [Google Scholar]
- 15. Takakuwa T, Li T, Kanno H, Nakatsuka S, Aozasa K. No evidence of replication error phenotype in nasal NK/T cell lymphoma. Int J Cancer 1999; 84: 623. [DOI] [PubMed] [Google Scholar]
- 16. Kodera T, Kohno T, Takakura S et al. Microsatellite instability in lymphoid leukemia and lymphoma cell lines but not in myeloid leukemia cell lines. Genes Chromosomes Cancer 1999; 26: 267–9. [DOI] [PubMed] [Google Scholar]
- 17. Inoue K, Kohno T, Takakura S, Hayashi Y, Mizoguchi H, Yokota J. Frequent microsatellite instability and BAX mutations in T cell acute lymphoblastic leukemia cell lines. Leuk Res 2000; 24: 255–62. Erratum in: Leuk Res 2001; 25: 275–8. [DOI] [PubMed] [Google Scholar]
- 18. Ejima Y, Yang L, Sasaki MS. Aberrant splicing of the ATM gene associated with shortening of the intronic mononucleotide tract in human colon tumor cell lines: a novel mutation target of microsatelitte instability. Int J Cancer 2000; 86: 262–8. [DOI] [PubMed] [Google Scholar]
- 19. Giannini G, Ristori E, Cerignoli F et al. Human MRE11 is inactivated in mismatch repair‐deficient cancers. EMBO Rep 2002; 3: 248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giannini G, Rinaldi C, Ristori E et al. Mutations of an intronic repeat induce impaired MRE11 expression in primary human cancer with microsatellite instability. Oncogene 2004; 23: 2640–7. [DOI] [PubMed] [Google Scholar]
- 21. Ottini L, Falchetti M, Saieva C et al. MRE11 expression is impaired in gastric cancer with microsatellite instability. Carcinogenesis 2004; 25: 2337–43. [DOI] [PubMed] [Google Scholar]
- 22. Kim NG, Choi YR, Baek MJ et al. Frameshift mutations at coding mononucleotide repeats of the hRAD50 gene in gastrointestinal carcinomas with microsatellite instability. Cancer Res 2001; 61: 36–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menoyo A, Alazzouzi H, Espin E, Armengol M, Yamamoto H, Schwartz S Jr. Somatic mutations in the DNA damage‐response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res 2001; 61: 7727–30. [PubMed] [Google Scholar]
- 24. Kanno H, Yasunaga Y, Ohsawa M et al. Expression of Epstein–Barr virus latent infection genes and oncogenes in lymphoma cell lines derived from pyothorax‐associated lymphoma. Int J Cancer 1996; 67: 86–94. [DOI] [PubMed] [Google Scholar]
- 25. Takakuwa T, Luo WJ, Ham MF, Mizuki M, Iuchi K, Aozasa K. Establishment and characterization of unique cell lines derived from pyothorax‐associated lymphoma which develops in long‐standing pyothorax and is strongly associated with Epstein–Barr virus infection. Cancer Sci 2003; 94: 858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daibata M, Taguchi T, Nemoto Y et al. Epstein–Barr virus (EBV)‐positive pyothorax‐associated lymphoma (PAL): chromosomal integration of EBV in a novel CD2‐positive PAL B‐cell line. Br J Haematol 2002; 117: 546–57. [DOI] [PubMed] [Google Scholar]
- 27. Matsuo Y, Drexler HG. Immunoprofiling of cell lines derived from natural killer‐cell and natural killer‐like T‐cell leukemia–lymphoma. Leuk Res 2003; 27: 935–45. [DOI] [PubMed] [Google Scholar]
- 28. Al Saati T, Delecluze HJ, Chittal S et al. A novel human lymphoma cell line (Deglis) with dual B/T phenotype and gene rearrangements and containing Epstein–Barr virus genomes. Blood 1992; 80: 209–16. [PubMed] [Google Scholar]
- 29. Hoang JM, Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R. BAT‐26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res 1997; 57: 300–3. [PubMed] [Google Scholar]
- 30. Zhou XP, Hoang JM, Li YJ et al. Determination of the replication error phenotype in human tumors without the requirement for matching normal DNA by analysis of mononucleotide repeat microsatellites. Genes Chromosomes Cancer 1998; 21: 101–7. [DOI] [PubMed] [Google Scholar]
- 31. Duval A, Raphael M, Brennetot C et al. The mutator pathway is a feature of immunodeficiency‐related lymphomas. Proc Natl Acad Sci USA 2004; 101: 5002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takakuwa T, Luo WJ, Ham MF, Sakane‐Ishikawa E, Wada N, Aozasa K. Integration of Epstein–Barr virus into chromosome 6q15 of Burkitt lymphoma cell line (Raji) induces loss of BACH2 expression. Am J Pathol 2004; 164: 967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abbott DW, Freeman ML, Holt JT. Double‐strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst 1998; 90: 978–85. [DOI] [PubMed] [Google Scholar]
- 34. Liu A, Takakuwa T, Fujita S et al. Alterations of DNA damage‐response genes ATM and ATR in pyothorax‐associated lymphoma. Lab Invest 2005; 85: 436–46. [DOI] [PubMed] [Google Scholar]
- 35. Takeuchi S, Takeuchi N, Fermin AC, Taguchi H, Koeffler HP. Frameshift mutations in caspase‐5 and other target genes in leukemia and lymphoma cell lines having microsatellite instability. Leuk Res 2003; 27: 359–61. [DOI] [PubMed] [Google Scholar]
- 36. Matheson EC, Hall AG. Assessment of mismatch repair function in leukaemic cell lines and blasts from children with acute lymphoblastic leukaemia. Carcinogenesis 2003; 24: 31–8. [DOI] [PubMed] [Google Scholar]
- 37. Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 2004; 5: 389–96. [DOI] [PubMed] [Google Scholar]
- 38. Luo G, Yao MS, Bender CF et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA 1999; 96: 7376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]