

Abstract
Although adhesive interactions between metastasizing cancer cells and vascular endothelial cells are critical in hematogenous metastasis, the early molecular events of the cancer–endothelial interaction remain largely obscure. Here we investigated the functional impact of cancer cells on endothelial permeability. We examined the binding of human pancreatic carcinoma cells MIA PaCa‐2, PANC‐1 and PSN‐1 to a human umbilical vein endothelial cell (HUVEC) monolayer and the subsequent changes in the transendothelial electronic resistance (TEER) of the HUVEC. We found that MIA PaCa‐2 and PANC‐1 cells preferentially bound to the tri‐cellular corners of HUVEC and induced a rapid and irreversible reduction of TEER. The reduction of HUVEC TEER was associated with the focal disengagement of endothelial junctional adhesion molecules VE‐cadherin and CD31. Blocking antibodies to integrin β1, CD44, or CD9 affected neither the MIA PaCa‐2 binding to HUVEC nor the reduction of TEER. Specific inhibitors for metalloproteinases, tyrosine‐kinases and lipoxigenases, and a neutralizing anti‐vascular endothelial growth factor antibody failed to affect the MIA PaCa‐2‐induced reduction of HUVEC TEER, whereas treatment of the cells with paraformaldehyde or cytochalasin B abrogated the TEER reduction. These findings indicate that the MIA PaCa‐2 cells bind selectively to endothelial tri‐cellular corners, triggering a reduction of HUVEC TEER, which requires the active metabolism and intact actin cytoskeleton of the carcinoma cells, and is apparently unrelated to previously described cell adhesion and soluble factor pathways. Our data indicate a novel cell‐contact‐dependent mechanism for the cancer cell‐mediated breakdown of endothelial barrier functions, which may be important in hematogenous cancer metastasis. (Cancer Sci 2005; 96: 766–773)
Abbreviations:
- CMFDA
5‐chloromethylfluorescein diacetate
- HUVEC
human umbilical vein endothelial cell
- mAb
monoclonal antibody
- PFA
paraformaldehyde
- PBS
phosphate‐buffered saline
- TEER
transendothelial electronic resistance
- TIMP
tissue inhibitor of metalloproteinase
- VEGF
vascular endothelial growth factor
The process of cancer metastasis consists of a series of sequential steps, all of which must be successfully completed to give rise to new metastatic foci.( 1 , 2 , 3 ) In hematogenous metastasis, cancer cells that have escaped from the primary sites into the circulation are transported to distant organs and entrapped in the capillary beds by adhering to capillary endothelial cells.( 3 , 4 , 5 ) The metastasizing cancer cells need to extravasate through the vascular wall to invade the surrounding host tissues and establish secondary cancer sites.( 4 , 5 ) The extravasation process involves the breakdown of impermeable endothelial monolayers and is believed to be one of the major rate‐limiting steps in hematogenous metastasis.( 1 , 4 , 5 )
Recent intravital imaging studies visualized the early events of cancer–endothelial cell interactions. In a liver metastasis model, cancer cells entrapped in sinusoidal capillaries induce rapid endothelial cell retraction in situ ( 6 ) and extravasate with high efficiency.( 7 , 8 ) In a pulmonary metastasis model, endothelium‐attached cancer cells proliferate within the blood vessels for a few days and subsequently penetrate the surrounding host tissues by destroying the vascular walls.( 9 , 10 ) Endothelial cells lining the blood vessels control the passage of cells attached to their luminal surface by forming physiological barriers using endothelial junctions.( 4 , 5 ) Although complex interactions between the metastasizing cancer cells and vascular endothelial cells are critical for establishing metastasis,( 3 , 4 , 5 ) the early molecular events of the cancer–endothelial cell interactions remain largely obscure.
The endothelial junctional complexes are comprised of three distinct junctional structures, namely tight junctions, adherens junctions and gap junctions.( 11 ) Tight junctions mainly regulate the paracellular permeability by forming a ‘barrier’ and ‘fence’ within the plasma membrane of adjacent endothelial cells. The adherens junctions also regulate vascular permeability and add mechanical strength to the endothelial linkage, whereas gap junctions mediate communication between endothelial cells. Upon interaction with cancer cells, vascular permeability is often down‐regulated by modifications to the molecular organization of the endothelial junction complexes.( 12 , 13 ) Although a large array of cell adhesion molecules,( 14 , 15 , 16 , 17 , 18 , 19 ) metalloproteinases( 20 , 21 ) and soluble factors( 22 , 23 , 24 , 25 , 26 ) have been implicated in the active cross‐talk between cancer cells and blood vessels, the mechanisms underlying the breakdown of endothelial barrier functions remain to be fully elucidated.
Previous studies demonstrated that the molecular architecture of endothelial junctional zones appears heterogeneous and complex.( 11 ) In particular, at tri‐cellular corners, where the borders of three cells converge, the tight junctions are reported to be partially discontinuous,( 27 , 28 ) yet the barrier properties are preserved. Due to these unique structural and functional features, endothelial tri‐cellular corners are hypothesized to be potential sites for the opening and closing of paracellular transmigration pathways, which allow the passage of circulating cells to the extravascular tissue.( 27 , 29 , 30 ) In support of this hypothesis, Burns et al. demonstrated that endothelial tri‐cellular corners are the preferred sites for neutrophil firm adhesion and extravasation under physiological flow conditions in vitro and in vivo.( 31 , 32 , 33 )
Pancreatic cancers often form hematogenous metastases in the early phase of the disease, resulting in a poor prognosis for patients.( 34 ) In the present study, we used highly invasive human pancreatic carcinoma cells( 22 , 35 , 36 ) and human vascular endothelial cells to investigate the influence of cancer cells on the permeability of vascular endothelial cells. Transendothelial electronic resistance (TEER) and cell binding analysis showed that pancreatic carcinoma cells could induce a rapid and irreversible reduction of TEER and preferentially bound to endothelial tri‐cellular corners in vitro. The cancer cell binding to endothelial cells disrupted the endothelial junction complex at the site of cell contact, which was associated with the focal disappearance of VE‐cadherin and CD31. Our results suggest that endothelial tri‐cellular corners are preferred sites for invasive human pancreatic cancer cell binding and that the cancer cells binding to these particular sites appear to induce potently the disengagement of endothelial junctions. Our data also indicate a novel cell‐contact‐dependent mechanism for the cancer‐cell‐mediated breakdown of endothelial barrier functions, which may be important in hematogenous cancer metastasis.
Materials and Methods
Reagents
Recombinant human vascular endothelial growth factor (VEGF) and goat anti‐human VEGF antibody were purchased from R&D Systems (Minneapolis, MN, USA). Tissue inhibitor of metalloproteinase (TIMP)‐1 and TIMP‐2 were purchased from Daiichi Fine Chemical (Takaoka, Japan). Monoclonal antibodies (mAbs) against human CD31 (hec 7) and CD29/integrin β1 (Lia1/2) were obtained through the VIth Human Leukocyte Differentiation Antigen Workshop (Kobe, Japan, 1996). Anti‐human CD44 mAb (BRIC235) was from the International Blood Group Reference Laboratory (Bristol, UK). Anti‐human CD9 mAb (MM2/57) was from Biodesign (Saco, ME, USA). Unless otherwise noted, reagents were obtained from Sigma Aldrich (St Louis, MO, USA).
Cell culture
Human pancreatic carcinoma cell lines MIA PaCa‐2 and PANC‐1 were obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan) and PSN‐1 was a kind gift from Dr F. Nakata (Osaka City University Medical School, Osaka, Japan). The pancreatic carcinoma cells were cultured in RPMI‐1640 medium supplemented with 10% fetal calf serum, 1% (v/v) 100 × nonessential amino acids, 1 mM sodium pyruvate, 2 mM L‐glutamine, 50 µM 2‐mercaptoethanol, 100 units/mL penicillin and 100 µg/mL streptomycin. Passage 1 human umbilical vein endothelial cells (HUVEC) were obtained from BioWhittaker (Walkersville, MD, USA). HUVEC cultures were serially passaged, maintained in EGM‐2 medium (BioWhittaker) and used before the fifth passage. CHO cells were cultured in Ham's F‐12 medium containing the same supplements as above.
Measurement of TEER
HUVEC (0.5 × 105 cells) were cultured for 5–7 days to form a monolayer on a polycarbonate filter insert (Transwell, pore size 0.4 µm, 6.5 mm diameter; Corning, Corning, NY, USA) coated with human plasma fibronectin (Invitrogen, Carlsbad, CA, USA) (10 µg/mL). To measure TEER, the filter insert with HUVEC was transferred to a resistance measurement chamber (ENDOHM‐12; World Precision Instruments, Sarasota, FL, USA) and the upper luminal compartment and lower abluminal compartment were filled with 100 µL and 600 µL of culture medium, respectively. Cancer cells (1.0 × 105 cells) were added to the upper compartment of the resistance measurement chamber, and TEER across the HUVEC monolayer was measured at 37°C with an electrode placed in each compartment, at various time points.( 37 ) The electrical resistance of individual HUVEC monolayers was obtained by subtracting the resistance of a corresponding naked filter coated with fibronectin from that of the filter on which HUVEC were grown. Changes in TEER are presented as a percentage of the control value. Thrombin, which induces a transient reduction of HUVEC TEER, was used as a positive control.( 38 ) For inhibition studies, cancer cells were pretreated for 2 h with TIMP‐1 (10 µg/mL), TIMP‐2 (10 µg/mL), KB‐R7785( 39 ) (10 µM), marimastat( 40 ) (1 µM), or brefeldin A (10 µg/mL), or for 6 h with nordihydroguaiaretic acid (10 µM) and used without washing. Cancer cells pretreated with 1% paraformaldehyde (PFA) (2 h) or 100 µM cytochalasin B (3 h) were used after extensive washing with the culture medium. To study the effects of neutralizing mAbs, cancer cells were pretreated with anti‐CD29 mAb (10 µg/mL) and/or anti‐CD44 mAb (10 µg/mL) or anti‐CD9 mAb (10 µg/mL) for 30 min and used after washing. HUVEC monolayers pretreated with herbimycin A (1 µM, 30 min) were used after washing with the culture medium. All experiments were performed at least three times and the representative data are shown.
Preparation of culture supernatant
MIA PaCa‐2 cells (5.0 × 105 cells) were seeded on 75 cm2 flasks and cultured for 4 days to reach confluence. After being washed three times with phosphate‐buffered saline (PBS) to remove the serum components, the cells were further incubated in 10 mL serum‐free RPMI‐1640 medium. The culture supernatants were collected at 2, 24 and 48 h, and concentrated 100‐fold by ultrafiltration using a Centricon (CentriPlus YM‐3; Millipore, Milford, MA, USA). The concentrated culture supernatants were dialyzed against PBS and used in the TEER assay (10 µL/well).
Adhesion assays
Cancer cells (1.0 × 105 cells) were added to the HUVEC monolayer grown on fibronectin‐coated transwell inserts and incubated for the indicated times. After unbound cancer cells were removed by gentle washing, the cells were fixed with 0.05% glutaraldehyde in PBS for 10 min at room temperature, washed twice with PBS and incubated for 30 s with 0.25% AgNO3. After being washed with PBS, they were mounted on a glass slide using Gel/Mount (Biomeda, Foster, CA, USA), and the silver staining was developed under UV light for 5 min. Bound cells were counted manually under the microscope (BX50; Olympus, Tokyo, Japan). In some experiments, MIA PaCa‐2 cells were treated with cytochalasin B as above and added to the HUVEC monolayer.
Immunofluorescence microscopy
MIA PaCa‐2 cells were labeled with 4 µM 5‐chloromethylfluorescein diacetate (CMFDA; Molecular Probes, Eugene, OR, USA) for 10 min at 37°C. The fluorescently labeled MIA PaCa‐2 cells (1.0 × 105 cells/well) were added to HUVEC monolayers and incubated for the indicated times. After unbound tumor cells were removed by gentle washing, the cells on the membranes were fixed in 3% PFA/PBS for 15 min at room temperature, followed by treatment with 0.2% Triton X‐100/PBS for 5 min, and blocked in PBS containing 10% fetal calf serum for 30 min at room temperature. After being washed with PBS, the cells were incubated with an anti‐VE‐cadherin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti‐CD31 antibody and then with biotin‐conjugated donkey anti‐goat IgG (Chemicon, Temecula, CA, USA) or biotinylated goat anti‐mouse IgG (H + L) antibody (Vector Laboratories, Burlingame, CA, USA), followed by Alexa Fluor 594 Streptavidin (Molecular Probes). After being washed with PBS, the samples were mounted in Prolong Antifade (Molecular Probes) and observed using an LSM 410 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Results
Pancreatic carcinoma MIA PaCa‐2 and PANC‐1 cells induce a rapid and irreversible reduction of the TEER of HUVEC
HUVEC grown to confluence on a polycarbonate transwell insert were placed in a resistance measurement chamber. Invasive human pancreatic carcinoma cells, MIA PaCa‐2, PANC‐1, or PSN‐1 cells, were added to the upper compartment of the transwell to allow direct contact with the HUVEC, and the changes in HUVEC TEER were monitored for 2 h. As shown in Fig. 1, MIA PaCa‐2 and PANC‐1 induced a strong reduction in HUVEC TEER, whereas PSN‐1 induced only a modest reduction. After the addition of MIA PaCa‐2 or PANC‐1 cells, the reduction of HUVEC TEER was evident within 30 min and reached ∼50% of the value for untreated HUVEC after 2 h. Prolonged incubation with MIA PaCa‐2 cells further decreased the HUVEC TEER (< 20% of untreated HUVEC at 4 h). In contrast to the effects of MIA PaCa‐2 and PANC‐1 cells, thrombin elicited only a modest and transient decrease in HUVEC TEER. CHO cells failed to affect the HUVEC TEER. Together, these results indicate that certain pancreatic carcinoma cells rapidly induce a strong and irreversible reduction of HUVEC TEER. In the following experiments, we mainly used MIA PaCa‐2 cells to investigate the mechanisms underlying the reduction in TEER.
Figure 1.
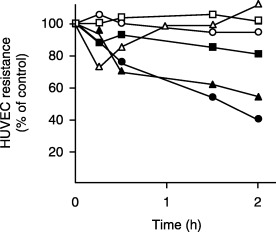
Certain pancreatic cancer cells decreased the transendothelial electronic resistance (TEER) of a human umbilical vein endothelial cell (HUVEC) monolayer. Human pancreatic cancer cells (MIA PaCa‐2 [•], PANC‐1 [▴] and PSN‐1 [▪]) or CHO cells (□) (1 × 105 cells/well) were added to HUVEC monolayers formed on fibronectin‐coated polycarbonate membranes in the upper chamber of a transwell chamber. The TEER of the HUVEC monolayer was measured at various time points. Thrombin (▵) (3 U/mL) and culture medium (○) were used as a positive and negative control, respectively.
Direct contact between MIA PaCa‐2 cells and HUVEC is important for the reduction of HUVEC TEER
We next sought to determine the relative contributions of direct cell contact and soluble factors to the MIA PaCa‐2‐induced reduction of HUVEC TEER. As shown in Fig. 2a, when MIA PaCa‐2 cells were added to the lower chamber so that they could not make direct contact with the HUVEC, they failed to decrease TEER. Concentrated MIA PaCa‐2 cell culture supernatants also failed to affect HUVEC TEER (Fig. 2b). The pretreatment of MIA PaCa‐2 cells with brefeldin A, which inhibits the secretion of various soluble factors,( 41 ) did not affect the MIA PaCa‐2 cell‐induced reduction of HUVEC TEER (Table 1). Because MIA PaCa‐2 cells produce VEGF,( 42 ) which can increase vascular permeability,( 25 , 26 ) we next tested the effects of a neutralizing anti‐VEGF antibody. As shown in Fig. 2c, an anti‐VEGF antibody that could abrogate the activities of exogenously added VEGF (10 nM) did not elicit detectable changes in the MIA PaCa‐2 cell‐induced reduction of HUVEC TEER (Fig. 2c). These observations collectively indicate that the direct cell–cell contact between MIA PaCa‐2 cells and HUVEC plays a major role in the reduction of HUVEC TEER and that soluble factors, such as VEGF, which may be produced by MIA PaCa‐2 cells, appear to be little involved in the reduction of HUVEC TEER under the conditions used in this study.
Figure 2.
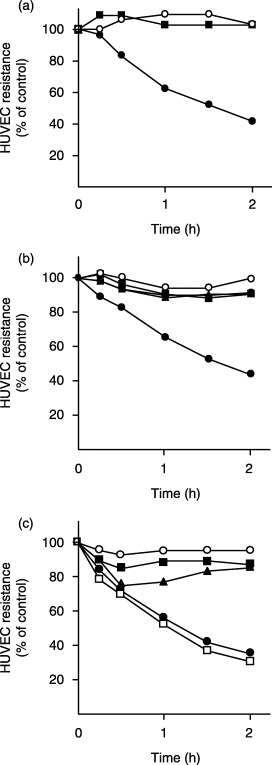
Direct cell contact between MIA PaCa‐2 cells and human umbilical vein endothelial cells (HUVEC) was important for the reduction of HUVEC transendothelial electronic resistance (TEER). (a) MIA PaCa‐2‐induced reduction of HUVEC TEER. MIA PaCa‐2 cells (1 × 105 cells/well) were added either to the upper (•) or lower (▪) well of a transwell chamber, and HUVEC TEER was measured at various time points. Culture medium (○) was used as a negative control. (b) Effects of MIA PaCa‐2 culture supernatants on HUVEC TEER. MIA PaCa‐2 cells (1 × 105 cells/well) (•) and concentrated culture supernatants of MIA PaCa‐2 cells harvested at 2 h (□), 24 h (▪) and 48 h (▴) were added to HUVEC monolayers and TEER was measured. Fresh culture medium (○) was used as a control. (c) Effects of anti‐ vascular endothelial growth factor (VEGF) antibody on the MIA PaCa‐2 induced reduction of HUVEC TEER. MIA PaCa‐2 cells (1 × 105 cells/well) were added to HUVEC in the presence (□) or absence (•) of an anti‐VEGF antibody (20 µg/mL), and HUVEC TEER was examined. In parallel, the effects of exogenously added VEGF (10 nM) on HUVEC TEER were tested in the presence (▪) or absence (▴) of the anti‐VEGF antibody. Culture medium (○) was used as a negative control.
Table 1.
Effects of various inhibitors on MIA PaCa‐2‐induced reduction of human umbilical vein endothelial cell (HUVEC) transendothelial electronic resistance (TEER)
| Inhibitors | Concentration | Inhibition of TEER reduction † |
|---|---|---|
| Paraformaldehyde | 1% | +++ |
| Cytochalasin B | 100 µM | ++ |
| Brefeldin A | 10 µg/mL | – |
| Anti‐β1 integrin | 10 µg/mL | – |
| Anti‐CD44 | 10 µg/mL | – |
| Anti‐β1 integrin | 10 µg/mL | – |
| + Anti‐CD44 | 10 µg/mL | |
| Anti‐CD9 | 10 µg/mL | – |
| KB‐R7785 | 10 µM | – |
| Marimastat | 1 µM | – |
| TIMP‐1 | 10 µg/mL | – |
| TIMP‐2 | 10 µg/mL | – |
| Herbimycin A | 1 µM | – |
| Nordihydroguaiaretic acid | 10 µM | – |
TEER of HUVEC was measured at 2 h after the addition of MIA PaCa‐2 cells.
70–100% of the reduction of TEER was inhibited;
, 30–70%; –, 0%.
MIA PaCa‐2 cell‐induced reduction of HUVEC TEER is dependent on an active metabolism and intact cytoskeleton of MIA PaCa‐2 cells
Cancer cell adhesion to vascular endothelium is regulated by the coordinated actions of cell‐adhesion molecules and cytoskeletal components. We found that cytochalasin B, which disrupts the actin cytoskeleton, significantly inhibited the binding of MIA PaCa‐2 cells to the HUVEC monolayer (Fig. 3a) and the TEER reduction (Fig. 3b). MIA PaCa‐2 cells express high levels of α3β1 and α5β1 integrins, CD44 and CD9 (data not shown), which have been implicated in various cancer–endothelial cell interactions. These adhesion molecules, however, did not appear to play significant roles in the MIA PaCa‐2 cell binding to HUVEC, given that neutralizing anti‐integrin β1 and anti‐CD44 mAbs, used either alone or in combination, or a neutralizing anti‐CD9 mAb, showed no apparent effects (Table 1). Other adhesion molecules implicated in tumor–host interactions, such as α1β1, αLβ2, αMβ2, α4β1, α4β7, αvβ3 integrins, nectin‐2 and DNAM‐1, were not detectable on the surface of MIA PaCa‐2 cells (data not shown).
Figure 3.
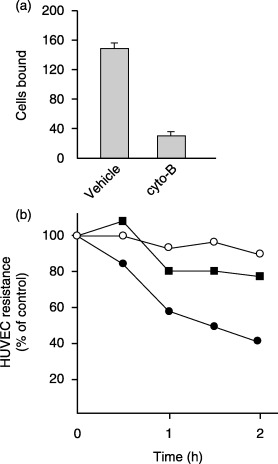
The intact actin cytoskeleton of MIA PaCa‐2 cells was required for both binding to human umbilical vein endothelial cells (HUVEC) and the reduction of HUVEC transendothelial electronic resistance (TEER). (a) MIA PaCa‐2 cell binding to the HUVEC monolayer. Cytochalasin B‐treated (100 µM, 3 h) or vehicle‐treated MIA PaCa‐2 cells were added to the HUVEC monolayer, and incubated for 2 h at 37°C. After washing, the number of MIA PaCa‐2 cells that bound to HUVEC was counted. The number of bound MIA PaCa‐2 cells was scored for three 200× microscopic fields at the indicated time points. The data are presented as the number of bound cells in a 0.658 mm2 field (mean ± SD). (b) Effects of treating MIA PaCa‐2 cells with cytochalasin B on the reduction of HUVEC TEER. Cytochalasin B‐treated (▪) or vehicle‐treated (•) MIA PaCa‐2 cells were added to the HUVEC monolayer, and HUVEC TEER was measured. Culture medium was added as a negative control (○).
We next asked if MIA PaCa‐2 cells require an intact metabolism to reduce HUVEC TEER. Pretreatment of MIA PaCa‐2 cells with PFA abrogated the TEER reduction, although it affected MIA PACA‐2 cell binding only marginally (Table 1 and data not shown). We also investigated the potential contribution of metalloproteinases, tyrosine‐kinases and lipoxigenases in the reduction of TEER using specific inhibitors (KB‐R7785, marimastat, TIMP‐1, TIMP‐2, herbimycin A and nordihydroguaiaretic acid) at concentrations sufficient to block their target enzymes,( 24 , 40 , 43 , 44 , 45 ) but no significant effects were observed (Table 1). Together, these results indicate that an active metabolism of MIA PaCa‐2 cells is indispensable for the reduction of HUVEC TEER and that an intact actin cytoskeleton of the MIA PaCa‐2 cells is also required for this process.
MIA PaCa‐2 cells and PANC‐1 cells preferentially adhere to HUVEC at tri‐cellular corners
Previous studies by Burns et al.( 31 , 32 , 33 ) demonstrated that neutrophils selectively adhere to the tri‐cellular corners of HUVEC and transmigrate thereafter. We thus examined whether invasive cancer cells also preferentially bind to endothelial tri‐cellular corners. In this experiment, the binding of MIA PaCa‐2 cells to HUVEC was differentially scored as binding to tri‐cellular corners or to the region between two adjacent endothelial cells, and the results were compared with those obtained using PANC‐1 and PSN‐1 cells. As shown in Fig. 4a, silver nitrate staining allowed us to detect the precise position of the bound cancer cells in the HUVEC cell junction. Although MIA PaCa‐2, PANC‐1 and PSN‐1 showed comparable levels of binding (Fig. 4b), MIA PaCa‐2 and PANC‐1 cells, but not PSN‐1 cells, preferentially bound to HUVEC tri‐cellular corners; such binding accounted for approximately 75% and 70% of the MIA PaCa‐2 and PANC‐1 binding, respectively, but only 30% of the PSN‐1 binding (Fig. 4c). These observations indicate that MIA PaCa‐2 and PANC‐1 cells, which could induce a large reduction of TEER, preferentially bound to the tri‐cellular corners of HUVEC.
Figure 4.
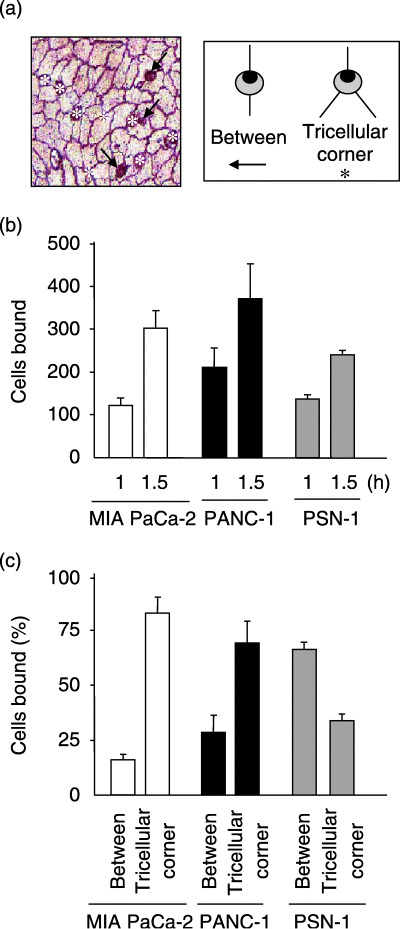
MIA PaCa‐2 cells and PANC‐1 cells preferentially adhered to human umbilical vein endothelial cells (HUVEC) at tri‐cellular corners. (a) MIA PaCa‐2 cells adhering to HUVEC were scored as adhering at tri‐cellular corners (asterisks), or between two adjacent endothelial cells (arrows). The photograph represents results obtained at 60 min. (b) Quantitative analysis of the MIA PaCa‐2, PANC‐1 and PSN‐1 cell binding to the HUVEC monolayer. The number of bound tumor cells was scored for three 200× microscopic fields at the indicated time points. The data are presented as the number of bound cells in a 0.658 mm2 field (mean ± SD). (c) MIA PaCa‐2 and PANC‐1 cells preferentially bound to the HUVEC monolayer at tri‐cellular corners. MIA PaCa‐2, PANC‐1, and PSN‐1 cells were added to the HUVEC monolayer and incubated for 1 h. After the unbound tumor cells were removed by gentle washing, the bound tumor cells and endothelial cell borders were stained with silver nitrate. The bound tumor cells were easily distinguishable from HUVEC due to their round shape. The bar graph displays the percentages of the tumor cells that bound to tri‐cellular corners or between two adjacent HUVEC. At each time point, at least 80 bound tumor cells were examined.
MIA PaCa‐2 cell binding induces the focal disappearance of VE‐cadherin or CD31 from HUVEC junctions
We next investigated whether MIA PaCa‐2 cell binding affects the integrity of the endothelial junctions of HUVEC. To this end, CMFDA‐labeled MIA PaCa‐2 cells were added to confluent HUVEC monolayers, and incubated for 30, 60 and 90 min. After unbound MIA PaCa‐2 cells were removed, the HUVEC monolayers were stained with an anti‐VE‐cadherin or anti‐CD31 mAb and analyzed by confocal microscopy. Before the addition of MIA PaCa‐2, the HUVEC retained well‐developed endothelial junctions and showed no gaps at the tri‐cellular corners as revealed by immunofluorescence staining with anti‐VE‐cadherin (Fig. 5a) or anti‐CD31 (Fig. 5b) antibodies. As shown in Fig. 5, 30 min after their addition, MIA PaCa‐2 cells showed preferential binding to the tri‐cellular corners of the HUVEC monolayers, but the HUVEC junctions remained occluded. At 60 min, focal losses of anti‐VE‐cadherin and anti‐CD31 staining were observed in the immediate vicinity of the MIA PaCa‐2 cell‐binding sites (arrows), whereas the VE‐cadherin and CD31 staining was apparently unaffected around the neighboring endothelial cells. At 90 min, the disruption of cell junctions was seen in the HUVEC monolayer exclusively around the adherent MIA PaCa‐2 cells but not at sites distant from the tumor cell adhesion. Disengagement of the HUVEC junctions was confirmed by the examination of vertical confocal images. As shown in Fig. 5c, bound MIA PaCa‐2 cells were observed within the gap formed between HUVEC at 60 min or later, and appeared to be in direct contact with the subendothelial matrices. These observations indicate that MIA PaCa‐2 cells that had bound to the tri‐cellular corners of HUVEC induced the focal disappearance of endothelial VE‐cadherin and CD31 at an early stage of cell adhesion and subsequently disrupted the junctions of HUVEC to infiltrate the subendothelial layer.
Figure 5.
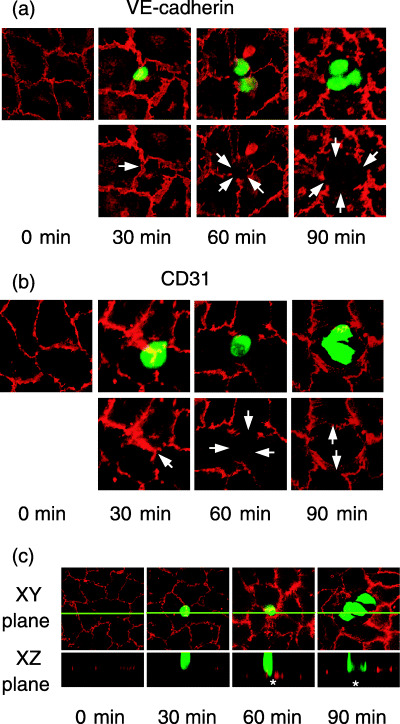
MIA PaCa‐2 cell binding induced the redistribution of VE‐cadherin and CD31 between adjacent human umbilical vein endothelial cells (HUVEC). (a, b) Immunohistochemical analysis of VE‐cadherin and CD31 in the HUVEC monolayer after the addition of MIA PaCa‐2 cells. MIA PaCa‐2 cells labeled with 5‐chloromethylfluorescein diacetate were added to a HUVEC monolayer and incubated for the indicated times. After the unbound MIA PaCa‐2 cells were removed by gentle washing, the HUVEC monolayers were stained with an anti‐VE‐cadherin antibody (a, red) or anti‐CD31 antibody (b, red) and observed under a confocal microscope. Overlaid images of MIA PaCa‐2 cells (green) and VE‐cadherin or CD31 (red) staining are shown in the upper panels. VE‐cadherin or CD31 staining alone is shown in red (lower panels). At 60 and 90 min, MIA PaCa‐2 cells (green) adhered to the HUVEC monolayer and induced the redistribution of VE‐cadherin and CD31 (red) between adjacent HUVEC (lower panels, arrows). Arrows indicate the positions of MIA PaCa‐2 cell binding. (c) Cross‐sectional view of MIA PaCa‐2 cells bound to the HUVEC monolayer. After the addition of MIA PaCa‐2 cells (green), HUVEC monolayers were stained with anti‐CD31 (red) as in Fig. 5b. The XZ plane cross‐sectional views (lower panels) according to the green line in the XY plane (upper panels) were obtained at the indicated time points. At 60 and 90 min, bound MIA PaCa‐2 cells were observed in the gaps (asterisks) formed between HUVEC.
Discussion
In hematogenous metastasis, cancer cells bind to the vascular wall and disengage the endothelial junctions to invade the surrounding tissues by mechanisms as yet unknown. In the present study, we showed that highly invasive human pancreatic carcinoma cells, such as MIA PaCa‐2 and PANC‐1, preferentially bound to the tri‐cellular corners of HUVEC and induced a rapid and irreversible reduction of TEER. The reduction of HUVEC TEER required direct cell adhesion between the tumor cells and HUVEC and the active metabolism and intact cytoskeleton of the tumor cells, which apparently induced the focal disengagement of endothelial junctional adhesion molecules, including VE‐cadherin and CD31. Although cancer cell attachment may occur randomly to the cell surface and cell‐to‐cell junctions of HUVEC, our results collectively indicate that the binding of certain pancreatic cancer cells to endothelial tri‐cellular corners plays a critical role in increasing endothelial permeability.
Certain cell‐adhesion molecules, such as β1 integrins, CD44, CD9 and selectins,( 14 , 15 , 18 , 19 ) have been implicated in cancer–endothelial interactions; however, these adhesion receptors did not seem to play important roles in the binding of MIA PaCa‐2 cells to HUVEC under our experimental conditions. Blocking mAbs to the integrin β1 chain and/or CD44 or CD9 did not show any detectable effects on the MIA PaCa‐2 cell binding to HUVEC (data not shown). In addition, the MIA PaCa‐2 cells and HUVEC used in this study were devoid of the expression of sialyl Lewisx epitopes and E‐/P‐selectins. Other adhesion molecules implicated in tumor–host interactions, such as αLβ2, αMβ2 α4β7 and αvβ3 integrins,( 15 , 16 , 17 ) nectin‐2( 46 ) and DNAM‐1( 47 ) were also undetectable in the MIA PaCa‐2 cells. The cell adhesion molecules mediating the MIA PaCa‐2 cell binding to HUVEC need to be identified in future investigations.
The MIA PaCa‐2 cell‐induced reduction of HUVEC TEER appeared to be cell‐contact‐dependent, and endothelial gap formation was selectively observed at the sites of MIA PaCa‐2 cell binding. These observations appear incompatible with those reported by Nakamori et al.,( 22 ) in which both MIA PaCa‐2 and PSN‐1 cells could induce an increase in endothelial permeability by secreting humoral factors. However, induction of the endothelial retraction in their study was observed 3 h or more (most significantly at 48 h) after stimulation with tumor‐derived factors, whereas in our study the disruption of endothelial junctions was already detectable at 30 min and prominent 2 h after the tumor cells were added. Therefore, it is likely that these two studies examined distinct phenomena at different time points, and pancreatic carcinoma cells may have two distinct mechanisms for disrupting endothelial junctions: one operating early using a contact‐dependent mechanism; the other operating late using a humoral mechanism. Previous studies using other types of cancers have reported a similar contact‐dependent disengagement of endothelial junctions and suggest that VEGF( 25 , 26 ) or eicosanoid 12(S)‐hydroxyeicosateraenoic acid( 23 , 24 ) produced by the cancer cells plays a critical role in the endothelial disengagement. In contrast, our data appear to negate the involvement of at least these soluble factors in the MIA PaCa‐2‐induced reduction of HUVEC TEER under the conditions and in the time frame we examined, because neither a neutralizing anti‐VEGF antibody nor the lipoxygenase inhibitor nordihydroguaiaretic acid affected the reduction of TEER. We found, instead, that pretreatment of MIA PaCa‐2 cells with PFA, but not with specific inhibitors for metalloproteinases and tyrosine‐kinases, abrogated the reduction of TEER. Thus, while the potential contribution of cancer‐cell‐derived soluble factors cannot be ruled out formally, our results favor a model in which MIA PaCa‐2 cells transmit signals to the HUVEC by way of PFA fixation‐sensitive mechanisms in a cell‐contact‐dependent manner, leading to the disengagement of endothelial junctions at early time points after cell adhesion.
An unexpected finding of this study was that invasive human pancreatic carcinoma MIA PaCa‐2 and PANC‐1 cells preferentially bound to HUVEC tri‐cellular corners. Previous studies reported that neutrophils( 31 , 32 , 33 ) as well as melanoma A375 cells( 14 ) also preferentially bind to the tri‐cellular corners of HUVEC. Unlike MIA PaCa‐2 cells, these cells migrated across the HUVEC monolayers without, apparently, disrupting the endothelial junctions.( 14 , 31 , 33 ) Thus, cell binding to the tri‐cellular corners per se is not sufficient to trigger the irreversible dissociation of cell junctions in HUVEC but probably requires one or more additional factors. Although the mechanisms underlying the selective binding to tri‐cellular corners remain unclear, one possible explanation is that the tight junction strands are partly discontinuous at tri‐cellular corners, exposing the pro‐adhesive subendothelial matrices, resulting in the matrix binding of the pancreatic carcinoma cells at these specific regions. Alternatively, the endothelial tri‐cellular corners may selectively express certain cell‐adhesion molecules that support the selective binding of the pancreatic cancer cells. This possibility is supported by recent studies showing that the epithelial cells of developing Drosophila express a cell adhesion molecule that is preferentially localized to tri‐cellular corners.( 48 , 49 )
In the present study, we showed that certain highly invasive human pancreatic cancer cells preferentially bound to tri‐cellular corners and reduced TEER. The reduction of TEER was accompanied by the local disjunction of VE‐cadherin and CD31, thus facilitating the cancer cells’ access to the subendothelial matrix and invasion into surrounding tissues. Our data provide a novel cell‐contact‐dependent mechanism for the cancer cell mediated breakdown of endothelial barrier functions, which deserves further investigation.
Acknowledgments
We thank Dr F. Nakata for PSN‐1 cells and Kanebo, Ltd for KB‐R7785. We thank Drs T. Hirata, H. Hayasaka and M. H. Jang for critically reading the manuscript. We also thank Ms. S. Yamashita and M. Komine for secretarial assistance and Ms. S. Kuroda for technical help. This work was supported by a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a grant for Advanced Research on Cancer from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 2003; 3: 453–8. [DOI] [PubMed] [Google Scholar]
- 2. Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer 1997; 80: 1529–37. [DOI] [PubMed] [Google Scholar]
- 3. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2: 563–72. [DOI] [PubMed] [Google Scholar]
- 4. Nicolson GL. Metastatic tumor cell interactions with endothelium, basement membrane and tissue. Curr Opin Cell Biol 1989; 1: 1009–19. [DOI] [PubMed] [Google Scholar]
- 5. Orr FW, Wang HH. Tumor cell interactions with the microvasculature: a rate‐limiting step in metastasis. Surg Oncol Clin N Am 2001; 10: 357–81, ix–x. [PubMed] [Google Scholar]
- 6. Mook OR, Van Marle J, Vreeling‐Sindelarova H, Jonges R, Frederiks WM, Van Noorden CJ. Visualization of early events in tumor formation of eGFP‐transfected rat colon cancer cells in liver. Hepatology 2003; 38: 295–304. [DOI] [PubMed] [Google Scholar]
- 7. Luzzi KJ, MacDonald IC, Schmidt EE et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 1998; 153: 865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haier J, Korb T, Hotz B, Spiegel HU, Senninger N. An intravital model to monitor steps of metastatic tumor cell adhesion within the hepatic microcirculation. J Gastrointest Surg 2003; 7: 507–14. [DOI] [PubMed] [Google Scholar]
- 9. Al‐Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium‐attached tumor cells: a new model for metastasis. Nat Med 2000; 6: 100–02. [DOI] [PubMed] [Google Scholar]
- 10. Wong CW, Song C, Grimes MM et al. Intravascular location of breast cancer cells after spontaneous metastasis to the lung. Am J Pathol 2002; 161: 749–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dejana E. Endothelial cell‐cell junctions: happy together. Nat Rev Mol Cell Biol 2004; 5: 261–70. [DOI] [PubMed] [Google Scholar]
- 12. Lewalle JM, Bajou K, Desreux J et al. Alteration of interendothelial adherens junctions following tumor cell‐endothelial cell interaction in vitro. Exp Cell Res 1997; 237: 347–56. [DOI] [PubMed] [Google Scholar]
- 13. Cai J, Jiang WG, Mansel RE. Phosphorylation and disorganization of vascular‐endothelial cadherin in interaction between breast cancer and vascular endothelial cells. Int J Mol Med 1999; 4: 191–5. [DOI] [PubMed] [Google Scholar]
- 14. Longo N, Yanez‐Mo M, Mittelbrunn M et al. Regulatory role of tetraspanin CD9 in tumor‐endothelial cell interaction during transendothelial invasion of melanoma cells. Blood 2001; 98: 3717–26. [DOI] [PubMed] [Google Scholar]
- 15. Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med 1999; 222: 124–38. [DOI] [PubMed] [Google Scholar]
- 16. St‐Pierre Y, Aoudjit F, Lalancette M, Potworowski EF. Dissemination of T cell lymphoma to target organs: a post‐homing event implicating ICAM‐1 and matrix metalloproteinases. Leuk Lymphoma 1999; 34: 53–61. [DOI] [PubMed] [Google Scholar]
- 17. Holzmann B, Gosslar U, Bittner M. α4 integrins and tumor metastasis. Curr Top Microbiol Immunol 1998; 231: 125–41. [DOI] [PubMed] [Google Scholar]
- 18. Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate‐mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci 2004; 95: 377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gunthert U, Hofmann M, Rudy W et al. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991; 65: 13–24. [DOI] [PubMed] [Google Scholar]
- 20. Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 2001; 17: 463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seiki M, Yana I. Roles of pericellular proteolysis by membrane type‐1 matrix metalloproteinase in cancer invasion and angiogenesis. Cancer Sci 2003; 94: 569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakamori S, Okamoto H, Kusama T et al. Increased endothelial cell retraction and tumor cell invasion by soluble factors derived from pancreatic cancer cells. Ann Surg Oncol 1997; 4: 361–8. [DOI] [PubMed] [Google Scholar]
- 23. Honn KV, Tang DG, Grossi I et al. Tumor cell‐derived 12 (S)‐hydroxyeicosatetraenoic acid induces microvascular endothelial cell retraction. Cancer Res 1994; 54: 565–74. [PubMed] [Google Scholar]
- 24. Honn KV, Grossi IM, Diglio CA, Wojtukiewicz M, Taylor JD. Enhanced tumor cell adhesion to the subendothelial matrix resulting from 12 (S)‐HETE‐induced endothelial cell retraction. FASEB J 1989; 3: 2285–93. [DOI] [PubMed] [Google Scholar]
- 25. Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF‐mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol 2004; 167: 223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE‐cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998; 111: 1853–65. [DOI] [PubMed] [Google Scholar]
- 27. Walker DC, MacKenzie A, Hosford S. The structure of the tricellular region of endothelial tight junctions of pulmonary capillaries analyzed by freeze‐fracture. Microvasc Res 1994; 48: 259–81. [DOI] [PubMed] [Google Scholar]
- 28. Burns AR, Walker DC, Smith CW. Relationship between tight junctions and leukocyte transmigration. In: Cereijido M, Anderson J, eds. Tight Junctions, 2nd edn. Florido: CRC Press, 2001; 629–52. [Google Scholar]
- 29. Dejana E, Corada M, Lampugnani MG. Endothelial cell‐to‐cell junctions. FASEB J 1995; 9: 910–8. [PubMed] [Google Scholar]
- 30. Dejana E, Del Maschio A. Molecular organization and functional regulation of cell to cell junctions in the endothelium. Thromb Haemost 1995; 74: 309–12. [PubMed] [Google Scholar]
- 31. Burns AR, Walker DC, Brown ES et al. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J Immunol 1997; 159: 2893–903. [PubMed] [Google Scholar]
- 32. Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev 2003; 83: 309–36. [DOI] [PubMed] [Google Scholar]
- 33. Burns AR, Bowden RA, MacDonell SD et al. Analysis of tight junctions during neutrophil transendothelial migration. J Cell Sci 2000; 113: 45–57. [DOI] [PubMed] [Google Scholar]
- 34. DiMagno EP, Reber HA, Tempero MA. AGA technical review on the epidemiology, diagnosis, and treatment of pancreatic ductal adenocarcinoma. Gastroenterology 1999; 117: 1464–84. [DOI] [PubMed] [Google Scholar]
- 35. Hotz HG, Reber HA, Hotz B et al. An orthotopic nude mouse model for evaluating pathophysiology and therapy of pancreatic cancer. Pancreas 2003; 26: e89–98. [DOI] [PubMed] [Google Scholar]
- 36. Schwarz RE, McCarty TM, Peralta EA, Diamond DJ, Ellenhorn JD. An orthotopic in vivo model of human pancreatic cancer. Surgery 1999; 126: 562–7. [PubMed] [Google Scholar]
- 37. Gautam N, Hedqvist P, Lindbom L. Kinetics of leukocyte‐induced changes in endothelial barrier function. Br J Pharmacol 1998; 125: 1109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wojciak‐Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci 2001; 114: 1343–55. [DOI] [PubMed] [Google Scholar]
- 39. Morimoto Y, Nishikawa K, Ohashi M. KB‐R7785, a novel matrix metalloproteinase inhibitor, exerts its antidiabetic effect by inhibiting tumor necrosis factor‐α production. Life Sci 1997; 61: 795–803. [DOI] [PubMed] [Google Scholar]
- 40. Wojtowicz‐Praga SM, Dickson RB, Hawkins MJ. Matrix metalloproteinase inhibitors. Invest New Drugs 1997; 15: 61–75. [DOI] [PubMed] [Google Scholar]
- 41. Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem 1986; 261: 11398–403. [PubMed] [Google Scholar]
- 42. Itakura J, Ishiwata T, Friess H et al. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin Cancer Res 1997; 3: 1309–16. [PubMed] [Google Scholar]
- 43. Goto T, Maeda H, Tanaka T. A selective inhibitor of matrix metalloproteinases inhibits the migration of isolated osteoclasts by increasing the life span of podosomes. J Bone Miner Metab 2002; 20: 98–105. [DOI] [PubMed] [Google Scholar]
- 44. Kajita M, Itoh Y, Chiba T et al. Membrane‐type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol 2001; 153: 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uehara Y, Fukazawa H. Use and selectivity of herbimycin A as inhibitor of protein‐tyrosine kinases. Meth Enzymol 1991; 201: 370–9. [DOI] [PubMed] [Google Scholar]
- 46. Takai Y, Irie K, Shimizu K, Sakisaka T, Ikeda W. Nectins and nectin‐like molecules. Roles in cell adhesion, migration, and polarization. Cancer Sci 2003; 94: 655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reymond N, Imbert AM, Devilard E et al. DNAM‐1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med 2004; 199: 1331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schulte J, Tepass U, Auld VJ. Gliotactin, a novel marker of tri‐cellular junctions, is necessary for septate junction development in Drosophila. J Cell Biol 2003; 161: 991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Venema DR, Zeev‐Ben‐Mordehai T, Auld VJ. Transient apical polarization of Gliotactin and Coracle is required for parallel alignment of wing hairs in Drosophila. Dev Biol 2004; 275: 301–14. [DOI] [PubMed] [Google Scholar]