

Abstract
The mucin‐type sialoglycoprotein podoplanin (aggrus) is involved in tumor cell‐induced platelet aggregation and tumor metastasis. C‐type lectin‐like receptor‐2 (CLEC‐2) was recently identified as an endogenous receptor of podoplanin on platelets. However, the pathophysiological importance and function of CLEC‐2 have not been elucidated. Here we clarified the pathophysiological interaction between podoplanin and CLEC‐2 in vitro and in vivo. Using several deletion mutants of CLEC‐2 expressed as Fc chimeras, we first identified an important podoplanin‐recognition domain in CLEC‐2. Furthermore, the podoplanin–CLEC‐2 interaction was confirmed using several deletion mutants of podoplanin expressed as Fc chimeras. Not only the disialyl‐core1‐attached glycopeptide but also the stereostructure of the podoplanin protein was found to be critical for the CLEC‐2‐binding activity of podoplanin. We next synthesized various glycopeptides of podoplanin that included both the platelet aggregation‐stimulating domain and O‐glycan on Thr52. Interestingly, a disialyl‐core1‐attached glycopeptide was recognized specifically by CLEC‐2. Moreover, the anti‐podoplanin monoclonal antibody NZ‐1 suppressed both the podoplanin–CLEC‐2 interaction and podoplanin‐induced pulmonary metastasis, suggesting that CLEC‐2 is the first pathophysiological receptor of podoplanin to be identified. In summary, we clarified the molecular interaction in vitro and in vivo between a platelet aggregation‐inducing factor, podoplanin, and its specific pathophysiological receptor on platelets, CLEC‐2. Podoplanin and CLEC‐2 might represent promising therapeutic targets in cancer metastasis. (Cancer Sci 2008; 99: 54–61)
Tumor metastasis is known to be associated with a platelet‐aggregating activity possessed by human and other mammalian cancers, implying that aggregation is important in tumor‐cell nestling, whereas released growth factors are important in angiogenesis or tumor growth.( 1 ) A previous study has clarified that podoplanin (also known as aggrus), a membranous sialoglycoprotein in cancer cells of mice (44 kDa) and humans (36 kDa), aggregates platelets with no relation to plasma components.( 2 ) Podoplanin is also known to be a lymphatic‐specific marker,( 3 ) and its expression has been reported in several tumor cells, including squamous cell carcinoma,( 4 , 5 , 6 ) mesothelioma,( 7 ) testicular seminoma,( 8 ) and brain tumors,( 9 , 10 ) although podoplanin is not expressed in gastrointestinal, pulmonary, or mammary adenocarcinomas, which frequently undergo metastasis.( 7 ) Recent investigations have reinforced the notion that the expression of podoplanin in several tumors is associated with tumor metastasis or progression.( 6 , 10 , 11 ) Podoplanin belongs to the family of type‐I transmembrane sialomucin‐like glycoproteins that comprise an extracellular domain with abundant Ser and Thr residues as potential O‐glycosylation sites, a single transmembrane portion, and a short cytoplasmic tail with putative sites for protein kinase C and cAMP phosphorylation.( 12 )
We previously showed that the EDxxVTPG segment in the extracellular domain, designated as the platelet aggregation‐stimulating (PLAG) domain, is critical for the activity of podoplanin.( 2 ) In particular, this motif, which is highly conserved across species, is triplicated in tandem.( 12 ) In a study of targeted mutagenesis of podoplanin molecules, we obtained evidence that Thr residues in the PLAG domain play an important role in platelet aggregation.( 2 ) Furthermore, the unique characteristics of the Chinese hamster ovary (CHO) mutant cell lines Lec1 (N‐glycan‐deficient), Lec2 (CMP‐sialic acid transporter‐deficient), and Lec8 (UDP‐galactose transporter‐deficient) revealed that sialylated O‐glycan is critical for the platelet aggregation‐inducing activity of podoplanin.( 13 ) Recently, we purified human podoplanin from the glioblastoma cell line LN319 and from podoplanin‐transfected CHO cells.( 14 ) Using a lectin microarray, Edman degradation, and matrix‐assisted laser desorption–ionization time‐of‐flight mass spectrometry (MALDI‐TOF‐MS), we showed that podoplanin possesses a disialyl‐core1 structure at Thr52 in the PLAG domain. It remains to be clarified, however, whether the disialyl‐core1 structure at Thr52 is essential for its platelet‐aggregating activity. In addition, the minimum unit of glycopeptide necessary for this activity has not been identified. Furthermore, several lines of evidence obtained using podoplanin knockout mice suggest that podoplanin is crucially involved in lymphatic vessel formation.( 15 ) However, how podoplanin regulates the formation of lymphatic vessels or the tumor–platelet interaction remains unknown, and identification of the pathophysiological target with which podoplanin interacts is ardently awaited among research workers in many scientific fields. Recently, C‐type lectin‐like receptor‐2 (CLEC‐2), which is a non‐classical C‐type lectin,( 16 , 17 ) was identified as an endogenous receptor of podoplanin on platelets.( 18 )
In the present work, the specific binding activity between podoplanin and CLEC‐2 was confirmed using several deletion mutants of podoplanin and CLEC‐2 expressed as Fc chimeras. Furthermore, using several glycosyltransferases, we synthesized glycopeptides including both the PLAG domain and the O‐glycan on Thr52 to identify the minimum unit necessary for binding to CLEC‐2. Finally, we verified that the podoplanin–CLEC‐2 interaction is pathophysiologically important in an experimental metastasis model using an anti‐podoplanin antibody, NZ‐1.( 19 )
Materials and Methods
Cell lines and stable transfectants. Chinese hamster ovary cells (CHO) transfected with human podoplanin (hPod) were established as described previously.( 2 ) CHO/hPod cells were cultured in RPMI‐1640 medium (Wako, Tokyo, Japan) supplemented with 10% heat‐inactivated fetal bovine serum (FBS; Sigma, St Louis, MO, USA), 2 mM l‐glutamine (Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin, 100 µg/mL streptomycin (Invitrogen), and 1 mg/mL geneticin (G418; Sigma) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. 293T‐REx/CLEC‐2 cells were established as described previously,( 20 ) and were cultured in Dulbecco's Modified Eagle's Medium (Wako) supplemented with 10% heat‐inactivated FBS, 2 mM l‐glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, 100 µg/mL zeocin (Invitrogen), and 5 µg/mL blasticidin (Invitrogen). Expression of CLEC‐2 was induced by the addition of 1 µg/mL doxycycline (Takara Bio, Shiga, Japan) to the medium 24 h before experimentation. 293T‐REx/CLEC‐2 cells without doxycycline were used as a control. The glioblastoma cell line LN319 was donated by Dr Webster K. Cavenee (Ludwig Institute for Cancer Research, San Diego, CA, USA) and cultured as described.( 14 , 19 )
Animals. Female BALB/c nude (nu/nu) mice (7 weeks old) were purchased from Charles River Japan (Kanagawa, Japan). Animals were housed under pathogen‐free conditions. The Animal Care and Use Committee of the National Institute of Advanced Industrial Science and Technology (Tsukuba, Japan) approved the animal experiments described herein.
Expression and purification of soluble podoplanin and CLEC‐2. cDNA of human podoplanin and human CLEC‐2 (AF124841) containing the extracellular domains of these proteins was obtained by polymerase chain reaction (PCR) as described previously.( 18 ) PCR was carried out using HotStarTaq polymerase (Qiagen, Hilden, Germany). The following oligonucleotides were used as primers (the EcoRV or EcoRI site in the forward primer and the BglII site in the reverse primer are underlined): hCL2ΔN58 (forward CCGATTACACAGCGCAATTACCT, reverse GAAGATCTAGGTAGTTGGTCCAC), hCL2ΔN87 (forward acgaattcgCAATCAGAACTAAAGGGCAC, reverse [hCL2.R688‐BglII] acagatctAGGTAGTTGGTCCACCTTGG), hCL2ΔN147 (forward acgaattcgGAGTACATCAAAGCCAGGAC, reverse hCL2. R688‐BglII), hCL2ΔN177 (forward acgaattcgGTTATCTCAGAAAATATGTT, reverse hCL2.R688‐BglII), PodΔC128 (forward GCGATATCAGAAGGAGCCAGCACAGG, reverse GGCAGATCTTGTTGACAAACCATCTTTC), PodΔC103 (forward [EcoRI‐hPod.F1] acgaattcgATGTGGAAGGTGTCAGCTCT, reverse acagatctGTTTGAGGCTGTGGCGCTTG), PodΔC80 (forward EcoRI‐hPod.F1, reverse acagatctGATGCGAATGCCTGTTACAC), and PodΔC57 (forward EcoRI‐hPod.F1, reverse acagatctTTCGCTGGTTCCTGGAGTCA). The PCR products were purified using a Rapid PCR Purification Kit (Marligen Bioscience, Ijamsville, MD, USA), digested with EcoRV or EcoRI and BglII, purified again, and ligated into the pFUSE‐hFc2 (IL2ss) vector (InvivoGen, San Diego, CA, USA), which contains human IgG Fc after the ligation site and the interleukin 2 signal sequence before the ligation site to allow secretion of Fc fusion proteins. CHO cells were transfected with plasmids using Lipofectamine 2000. For purification of the fusion proteins, the medium was centrifuged, and the supernatant obtained was applied to a column of protein A–sepharose (Pierce, Rockford, IL, USA). After extensive washing with phosphate‐buffered saline (PBS), the fusion proteins were eluted with 0.1 M glycine and 0.15 M NaCl (pH 2.8), and then neutralized with 1 M Tris pH 10.0. The proteins were dialyzed against PBS. The expression and purity of the proteins were confirmed by sodium dodecylsulfate–polyacrylamide gel electrophoresis using 10–20% gradient gels (Wako). Biotinylation of the fusion proteins was carried out using a SureLINK Chromophoric Biotin Labeling Kit (KPL, Gaithersburg, MD, USA).
Synthesis of O‐linked glycosylated glycopeptides. Human podoplanin glycopeptide (hpp3854) with a GalNAc residue was purchased from Peptide Institute (Osaka, Japan) and used as an acceptor substrate. For synthesis of the core1 structure on hpp3854, 25 mM HEPES (pH 7.0) containing 4 mg acceptor substrate, 10 mM MnCl2, and an appropriate concentration of UDP‐Gal was used. A 100‐µL volume of purified Core1GalT was added to the reaction mixture, which was then incubated at 37°C for 17 h. The sialylation experiments have been described previously.( 14 ) For the preparation of hpp3854 with the a sialyl core1 (NeuAcα2‐3Galβ1‐3GalNAcα1‐ or NeuAcα2‐6Galβ1‐3GalNAcα1‐) residue, 25 mM HEPES (pH 7.0) containing 1 mg core1‐hpp3854, 10 mM MnCl2, and an appropriate concentration of CMP‐NeuAc was used. A 50‐µL volume of purified ST3GalI or ST6GalNAcI enzyme was added to the reaction mixture and incubated at 37°C for 24 h. For the preparation of hpp3854 peptide with the disialyl‐core1 (NueAcα2‐3Galβ1‐3[NeuAcα2‐6]GalNAcα1‐) residue, both enzymes were added to the reaction mixture and incubated at 37°C for 24 h. After removing the resin by filtration using an Ultrafree‐MC column (Millipore, Billerica, MA, USA), the glycopeptides were desalted with a reverse‐phase column (Oasis HLB column; Waters, Milford, MA, USA).
Western blot analysis. Proteins were electrophoresed under reducing conditions on 10–20% polyacrylamide gels (Wako). The separated proteins were transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking with 4% skim milk in PBS, the membrane was incubated with NZ‐1 (a rat monoclonal antibody; 1 µg/mL), rabbit anti‐human Fc (5 µg/mL; Rockland, Gilbertsville, PA, USA), and then with peroxidase‐conjugated anti‐rat or anti‐rabbit antibodies (1/1000 dilution; Dako, Glostrup, Denmark) and developed for 1 min with ECL reagents (GE Healthcare UK, Buckinghamshire, UK) using X‐Omat AR film (Kodak, Rochester, NY, USA).
Enzyme‐linked immunosorbent assay. Glycopeptides corresponding to amino acids of the human podoplanin sequence, podoplanin–Fc chimeras, CLEC‐2–Fc chimeras, and antibodies were immobilized on 96‐well plates at 10 µg/mL for 1 h. The polyclonal anti‐CLEC‐2 antibody was from R & D Systems (Minneapolis, MN, USA). After blocking with 1% bovine serum albumin (BSA) in PBS, the plates were incubated with NZ‐1, biotinylated NZ‐1, biotinylated podoplanin–Fc chimera, or biotinylated CLEC‐2–Fc chimera at 10 µg/mL for 1 h. After washing with 0.05% Tween 20 in PBS, the plates were incubated with peroxidase‐conjugated antirat IgG (Dako), or peroxidase‐conjugated biotin–streptavidin complex (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA, USA) for 1 h. After further washing, the enzymatic reaction was conducted with a substrate solution containing TMB (TMB‐Turbo or TMB‐Ultra; Pierce). The reaction was stopped with 1 M H2SO4, and the optical density was measured at 450 nm with an autoplate reader. These reactions were carried out using a volume of 50 µL at room temperature.
Flow cytometry. Parental CHO, CHO/hPod, or 293T‐REx/CLEC‐2 cells were harvested by brief exposure to 1 mM ethylenediaminetetraacetic acid. After washing with PBS, the cells were treated with CLEC‐2–Fc chimeras or podoplanin–Fc chimeras for 1 h at 4°C followed by treatment with anti‐human Fc (Rockland) and Oregon green‐conjugated anti‐rabbit IgG (Invitrogen). Fluorescence data were collected using a FACS Caliber flow cytometer (BD Biosciences, Barintree, MA, USA).
Mass spectrometry. Each synthetic glycopeptide was analyzed by MALDI‐TOF‐MS (Reflex IV; Bruker‐Daltonik) in reflectron negative ion mode. For sample preparation, 0.5 µL matrix solution (10 mg 2,5‐dihydroxybenzoic acid dissolved in 1 mL 30% ethanol) was deposited on the stainless steel target plate and allowed to dry in air. Next, 0.5 µL of appropriately diluted analyte solution was used to cover the matrix on the target plate and allowed to dry in air.
Platelet aggregation assay. Heparinized mouse or human whole blood was drawn from BALB/c mice or healthy drug‐free volunteers. Platelet aggregation was measured according to the screen filtration pressure method using WBA Carna (IMI, Saitama, Japan).( 14 , 19 , 21 ) Two hundred microliters each of the whole‐blood samples and NZ‐1 or control rat IgG in four reaction tubes were stirred at 1000 r.p.m. at 37°C and preincubated for 2 min, followed by the addition of 12 µL of each of the samples. Using a 3.7‐mm‐diameter syringe containing screen microsieves made of nickel, with 300 openings of 20 × 20 µm2 in a 1‐mm‐diameter area, whole‐blood samples were sucked to detect aggregation pressure at a rate of 200 µL/6.4 s 7–10 min later. The final platelet aggregation pressure of each reaction tube was determined at the pressure rate (%) of a pressure sensor connected to the syringe.
Experimental lung metastasis. CHO/hPod cells were harvested, washed, and resuspended in Hanks’ Balanced Solutions (HBSS; 2.5 × 106 cells/mL).( 11 ) Next, the cells were incubated with 150 µg NZ‐1, 150 µg control rat IgG, or 100 µg F(ab′)2 of NZ‐1, and inoculated intravenously (2.5 × 105 cells/mouse) into the lateral tail vein of female BALB/c‐nu/nu mice. After 20 days, the mice were killed and the surface lung metastatic foci were counted and measured.
Results
Binding activity of the CLEC‐2–Fc chimera to podoplanin. We first determined the podoplanin‐recognition domain of CLEC‐2. Several CLEC‐2–Fc N‐terminal deletion mutants were produced (Fig. 1a,b), and the binding activities of human podoplanin to these deletion mutants were assayed. Non‐classical C‐type lectins such as CLEC‐2 are known to possess a C‐type lectin‐like domain (CTLD), homologous to the carbohydrate‐recognition domain of classical C‐type lectins. As shown in Fig. 1c, hCL2ΔN58 and hCL2ΔN87 bound to both LN319 and CHO/hPod cells, whereas hCL2ΔN147 and hCL2ΔN177 did not. Similarly, hCL2ΔN58 and hCL2ΔN87 were recognized by the podoplanin–Fc chimera by enzyme‐linked immunosorbent assay (ELISA); however, podoplanin–Fc reacted strongly with hCL2ΔN58 compared with hCL2ΔN87 (Fig. 1d). Furthermore, we produced several CLEC‐2–Fc C‐terminal deletion mutants; however, none of them reacted with podoplanin (data not shown). These results indicate that the stereostructure of CLEC‐2 is important for recognizing podoplanin‐like classical C‐type lectins. Therefore, we used hCL2ΔN58 as a recombinant human CLEC‐2–Fc for the binding assay with podoplanin.
Figure 1.

Binding activity of C‐type lectin‐like receptor‐2 (CLEC‐2)–Fc chimeras to podoplanin. (a) CLEC‐2–Fc deletion mutants were produced as shown in this scheme. (b) Purified CLEC‐2–Fc chimeras (10 or 0.1 µg/lane) were electrophoresed using 10–20% gels and stained with Coomassie Brilliant Blue (CBB) or western blotted using anti‐Fc. (c) LN319 and CHO/hPod cells were harvested by brief exposure to 1 mM ethylenediaminetetraacetic acid. After washing with phosphate‐buffered saline (PBS), cells were treated with CLEC‐2–Fc chimeras or PBS for 1 h at 4°C followed by treatment with antihuman Fc (2 µg/mL) and Oregon green‐conjugated antirabbit IgG (2 µg/mL). Fluorescence data were collected using by flow cytometry. (d) CLEC‐2–Fc chimeras or an anti‐Fc were immobilized at a concentration of 10 µg/mL on a 96‐well plate. After blocking with 1% bovine serum albumin (BSA) in PBS, the plates were incubated with a biotinylated podoplanin–Fc chimera at 10 µg/mL, followed by incubation with a peroxidase‐conjugated biotin–streptavidin complex. The enzymatic reaction was conducted with a substrate solution containing TMB (TMB‐Ultra). After the reaction was stopped with 1 M H2SO4, the optical density was measured at 450 nm with an autoplate reader. *P < 0.01 compared with control, by t‐test.
Binding activity of the podoplanin–Fc chimera to CLEC‐2. We next produced several types of C‐terminal deletion mutants of human podoplanin as Fc chimeras (Fig. 2a,b) and investigated the binding activity of CLEC‐2 to these deletion mutants. Of the four types of C‐terminal deletion mutant (PodΔC128, PodΔC103, PodΔC80, and PodΔC57), PodΔC128 and PodΔC103 bound to 293T/REx/CLEC‐2 cells in flow cytometry when CLEC‐2 was induced by doxycycline (Fig. 2c). In contrast, PodΔC80 and PodΔC57 reacted weakly with 293T/REx/CLEC‐2 cells. CLEC‐2 binding to the podoplanin deletion mutants was also examined by ELISA. As shown in Fig. 2d, PodΔC128 was recognized strongly by CLEC‐2‐Fc compared with PodΔC103, and PodΔC80 and PodΔC57 were recognized weakly. These results suggest that not only the PLAG domain but also the C‐terminal region of podoplanin is necessary for the strong interaction between podoplanin and CLEC‐2, although the PLAG domain is essential for its platelet‐aggregating activity.( 2 ) Alternatively, the stereostructure of podoplanin might be important for its strong binding to CLEC‐2. However, PodΔC57, which possesses only the PLAG domain, was shown to be sufficient to bind to CLEC‐2; therefore, we moved on to identification of the minimum glycopeptides of podoplanin using our glycan‐synthetic technology.
Figure 2.
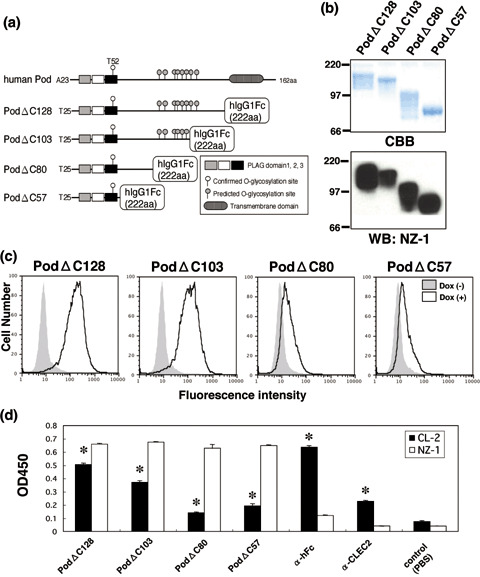
Binding activity of podoplanin–Fc chimeras to C‐type lectin‐like receptor‐2 (CLEC‐2). (a) Podoplanin–Fc deletion mutants were produced as shown in this scheme. (b) Purified podoplanin–Fc chimeras (10 or 0.1 µg/lane) were electrophoresed using 10–20% gels and either stained with Coomassie Brilliant Blue (CBB) or western blotted with NZ‐1. (c) Expression of CLEC‐2 was induced by the addition of 1 µg/mL doxycycline to 293T‐REx/CLEC‐2 cells 24 h before experimentation. Cells were harvested by brief exposure to 1 mM ethylenediaminetetraacetic acid. After washing with phosphate‐buffered saline (PBS), doxycycline+ or doxycycline− cells were treated with podoplanin–Fc chimeras for 1 h at 4°C followed by treatment with antihuman Fc (2 µg/mL) and Oregon green‐conjugated antirabbit IgG (2 µg/mL). Fluorescence data were collected using by flow cytometry. (d) Podoplanin–Fc chimeras, an anti‐Fc, or an anti‐CLEC‐2 polyclonal antibody were immobilized at a concentration of 10 µg/mL on 96‐well plates. After blocking with 1% bovine serum albumin (BSA) in PBS, the plates were incubated with biotinylated NZ‐1 or biotinylated CLEC‐2–Fc chimera at 10 µg/mL, followed by incubation with a peroxidase‐conjugated biotin–streptavidin complex. The enzymatic reaction was conducted with a substrate solution containing TMB (TMB‐Turbo). After the reaction was stopped with 1 M H2SO4, the optical density was measured at 450 nm with an autoplate reader. *P < 0.01 compared to control, by t‐test.
Binding activity of podoplanin glycopeptides to CLEC‐2. In our study of targeted podoplanin mutagenesis, we previously showed that Thr residues in the PLAG domain play an important role in platelet aggregation.( 2 ) However, the minimum unit of glycopeptide that is necessary for this activity has not been identified. As shown in Figure 3a, we synthesized five glycopeptides: hpp3854 (corresponding to amino acids 38–54 of human podoplanin) + GalNAc, hpp3854 + core1, hpp3854 + NeuAcα2‐3 core1, hpp3854 + NeuAcα2‐6 core1, and hpp3854 + disialyl‐core1 using our glycan‐synthetic technology. The glycans synthesized were confirmed by mass spectrometry (data not shown). We then investigated the binding activity of CLEC‐2 to these glycopeptides by ELISA. As a result, hpp3854 + disialyl‐core1 reacted strongly with CLEC‐2 compared with hpp3854 + GalNAc, whereas hpp3854 + core1, hpp3854 + NeuAcα2‐3 core1, and hpp3854 + NeuAcα2‐6 core1 showed no difference to hpp3854 + GalNAc, although weak reactions were observed (Fig. 3b). These results indicate that only one glycan on Thr52, the disialyl‐core1 structure, is enough for recognition of CLEC‐2. We also investigated the platelet‐aggregating activity of these glycopeptides. However, none of these glycopeptides induced platelet aggregation (data not shown), suggesting that only glycopeptide‐binding to CLEC‐2 is not enough to activate platelet.
Figure 3.
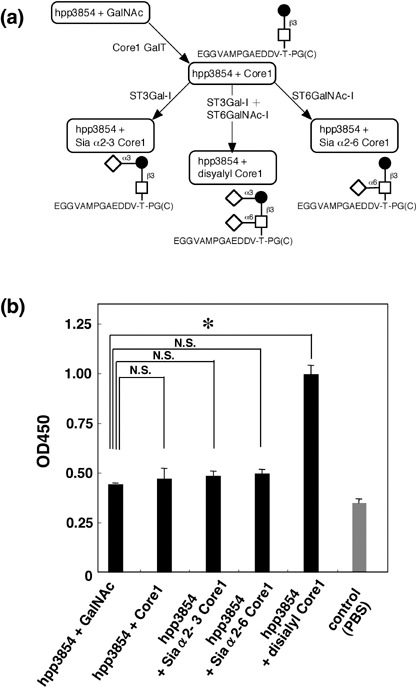
Binding activity of podoplanin glycopeptide to C‐type lectin‐like receptor (CLEC)‐2. (a) Podoplanin glycopeptides were synthesized as shown in this scheme. (b) The synthetic glycopeptides were immobilized at a concentration of 10 µg/mL on 96‐well plates. After blocking with 1% bovine serum albumin (BSA) in phosphate‐buffered saline, the plates were incubated with biotinylated CLEC‐2–Fc at 10 µg/mL, followed by a peroxidase‐conjugated biotin–streptavidin complex. The enzymatic reaction was conducted with a substrate solution containing TMB (TMB‐Ultra). After the reaction was stopped by 1 M H2SO4, the optical density was measured at 450 nm. *P < 0.01 compared to hpp3851 + GalNAc, by t‐test.
Platelet‐aggregation assay for the podoplanin–Fc chimera. We previously showed that endogenous or recombinant podoplanin proteins purified from LN319 glioblastoma cell lines or CHO/hPod cells, respectively, induce platelet aggregation.( 14 ) Here, we investigated the platelet‐aggregating activity of the podoplanin–Fc chimeras. As shown in Fig. 4, none of the podoplanin–Fc deletion mutants induced platelet aggregation at a dose of 10 µg/mL. In contrast, after preincubation of podoplanin–Fc with an anti‐human Fc antibody, platelet aggregation was induced by PodΔC128, but not by PodΔC103, PodΔC80, or PodΔC57. At a dose of 100 µg/mL, not only PodΔC128 but also PodΔC103 induced platelet aggregation. These results indicate that both crosslinking of the podoplanin–Fc chimera and strong binding to CLEC‐2 are necessary for inducing platelet aggregation.
Figure 4.

Platelet aggregation assay of podoplanin–Fc chimeras. Platelet aggregation was measured using WBA Carna with the screen filtration pressure method. Two hundred microliters each of mouse whole blood samples were preincubated for 2 min, followed by the addition of 12 µL each of podoplanin–Fc chimeras or podoplanin–Fc chimeras preincubated with anti‐Fc antibodies (10 µg/mL) for 30 min on ice. Whole blood samples were sucked 10 min later to detect aggregation.
Inhibition of podoplanin‐induced platelet aggregation by CLEC‐2–Fc deletion mutants. Of the four CLEC‐2–Fc deletion mutants, hCL2ΔN58 and hCL2ΔN87 were found to react with podoplanin; therefore, we investigated whether these CLEC‐2–Fc deletion mutants could inhibit podoplanin‐induced platelet aggregation. We found that hCL2ΔN58 strongly and hCL2ΔN87 moderately inhibited podoplanin‐induced platelet aggregation, whereas hCL2ΔN147 and hCL2ΔN177 did not (Fig. 5). This result indicates that CLEC‐2 is a physiological ligand of podoplanin.
Figure 5.
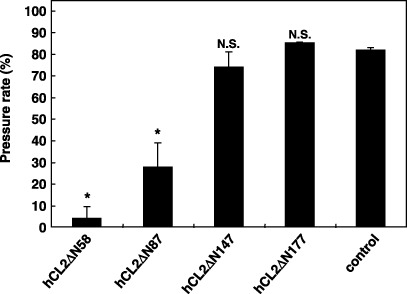
Inhibition of podoplanin‐induced platelet aggregation by C‐type lectin‐like receptor‐2 (CLEC‐2)–Fc deletion mutants. Heparinized human whole blood (WB) was drawn from healthy drug‐free volunteers. Platelet aggregation was measured using WBA Carna with the screen filtration pressure method. Two hundred microliters each of human WB samples were preincubated for 2 min, followed by the addition of 12 µL each of podoplanin–Fc chimera (PodΔC128; 2 µg/mL) preincubated with anti‐Fc antibodies (2 µg/mL) and CLEC‐2‐Fc chimeras (10 µg/mL) for 30 min on ice. WB samples were sucked 7 min later to detect aggregation. Black arrow, sucked blood; blue arrow, unsucked blood; arrowhead, clogged filter. Data are mean ± SD of three independent experiments. *P < 0.01, **P < 0.05 compared to control, by t‐test.
Inhibition of the podoplanin–CLEC‐2 interaction. CLEC‐2–Fc inhibited podoplanin‐induced platelet aggregation (Fig. 5). As shown in Fig. 6a, the PodΔC128–CLEC‐2 interaction was diminished by NZ‐1 in flow cytometry. Furthermore, the interaction between PodΔC57 and CLEC‐2 was inhibited completely by NZ‐1 (Fig. 6b). In ELISA, NZ‐1 also partially inhibited the PodΔC128–CLEC‐2 interaction (data not shown). In contrast, NZ‐1 completely inhibited podoplanin‐induced platelet aggregation,( 19 ) suggesting that the podoplanin–CLEC‐2 interaction might be pathophysiologically important in vivo.
Figure 6.
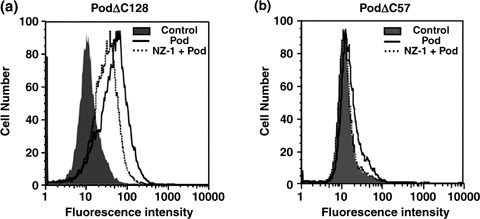
Inhibition of the podoplanin–C‐type lectin‐like receptor‐2 (CLEC‐2) interaction by NZ‐1. (a) Expression of CLEC‐2 was induced by the addition of 1 µg/mL doxycycline to 293T‐REx/CLEC‐2 cells 24 h before experimentation. Cells were harvested by brief exposure to 1 mM ethylenediaminetetraacetic acid. After washing with phosphate‐buffered saline (PBS), cells were treated with NZ‐1 (100 µg/mL) + PodΔC128 (1 µg/mL), PodΔC128 alone, or PBS for 1 h at 4°C followed by treatment with anti‐human Fc (2 µg/mL) and Oregon green‐conjugated anti‐rabbit IgG (2 µg/mL). (b) 293T‐REx/CLEC‐2 cells were treated with NZ‐1 (100 µg/mL) + PodΔC57 (10 µg/mL), PodΔC57 alone, or PBS for 1 h at 4°C followed by treatment with anti‐human Fc (2 µg/mL) and Oregon green‐conjugated anti‐rabbit IgG (2 µg/mL).
Inhibition of podoplanin‐induced metastasis by NZ‐1 or F(ab′)2 of NZ‐1. We then investigated whether NZ‐1 could suppress podoplanin‐induced pulmonary metastasis in an experimental metastasis model. Injection of CHO/hPod cells together with control IgG led to development of multiple lung metastatic foci (Fig. 7a). The number of metastatic lung nodules in mice injected with CHO/hPod + NZ‐1 was significantly lower than that in CHO/hPod + control IgG (P < 0.01; Fig. 7b). Consistent with this decrease in the number of metastatic foci, lung weight was lower in mice injected with CHO/hPod + NZ‐1 than in those injected with CHO/hPod + control IgG (Fig. 7c). Macroscopically, we did not observe metastatic foci in the liver, kidney, spleen, colon, or ovary in any of the mice (data not shown). Furthermore, we investigated whether F(ab′)2 of NZ‐1 could also suppress podoplanin‐induced pulmonary metastasis in the same model. As shown in Fig. 7a, F(ab′)2 of NZ‐1 also suppressed pulmonary metastasis. The number of metastatic lung nodules in mice injected with CHO/hPod + F(ab′)2 of NZ‐1 was significantly lower than that in CHO/hPod + control IgG in the same way with whole antibody of NZ‐1 (P < 0.01; Fig. 7b). Lung weight was also lower in mice injected with CHO/hPod + F(ab′)2 of NZ‐1 than in those injected with CHO/hPod + control IgG (Fig. 7c). These results indicate that compared with control IgG, NZ‐1 significantly inhibited pulmonary metastasis, suggesting that the podoplanin–CLEC‐2 interaction is pathophysiologically critical in podoplanin‐induced platelet aggregation and metastasis.
Figure 7.
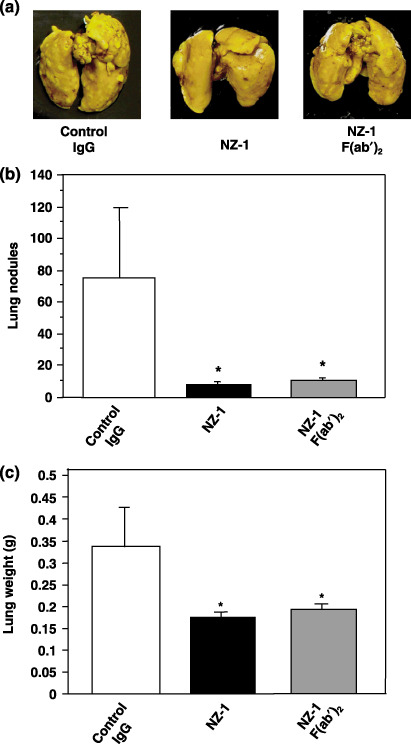
Inhibition of podoplanin‐induced metastasis by NZ‐1. (a) Chinese hamster ovary cells transfected with human podoplanin (CHO/hPod) were harvested, washed, and resuspended in HBSS (2.5 × 106 cells/mL). Then, the cells were incubated with 150 µg NZ‐1, 150 µg control rat IgG, or 100 µg F(ab′)2 of NZ‐1 and inoculated intravenously (2.5 × 105 cells/mouse) into lateral tail vein of female BALB/c‐nu/nµ mice (five mice in each group). After 20 days, the mice were killed, and surface lung metastatic foci were counted and measured. Detailed numbers of (b) the lung metastasis nodules and (c) the weight of the lung in each mouse are shown. Data are mean ± SD. *P < 0.01, by t‐test.
Discussion
The immunogloblin superfamily and G protein‐coupled receptors are known to stimulate platelet aggregation, but CLEC‐2, a C‐type lectin‐like receptor, is a completely new class of platelet‐aggregating receptor.( 17 , 22 ) Of interest, CLEC‐2 binds to its ligand in an unusual manner. C‐type lectins usually need Ca2+ to bind to ligand; however, CLEC‐2 does not need Ca2+ to bind to ligand (2, 3) as one of non‐classical C‐type lectins.( 16 ) CLEC‐2, with a single immunoreceptor tyrosine‐based activation motif (ITAM), activates Syk directly, whereas most C‐type lectins, such as MDL‐1 and DCAR, couple with Syk indirectly via association with ITAM‐bearing adaptor proteins such as FcRγ and DAP12.( 17 , 23 ) The powerful stimulatory activity of CLEC‐2 on platelets suggests that it may play an important role in vivo. Recently, CLEC‐2 was identified as a novel endogenous receptor of podoplanin on the cell surface.( 18 ) However, the pathophysiological importance and function of CLEC‐2 have not been elucidated. In the present study, the importance of the podoplanin–CLEC‐2 interaction has been clarified in vitro and in vivo.
Here, we first investigated the interaction between podoplanin and CLEC‐2 in detail. As shown in Fig. 3, only a disialyl‐core1 on Thr52 plus the PLAG domain (hpp3854 + disialyl‐core1) was recognized by CLEC‐2. Of the four podoplanin–Fc deletion mutants, PodΔC128 reacted most strongly with CLEC‐2 and induced platelet aggregation. In contrast, the others bound weakly to CLEC‐2 and did not induce platelet aggregation (Fig. 4). Moreover, hpp3854 + disialyl‐core1 also did not induce platelet aggregation (data not shown), suggesting that strong binding to CLEC‐2 may be essential for inducing platelet aggregation.
Using several CLEC‐2–Fc deletion mutants, the CTLD of the C‐type lectin family was consistent with the podoplanin‐recognition domain (Fig. 1a,b). These results indicate that the interaction between the CTLD of CLEC‐2 and a disialyl‐core1 in the PLAG domain of podoplanin is the most critical association in podoplanin‐induced platelet aggregation. Previous structure and mutational binding analysis of CLEC‐2 also suggested that its endogenous ligand is likely to be a protein with a predominantly negatively charged binding surface.( 24 ) Our data, showing that the disialyl‐core1 structure of podoplanin is essential for binding to CLEC‐2, supports this suggestion because sialic acid is negatively charged.
A recombinant podoplanin–Fc fusion protein stimulated more powerful platelet aggregation when it was crosslinked by anti‐Fc antibodies; PodΔC128 could induce platelet aggregation at a concentration of only 0.2 µg/mL (data not shown). Previously, Fuller et al. reported that an anti‐CLEC‐2 antibody also induced platelet aggregation.( 22 ) Furthermore, we previously showed that podoplanin‐expressing CHO cells strongly induce platelet aggregation in spite of their low expression of podoplanin.( 19 ) These findings suggest that clustering of CLEC‐2 might generate activation signals and platelet aggregation, although the podoplanin–CLEC‐2 interaction was observed using even a monomeric glycopeptide of podoplanin (Fig. 3). Furthermore, not only the PLAG domain–CTLD interaction but also the stereostructure of both podoplanin and CLEC‐2 might be essential for the platelet‐aggregating activity.
We showed that podoplanin on the surface of tumor cells induces platelet aggregation by interacting with CLEC‐2, because CLEC‐2–Fc inhibited podoplanin‐induced platelet aggregation (Fig. 5). A study using several clones possessing different metastatic abilities from a mouse colon adenocarcinoma 26 cell line revealed that a highly metastatic clone expresses more podoplanin and induces platelet aggregation to a greater extent,( 25 ) although podoplanin is not expressed in human colon adenocarcinoma.( 7 , 19 ) Moreover, an anti‐mouse podoplanin antibody (8F11) that inhibits podoplanin‐induced platelet aggregation suppressed lung colonization of mouse colon adenocarcinoma.( 26 ) We also showed that an anti‐human podoplanin antibody (NZ‐1), which neutralized platelet aggregation, inhibited the experimental metastasis of CHO/hPod cells (Fig. 7). The podoplanin–CLEC‐2 interaction was diminished by NZ‐1 (Fig. 6a); however, the interaction was not inhibited completely even at a dose of 1 mg/mL of NZ‐1 (data not shown), indicating that CLEC‐2 might be able to recognize sialic acid on threonine residues other than Thr52, because podoplanin is highly glycosylated. Indeed, the interaction between PodΔC57 and CLEC‐2 was inhibited completely by NZ‐1, because PodΔC57 possesses only one disialyl‐core1 (Fig. 6b). In the experimental metastasis model, F(ab′)2 fragments of NZ‐1 also suppressed pulmonary metastasis (Fig. 7), suggesting that suppression of tumor metastasis by NZ‐1 is not only dependent on antibody dependent cell‐mediated cytotoxity, but is also caused by neutralization of the interaction between podoplanin and CLEC‐2. These findings, taken together, suggest that CLEC‐2 is a critical pathophysiological ligand of podoplanin in vivo, and that podoplanin‐induced platelet aggregation through CLEC‐2 is one of the important mechanisms of tumor metastasis, although it might be true in limited human tumors. The importance of the podoplanin–CLEC‐2 interaction has been elucidated for the first time to our knowledge in a pathophysiological condition.
In conclusion, we have clarified the molecular interaction between a platelet aggregation‐inducing factor, podoplanin, and its specific receptor on platelets, CLEC‐2, and have shown that a glycopeptide including only one disialyl‐core1 structure is necessary for the binding of podoplanin to CLEC‐2. It is noteworthy that a glycopeptide containing only one disialyl‐core1 is functional and essential for platelet aggregation‐inducing activity. The interaction between podoplanin and CLEC‐2 may regulate tumor invasion and metastasis. Therefore, podoplanin and CLEC‐2 might represent promising therapeutic targets in cancer metastasis.
Acknowledgments
This study was supported in part by the Medical Glycomics Project supported by the New Energy and Industrial Technology Development Organization, and by a grant for Research Fellowships from the Japan Society for the Promotion of Science for Young Scientists, Japan (Y. Kato).
References
- 1. Gupta GP, Massague J. Platelets and metastasis revisited: a novel fatty link. J Clin Invest 2004; 114: 1691–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kato Y, Fujita N, Kunita A et al . Molecular identification of Aggrus/T1α as a platelet aggregation‐inducing factor expressed in colorectal tumors. J Biol Chem 2003; 278: 51 599–605. [DOI] [PubMed] [Google Scholar]
- 3. Breiteneder‐Geleff S, Soleiman A, Kowalski H et al . Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol 1999; 154: 385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kato Y, Kaneko M, Sata M, Fujita N, Tsuruo T, Osawa M. Enhanced expression of Aggrus (T1α/podoplanin), a platelet‐aggregation‐inducing factor in lung squamous cell carcinoma. Tumor Biol 2005; 26: 195–200. [DOI] [PubMed] [Google Scholar]
- 5. Martin‐Villar E, Scholl FG, Gamallo C et al . Characterization of human PA2.26 antigen (T1α‐2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int J Cancer 2005; 113: 899–910. [DOI] [PubMed] [Google Scholar]
- 6. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial–mesenchymal transition: podoplanin‐mediated remodeling of the actin cytoskeleton. Cancer Cell 2006; 9: 261–72. [DOI] [PubMed] [Google Scholar]
- 7. Kimura N, Kimura I. Podoplanin as a marker for mesothelioma. Pathol Int 2005; 55: 83–6. [DOI] [PubMed] [Google Scholar]
- 8. Kato Y, Sasagawa I, Kaneko M, Osawa M, Fujita N, Tsuruo T. Aggrus: a diagnostic marker that distinguishes seminoma from embryonal carcinoma in testicular germ cell tumors. Oncogene 2004; 23: 8552–6. [DOI] [PubMed] [Google Scholar]
- 9. Mishima K, Kato Y, Kaneko MK et al . Podoplanin expression in primary central nervous system germ cell tumors: a useful histological marker for the diagnosis of germinoma. Acta Neuropathol 2006; 111: 563–8. [DOI] [PubMed] [Google Scholar]
- 10. Mishima K, Kato Y, Kaneko MK, Nishikawa R, Hirose T, Matsutani M. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol 2006; 111: 483–8. [DOI] [PubMed] [Google Scholar]
- 11. Kunita A, Kashima TG, Morishita Y et al . The platelet aggregation‐inducing factor aggrus/podoplanin promotes pulmonary metastasis. Am J Pathol 2007; 170: 1337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaneko MK, Kato Y, Kitano T, Osawa M. Conservation of a platelet activating domain of Aggrus/podoplanin as a platelet aggregation‐inducing factor. Gene 2006; 378: 52–7. [DOI] [PubMed] [Google Scholar]
- 13. Kaneko M, Kato Y, Kunita A, Fujita N, Tsuruo T, Osawa M. Functional sialylated O‐glycan to platelet aggregation on Aggrus (T1α/podoplanin) molecules expressed in Chinese Hamster Ovary cells. J Biol Chem 2004; 279: 38 838–43. [DOI] [PubMed] [Google Scholar]
- 14. Kaneko MK, Kato Y, Kameyama A et al . Functional glycosylation of human podoplanin: glycan structure of platelet aggregation‐inducing factor. FEBS Lett 2007; 581: 331–6. [DOI] [PubMed] [Google Scholar]
- 15. Schacht V, Ramirez MI, Hong YK et al . T1α/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J 2003; 22: 3546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colonna M, Samaridis J, Angman L. Molecular characterization of two novel C‐type lectin‐like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol 2000; 30: 697–704. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki‐Inoue K, Fuller GL, Garcia A et al . A novel Syk‐dependent mechanism of platelet activation by the C‐type lectin receptor CLEC‐2. Blood 2006; 107: 542–9. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki‐Inoue K, Kato Y, Inoue O et al . Involvement of the snake toxin receptor CLEC‐2 in podoplanin‐mediated platelet activation by cancer cells. J Biol Chem 2007; 282: 25 993–6001. [DOI] [PubMed] [Google Scholar]
- 19. Kato Y, Kaneko MK, Kuno A et al . Inhibition of tumor cell‐induced platelet aggregation using a novel anti‐podoplanin antibody reacting with its platelet‐aggregation‐stimulating domain. Biochem Biophys Res Commun 2006; 349: 1301–7. [DOI] [PubMed] [Google Scholar]
- 20. Pohlmann S, Zhang J, Baribaud F et al . Hepatitis C virus glycoproteins interact with DC‐SIGN and DC‐SIGNR. J Virol 2003; 77: 4070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kariyazono H, Nakamura K, Arima J et al . Evaluation of anti‐platelet aggregatory effects of aspirin, cilostazol and ramatroban on platelet‐rich plasma and whole blood. Blood Coagul Fibrinolysis 2004; 15: 157–67. [DOI] [PubMed] [Google Scholar]
- 22. Fuller GL, Williams JA, Tomlinson MG et al . The C‐type lectin receptors CLEC‐2 and Dectin‐1, but not DC‐SIGN, signal via a novel YXXL‐dependent signaling cascade. J Biol Chem 2007; 282: 12 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson MJ, Sancho D, Slack EC, LeibundGut‐Landmann S, Reis e Sousa C. Myeloid C‐type lectins in innate immunity. Nat Immunol 2006; 7: 1258–65. [DOI] [PubMed] [Google Scholar]
- 24. Watson AA, Brown J, Harlos K, Eble JA, Walter TS, O’Callaghan CA. The crystal structure and mutational binding analysis of the extracellular domain of the platelet‐activating receptor CLEC‐2. J Biol Chem 2007; 282: 3165–72. [DOI] [PubMed] [Google Scholar]
- 25. Tsuruo T, Yamori T, Naganuma K, Tsukagoshi S, Sakurai Y. Characterization of metastatic clones derived from a metastatic variant of mouse colon adenocarcinoma 26. Cancer Res 1983; 43: 5437–42. [PubMed] [Google Scholar]
- 26. Sugimoto Y, Watanabe M, Oh‐hara T, Sato S, Isoe T, Tsuruo T. Suppression of experimental lung colonization of a metastatic variant of murine colon adenocarcinoma 26 by a monoclonal antibody 8F11 inhibiting tumor cell‐induced platelet aggregation. Cancer Res 1991; 51: 921–5. [PubMed] [Google Scholar]