

Abstract
Bone morphogenetic protein (BMP) 7 counteracts physiological epithelial‐to‐mesenchymal transition, a process that is indicative of epithelial plasticity in developmental stages. Because epithelial‐to‐mesenchymal transition and its reversed process mesenchymal‐to‐epithelial transition (MET) are also involved in cancer progression, we investigated whether BMP7 plays a role in WM‐266‐4 melanoma cell growth and metastasis. An MTT assay was conducted in WM‐266‐4 and HEK293T cell lines to show the cell growth inhibition ability of BMP7 and cisplatin. Semiquantitative RT‐PCR was used to determine MET in morphologically changed BMP7‐treated melanoma cells. MET‐induced cells expressed less a basic helix‐loop‐helix transcription factor (TWIST) in western blot analysis, and we confirm that BMP receptor (Alk2) siRNA transduction could restore TWIST protein expression via blocking of Smad 1, 5 and 8 signaling. Matrigel invasion and cell migration assays were done to investigate the BMP7‐induced metastasis inhibition ability. BMP7 treatment only slightly reduced cell growth rate, but induced apparent MET. BMP7 also reduced the invasion and migration ability. Furthermore, BMP7 reduced the resistance of WM‐266‐4 cells to cisplatin. Collectively, our findings indicate that the metastatis inhibition ability of BMP7 is involved in MET, and that BMP7 could be used as a potential metastasis inhibitor in human melanoma cells. (Cancer Sci 2009)
Although 90% of cancer deaths are caused by metastasis,( 1 ) most chemotherapeutic agents cannot prevent tumor metastasis. The pathogenesis and mechanisms underlying this event are still poorly understood.( 2 , 3 , 4 ) Metastasis is a ‘hidden’ event, which happens inside the body and is difficult to examine. It is believed to consist of four distinct steps: invasion, intravasation, extravasation, and metastatic colonization. Most carcinoma cells lose their cell–cell contact during the initial step of metastasis and move into the systemic circulation. For this, cells must acquire abilities of migration and invasion, together with cytoskeletal reorganization and active response to their microenvironment. In vivo video microscopy and quantitative approaches show that the first step, the acquisition of invasive ability and motility, is the rate‐limiting step in the metastatic cascade( 2 , 5 ) and clearly indicate that controlling this initial step of metastasis is critical for the development of novel strategies to prevent cancer metastasis.( 4 )
Epithelial‐to‐mesenchymal transition (EMT), a process vital for morphogenesis during embryonic development, is attracting attention as an important mechanism involved in the initial step of metastasis. During EMT, epithelial cells acquire fibroblast‐like properties and show reduced intercellular adhesion and increased motility.( 6 , 7 ) Conversely, mesenchymal cells possess remarkable plasticity and can eventually regain a fully differentiated epithelial phenotype via a mesenchymal‐to‐epithelial transition (MET).( 8 , 9 ) Because EMT involves the conversion of a sheet of tightly attached epithelial cells into highly mobile mesenchymal or neural crest cells, carcinoma cells undergo EMT to achieve local invasion and dissemination to distant organs.( 10 ) The ability to survive in the absence of normal matrix components was interpreted as an important property for cells undergoing EMT.( 11 , 12 , 13 ) Researchers are now extensively investigating the relationship between EMT and metastasis to identify potential targets for cancer therapy.
Bone morphogenetic protein (BMP) 7 is a 35‐kD homodimeric protein and a member of the transforming growth factor (TGF)‐β superfamily.( 14 ) Members of the TGF‐β superfamily are involved in the control of many different biological processes, including cell proliferation, differentiation, apoptosis, and regulation of invasiveness.( 15 , 16 , 17 ) TGF‐β is a well‐known inducer of EMT in the bone marrow and BMP7 counteracts TGF‐β1‐induced EMT.( 18 ) The discovery that perturbations in BMP pathways are genetically responsible for certain hereditary cancer syndromes such as familial juvenile polyposis and a subset of Cowden syndrome( 19 , 20 ) prompted the delineation of their significance in carcinogenesis.( 21 ) Because EMT is related to cancer metastasis, MET induced by BMP7 could inhibit prostate cancer bone metastasis, breast cancer bone metastasis, and renal fibrosis.( 18 , 22 , 23 ) BMP7 also inhibited tumor growth of human uveal melanoma in vivo,( 24 ) and EMT in melanoma was a major determinant of metastasis.( 25 ) Melanocytes originate from mesenchymal neural crest cells, thus melanoma cells easily spread from primary sites into the body.( 26 ) For this reason, melanomas are often fatal in humans. Despite the high relationship between metastatic melanoma cells and EMT, induction of MET and inhibition of metastasis by BMP7 in highly aggressive metastatic melanoma cells has not been studied.
To study the inhibitory effects of BMP7 on metastasis, we used the highly invasive human malignant metastatic melanoma cell line WM‐266‐4 and investigated the effects of BMP7 on cell morphology, migration, invasion, and resistance to cisplatin. We show that BMP7 induced MET in melanoma cells via upregulation of the specific receptor BMP receptor (Alk2) and downregulation of mesenchymal cell markers. In addition, BMP7 abolished cell migration and invasion abilities, and decreased resistance to cisplatin. Interestingly, cisplatin did not affect the cell migration and invasion capacity. Our results suggest that BMP7 administration could be an effective preventive therapy for metastatic melanomas.
Materials and Methods
Cell lines and BMP7 treatment. The human metastatic malignant melanoma cell line WM‐266‐4 and the human embryonic kidney cell line HEK293T were obtained from Korean Cell Line Bank (Cancer Research Institute, Seoul, Korea). WM‐266‐4 and HEK cell line were cultured in α‐MEM and DMEM (Gibco BRL, Long Island, NY, USA) supplemented with 10% FBS (Gibco), 100 U/mL penicillin (Gibco‐Invitrogen, Paisley, Scotland, UK), and 100 μg/mL streptomycin (Gibco‐Invitrogen). To determine the effect of human recombinant BMP7 protein (BMP7, Prospec‐Tany Techno Gene L Rehovnt Sctence Park, Israel) and cisplatin (CHOONGWAE Pharma Corporation, Dongjak‐gu, Seoul, Korea), WM‐266‐4 and HEK cells were grown in medium containing 0.5% serum. Cell morphology was examined under light microscopy (Olympus IX70, Olympus, Center Valley, PA, USA) and photographed using a Nikon D5000 digital camera (Nikon, Melville, NY, USA). In the cell survival assay, wound healing assay, and Matrigel invasion chamber assay, medium containing 0.1% serum was used. No attempts were made to start with a cell cycle‐synchronized cell population.
Cell survival assay. The MTT assay was carried out as previously described.( 27 ) Briefly, WM‐266‐4 and HEK cells (5 × 103) were seeded into 96‐well culture plates in triplicate in the absence of BMP7 and cisplatin, and incubated overnight in medium containing 0.1% serum. The plates were treated with various concentrations of BMP7 (0, 50, 100, 200, and 400 ng/mL) and cisplatin (0, 0.5, 1, 2, 4, 8, 16 μmol/L) for 8 days, with a media change every second day. MTT solution (20 μL of 5 mg/mL MTT in PBS) was added to each well, and plates were incubated for 4 h at 37°C. Following removal of the supernatant, 200 μL DMSO was added to each well, then transferred to 96‐well microplates and the optical density at 580 nm was measured with an EL800 microplate reader (BIO‐TEK Instruments, Winooski, VT, USA).
Immunostaining. Cells cultured on an eight‐well chamber slide (NUNC, Thermo Fisher Scientific, NY, USA) for 10 days under BMP7 treatment or not, were washed twice with PBS, fixed with 4% (w/v) paraformaldehyde in PBS for 15 min, and permeabilized with 0.2% Triton X‐100 for 2 min. Endogenous peroxidase was quenched with 1% H2O2 (v/v) in methanol for 30 min. After blocking in Tris‐buffered saline (TBS) buffer containing 0.1% gelatin, 5% normal horse serum, and 0.5% Triton X‐100, primary antibody was added and allowed to bind at 4°C overnight. Cells were washed three times in TBS with 1% Tween 20 and incubated with secondary antibody for 1 h before color development. Color was developed using 3,3′‐diaminobenzidine substrate (Sigma‐Aldrich, St Louis, MO, USA). The antibody used in the experiment was goat anti‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
RNA preparation and semiquantitative RT‐PCR. Total RNA was extracted using TRIzol reagent (Invitrogen, San Diego, CA, USA) following the protocol recommended by the manufacturer. cDNA was produced from 1 μg of each RNA sample using the Maxime RT PreMix Kit with an oligo dT Primer (iNtRON Biotechnology, Kyungki‐Do, Korea) in a 20‐μL reaction volume. PCR was carried out with AccuPower PCR Premix (Bioneer, Daejon, Korea) using the primer sets described in Table 1. The cycle that optimally reflected the amount of original template was determined and used in the semiquantitative PCR experiment. PCR products were visualized under ultraviolet light. Semiquantitative RT‐PCR band results were further analyzed using the Kodak Image Analysis Software (Rochester, NY, USA).
Table 1.
Primer sequences and product sizes for RT‐PCR
| Target | Primer sequence | Product size base pairs | Melting temperature (°C) | Amplification cycles |
|---|---|---|---|---|
| Alk2 | 5′‐GCATTCCCAGAGCACCAATC | 383 | 54 | 30 |
| 3′‐CTGTGAGTCTTGCGGATGGA | ||||
| Alk6 | 5′‐GCAGCACAGACGGATATTGT | 630 | 50 | 30 |
| 3′‐TTTCATGCCTCATCAACACT | ||||
| noggin | 5′‐CACTACGACCCAGGATTCAT | 213 | 55 | 25 |
| 3′‐CTCCGCAGCTTCTTGCTTAG | ||||
| SMA | 5′‐AGGAAGGACCTCTATGCTAACAAT | 355 | 54 | 25 |
| 3′‐AACACATAGGTAACGAGTCAGAGC | ||||
| Slug | 5′‐ACGCCTCCAAAAAGCCAAAC | 517 | 58 | 30 |
| 3′‐GGTAATGTGTGGGTCCGAAT | ||||
| TWIST | 5′‐GGAGTCCGCAGTCTTACGAG | 201 | 56 | 30 |
| 3′‐TCTGGAGGACCTGGTAGAGG | ||||
| snail | 5′‐CCTCCCTGTCAGATGAGGAC | 234 | 57 | 30 |
| 3′‐CCAGGCTGAGGTATTCCTTG | ||||
| GAPDH | 5′‐TCTAGACGGCAGGTCAGGTCCACC | 598 | 58 | 25 |
| 3′‐CCACCCATGGCAAATTCCATGGCA |
Preparation of whole cell lysates and Western blot analysis. To detect changes in Smad signaling caused by BMP7, cells were treated with various concentrations of BMP7 for 1 h and collected for western blotting. TWIST protein levels were detected after 8 days of BMP7 treatment. To prepare the whole cell lysates, cells were sonicated for 10 s in ice‐cold buffer containing 1% Triton X‐100, 150 mmol/L NaCl, 10 mmol/L Tris‐HCl (pH 7.4), 1 mmol/L EGTA, 1 mmol/L EDTA, and 0.5% NP‐40 with freshly added proteinase inhibitor (Roche, Mannheim, Germany) and phosphatase inhibitor cocktails (Sigma‐Aldrich). After 30 min on ice, cell debris was removed by centrifugation at 14 000 g for 20 min. The antibodies used in the western blot experiments were rabbit anti‐phospho‐Smad 1, 5 and 8 (Cell Signaling Technology, Beverly, MA, USA), rabbit anti‐TWIST (Santa Cruz Biotechnology), and goat anti‐actin (Santa Cruz Biotechnology).
siRNA transfection. The selected targeting siRNA duplexes for Alk2 as well as scrambled sequence siRNA were purchased from Sigma‐Aldrich and the sequences are indicated in Table 2. Cells were transfected with siRNA duplex in serum‐free culture medium using Oligofectamine reagent (Invitrogen). siRNA and Oligofectamine were diluted in separate tubes, combined, and incubated for 20 min at room temperature. The siRNA:Oligofectamin mixture was added to the medium and incubated for 6 h at 37°C followed by a medium change to complete serum and antibiotics. At 24 h after transfection, we confirmed that the transfection efficiency was over 80% by observing the siRNA 5′‐tagged green fluorescence under ultraviolet light. BMP7 treatments were conducted 12 h after transfection. In the case of BMP7 treatment for 8 days, transfection was done twice at 2‐day intervals.
Table 2.
Sequences for siRNA targeting
| Name | Sequence (5′–3′) | Size base pairs | Commercial name |
|---|---|---|---|
| Control | GAUCAUACGUGCGAUCAGA[dT][dT] | 21 | Scrambled‐S |
| UCUGAUCGCACGUAUGAUC[dT][dT] | 21 | Scrambled‐AS | |
| Si1 (Alk2) | GUUCUCAGACCCGACAUUA[dT][dT] | 21 | SASI‐Hs02_00325216_s |
| UAAUGUCGGGUCUGAGAAC[dT][dT] | 21 | SASI‐Hs02_00325216_as | |
| Si2 (Alk2) | CGUUGUACGACUAUCUUCA[dT][dT] | 21 | SASI‐Hs02_00325217_s |
| UGAAGAUAGUCGUACAACG[dT][dT] | 21 | SASI‐Hs02_00325217_as |
Matrigel invasion chamber assay. Inserts of 8‐μm pore‐sized membranes for 24‐well plates were prepared by coating with 100 μL of 1:4 diluted Matrigel basement membrane matrix high concentration (Becton Dickinson Labware, Bedford, MA, USA) following the manufacturer’s protocol. WM‐266‐4 cells (1 × 106) were left untreated or treated with 100 ng/mL BMP7 with or without 4 μmol/L cisplatin for 8 days then seeded in triplicate into inserts. Serum‐free α‐MEM containing 5 μg/mL fibronectin was added to the bottom chamber to serve as a chemoattractant. After 20 h of incubation, cells that migrated through the Matrigel were photographed and counted using Image Pro Plus software for analysis (Media Cybernetics, Inc., MD, USA).
Wound healing assay. WM‐266‐4 cells were treated with 100 ng/mL BMP7 or 4 μmol/L cisplatin for 8 days and grown to 90–100% confluency, then a wound line was made on the monolayer cell cultures with a microtip. Cells were washed twice with PBS, fresh medium was added, and photographs were taken at the indicated time points.
Determination of drug resistance by MTT assay. WM‐266‐4 cells (5 × 103) were seeded into 96‐well culture plates in triplicate and incubated overnight. After treatment with various concentrations of BMP7 (0, 100, and 200 ng/mL) for 3 days, cisplatin (0, 1, 2, 4, and 8 μmol/L) was added for an additional 8 days. The medium was changed every second day. The cell proliferation assay was carried out as described above.
Statistical analysis. The Dunn’s Multiple Comparison Test was used for comparison of gene expression levels of Alk2, smooth muscle actin (SMA), and TWIST. The non‐paired t‐test was used for comparison of cell migration in the wound healing assay, and the Mann–Whitney test was used for statistical evaluation of the cell survival assay, Matrigel invasion chamber assay, and drug resistance assay. All tests were applied using GraphPad Prism Version 3 (GraphPad Software, La Jolla, CA, USA). Data are presented as mean ± SD. A P‐value ≤ 0.05 was considered significant. Half maximal effective concentration (EC50) was obtained from Probit analysis in the Toxstat program (Western Eco System Technologies, Inc., WI, USA).
Results
BMP7 inhibited proliferation of melanoma cells only, whereas cisplatin inhibited growth of both melanoma and embryonic kidney cells. Human BMP7 inhibited WM‐266‐4 cell growth significantly at a concentration of 400 ng/mL (Fig. 1A). Lower concentrations of BMP7 showed a slight growth inhibition effect compared with control but could not attain statistical significance. Interestingly, BMP7 had no effect on the growth of human non‐cancer HEK293T cells even at the highest concentration of BMP7 tested. In contrast, cisplatin showed dose‐responsive growth inhibition of both WM‐266‐4 and HEK293T cells (Fig. 1B), with EC50 values of 5.64 μmol/L for the WM‐266‐4 cells and 3.87 μmol/L for HEK cells.
Figure ;1.
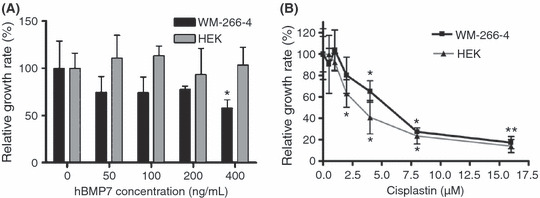
Growth curves of WM‐266‐4 and HEK293T cells treated with bone morphogenetic protein (BMP) 7 and cisplatin. (A) BMP7 at 400 ng/mL showed significant growth inhibition of melanoma cells whereas HEK293T cells did not respond even at the maximum concentration tested (*P < 0.015). (B) Both cell lines showed dose‐dependent survival rates in response to cisplatin. The EC50 of cisplatin in the melanoma cell line was approximately 1.5 times higher than in the HEK293T cell line (5.64 and 3.87 μmol/L, respectively). Bars, SD; *P < 0.04.
BMP7 induced MET transition in WM‐266‐4 cells. In normal culture conditions, WM‐266‐4 metastatic melanoma cells showed a highly dedifferentiated mesenchymal phenotype with an elongated spindle‐shaped cell body and three or more filopodia‐like cytoplasmic projections. Although the growth rate of WM‐266‐4 was not greatly affected by exposure to BMP7, we observed dramatic morphological changes in these cells as shown by the micrograph of cells treated with various concentrations of BMP7 for 8 days. With increasing concentrations of BMP7, cell morphology was changed into epithelial‐like cells with a polygonal cell body and short cytoplasmic projections (Fig. 2A). Actin immunostaining further confirmed these morphological changes more clearly (Fig. 2B). From this observation, we inferred that BMP7 induced MET in the melanoma cell line because morphological changes are key consequences of EMT or MET.( 28 ) Morphological changes were first evident in cells treated with 50 ng/mL BMP7 and obvious in cells treated with 100 and 200 ng/mL BMP7. Cisplatin caused cell death without apparent morphological changes, even in cells treated with 10 μmol/L cisplatin.
Figure 2.
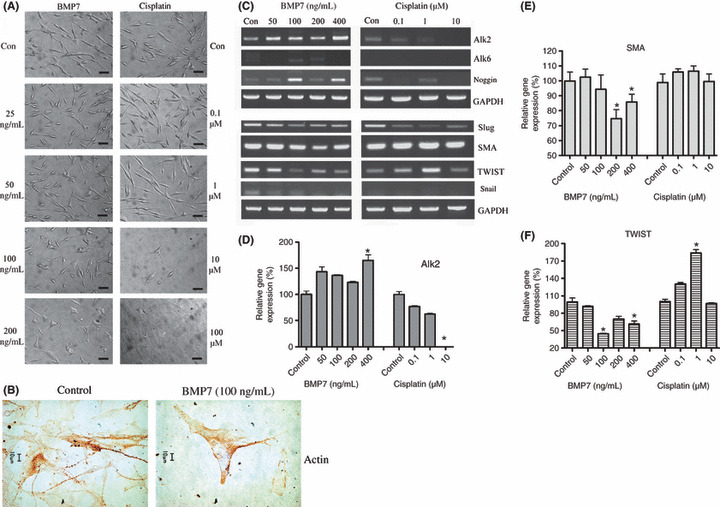
Mesenchymal‐to‐epithelial transition (MET) induced by bone morphogenetic protein (BMP) 7 in the WM‐266‐4 human melanoma cell line. (A) Cells were treated with various concentrations of BMP7 and cisplatin, and the monolayer morphology was photographed at ×40 magnification 8 days after treatment. Cells of the BMP7‐treated group showed morphological changes resembling cells undergoing MET, transitioning from mesenchymal to epithelial‐like cells in a dose‐responsive manner. In contrast, almost all of the cisplatin‐treated cells died without apparent MET at concentrations of 10 and 100 μmol/L. Scale bar = 50 μm. (B) Actin immunostaining revealed that normal WM‐266‐4 cells have numerous and long cytoplasmic projections (stained strong brown), but the cells under BMP7 100 ng/mL treatment for 10 days show short projections and polygonal cell body. Images are representatives of three replicate experiments. (C) Total RNA was prepared and semiquantitative RT‐PCR was conducted to confirm expression of genes related to BMP7‐responsive receptors. Expression of the BMP7‐specific receptor BMP receptor (Alk2) increased only in the BMP7‐treated group. Expression of Alk6 and noggin increased following BMP7 treatment, but did not show a dose‐dependent response. Expression of Slug, smooth muscle actin (SMA), TWIST, and snail were also decreased in the BMP7‐treated WM‐266‐4 cells. Cisplatin‐treated cells showed decreased levels of Slug mRNA, but not for SMA, TWIST, and snail. The experiments were repeated three times, each with triplicate samples. Semiquantitative RT‐PCR results for (D) Alk2, (E) SMA, and (F) TWIST are shown. P‐values for comparisons in the 95% significant level were analyzed by Dunn’s Multiple Comparison Test. Bars, SD; *P < 0.05.
Next, we confirmed expression of BMP receptors and their antagonist. WM‐266‐4 cells expressed Alk2 (ActRI), a specific BMP7 receptor, and the Alk2 expression level was significantly increased in the 400 ng/mL BMP7‐treated groups (Fig. 2C,D). This confirmed that the observed effects were directed via exogenous BMP7.( 28 , 29 ) Expression of Alk6 (BMPRIB) was not detected in the control group and was slightly detectable in the BMP7‐treated group. Levels of noggin, a known BMP7 antagonist that is upregulated following endogenous BMP7 expression,( 21 , 29 ) were also increased in the groups treated with 100 and 400 ng/mL BMP7. Cisplatin treatment decreased the expression levels of Alk2 in a dose‐dependent manner, and also decreased noggin expression level in the 0.1 and 10 μmol/L cisplatin treatment group.
We then tested whether expression of Slug, SMA, TWIST, and snail [key indicators of EMT( 30 , 31 )] was altered during the BMP7‐induced morphological changes in WM‐266‐4 cells. As shown in Figure 2(C,E,F), all four mesenchymal cell markers were downregulated by BMP7 treatment, further confirming that BMP7 induces MET. Because WM‐266‐4 cells are a malignant metastatic cell line, they did not express basal BMP7 and E‐cadherin (data not shown).
Alk2 gene knockdown blocks BMP7‐induced Smad signaling. BMP7 induced phosphorylation of Smad1/5/8 in a dose‐dependent manner (Fig. 3A). Although the decreasing extents seemed to be small compared with RT‐PCR data, we also confirmed that BMP7 treatment decreased TWIST protein expression levels (Fig. 3B). When two different sequences of siRNA, si1 and si2 in Table 1, were introduced into melanoma cells for Alk2 gene knockdown, Alk2 mRNA expression levels were decreased in both groups (Fig. 3C). Although si2 showed more decreased levels of Alk2 mRNA than si1, western blotting results revealed that only si1 could block Smad1/5/8 phosphorylation under BMP7 100 ng/mL treatment (Fig. 3D); thus, we selected si1 for further experiments. Si1 transfection under BMP7 could not decrease TWIST protein expression, compared with the scrambled siRNA‐treated control group (Fig. 3E). These results indicate that BMP7 induced MET via Alk2 on the cell surface and subsequent Smad1/5/8 phosphorylation. TWIST inhibition also seems to be related to MET induced by BMP7.
Figure ;3.
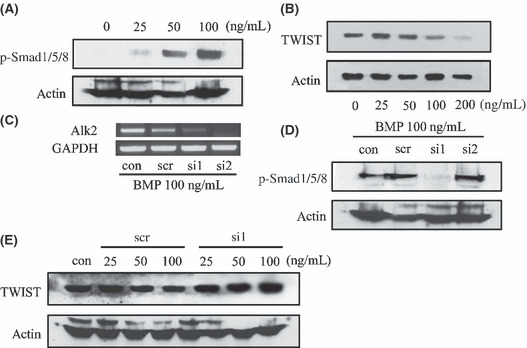
BMP receptor (Alk2) gene knockdown abrogated phosphorylation of Smad1/5/8 and a basic helix‐loop‐helix transcription factor (TWIST) inhibition. (A) Western blot analysis shows that bone morphogenetic protein (BMP) 7 treatment phosphorylates Smad1/5/8 in a dose‐dependent manner. Control cells express almost no p‐Smad‐1/5/8, but after 1 h of treatment with BMP7, the cells express p‐Smad1/5/8 increasingly. (B) WM‐266‐4 cells were treated with various concentrations of BMP7 for 8 days and collected for western blotting. TWIST expression was decreased in the 100 and 200 ng/mL BMP7‐treated groups. (C) Two different sequences of siRNA targeting Alk2 (si1 and si2) as well as scrambled sequence siRNA (scr) were transduced twice at 3‐day intervals. Cells were cotreated with BMP7 100 ng/mL for 8 days and the expression levels of Alk2 were examined. Alk2 mRNA expression were decreased in the si1‐ and si2‐transfected groups. (D) Only si1 siRNA inhibited phosphorylation of Smad1/5/8 under BMP7 100 ng/mL treatment. (E) TWIST protein expression decreased in the scrambled siRNA transfection group (scr) but not in the si1‐transfected group.
Transdifferentiated WM‐266‐4 cells showed reduced invasion ability. Following 8 days of treatment with 100 ng/mL BMP7, 4 μmol/L cisplatin, or both, WM‐266‐4 cells were applied onto 5 mg/mL gelled Matrigel and their invasion ability was investigated. As illustrated in Figure 4, cells that were invading into the basement membrane material barrier showed elongated downward cytoplasmic projections, and cell penetration into the bottom chamber was almost complete after 20 h. BMP7 treatment reduced the number of invading cells to 65% of that observed in the control group (Fig. 4B,C). Cisplatin alone did not affect cell invasion ability. No synergistic effect between BMP7 and cisplatin was observed.
Figure 4.
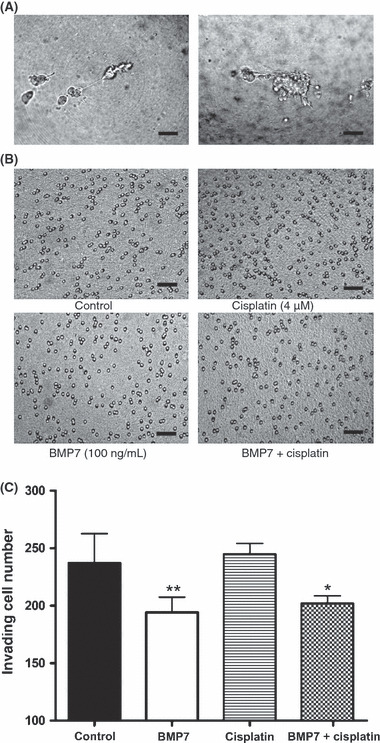
Bone morphogenetic protein (BMP) 7 inhibited melanoma cell invasion ability. (A) Cells invading through the extracellular basement membrane showed long filopodia‐like mesenchymal cell bodies (left). After invasion, round, non‐attached, single cells were present at the bottom of the Matrigel (right). Scale bar = 50 μm. (B) A 20‐h incubation time allowed complete invasion of most of the treated cells. The remnants covered the upper side of the Matrigel. Photographs were taken at the bottom site at 40 × magnification. Scale bar = 50 μm. (C) WM‐266‐4 cells were treated with 100 ng/mL BMP7, 4 μmol/L cisplatin, or both, for 8 days before the invasion assay. The BMP7‐treated group showed lower invaded cell numbers, which were reduced by approximately 65% invaded cell numbers compared with that of the control. Cisplatin did not affect cell invasion ability. The experiments were repeated three times, each with triplicate samples. Cell counts were analyzed using Image Pro Plus software. P‐values for comparisons in the 95% significant level were analyzed by Mann–Whitney two‐tailed test. Bars, SD; *P < 0.018; **P < 0.0042.
BMP7 reduced melanoma cell migration ability and resistance to cisplatin. WM‐266‐4 cells were subjected to wound healing assays following treatment with 100 ng/mL BMP7 or 4 μmol/L cisplatin for 8 days. As shown in Figure 5(A), BMP7 inhibited movement of the transdifferentiated cells into the wound line by 50% compared with untreated control cells, whereas cisplatin inhibited cell movement by only 10%. In addition, BMP7 affected resistance of melanoma cells to cisplatin (Fig. 5B); resistance to cisplatin was reduced following treatment with increasing concentrations of BMP7 (0, 100, and 200 ng/mL).
Figure 5.
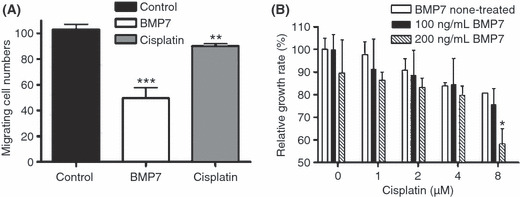
Bone morphogenetic protein (BMP) 7 inhibited melanoma cell migration ability and reduced resistance to cisplatin. (A) WM‐266‐4 cells were treated with 100 ng/mL BMP7 or 4 μmol/L cisplatin in 0.1% serum medium for 8 days and grown to 90% confluency. A wound line was made on the monolayer cultures with a microtip, cells were washed twice with PBS, and fresh medium was added. Photographs were taken at 12‐h time points. Migrating cell counts were analyzed using Image Pro Software. BMP7 reduced cell migration ability to <50% of that in the control, whereas cisplatin only slightly inhibited migration ability. Bars, SD. **P < 0.0074; ***P < 0.0006. (B) WM‐266‐4 cells were treated with BMP7 as indicated for 3 days, then with cisplatin at various concentrations for an additional 8 days. Cell resistance to cisplatin gradually decreased with increasing BMP7 concentrations. Bars, SD; *P < 0.005.
Discussion
This report provides strong evidence that BMP7 inhibits the metastatic potential of a selected metastatic melanoma cell line. Although BMP7 did not significantly inhibit growth of WM‐266‐4 cells, it changed the morphology of the melanoma cells and clearly reduced their migration and invasion abilities. We also showed that BMP7 induced MET in a melanoma cell line, which might be responsible for inhibition of metastasis‐related abilities. In contrast to BMP7, the chemotherapy drug cisplatin, which showed dose‐responsive growth inhibitory effects on WM‐266‐4 cells, did not induce MET. Moreover, cisplatin did not affect cell invasion abilities and only slightly reduced cell migration. Finally, we showed that BMP7 treatment decreased the resistance of melanoma cells to cisplatin. Together, these findings suggest that BMP7 might be a good candidate for the treatment of human melanoma metastasis in vivo.
The observation that implanted malignant human melanoma cells behave like host neural crest cells and migrate with host neural crest cells provided the initial hint of a possible relationship between embryonic morphogens and melanoma cells.( 26 , 32 ) These authors in the reference 26 and 32 suggested that a certain embryonic microenvironment might communicate with melanoma cells. In addition, several observations provided clues about EMT in malignant melanoma.( 25 , 33 , 34 , 35 ) Aggressive melanoma cells escape from BMP7‐mediated autocrine growth inhibition through coordinated upregulation of noggin, a secretory BMP antagonist.( 21 ) Consistent with this, our preliminary study revealed that WM‐266‐4 cells express noggin, but not E‐cadherin or BMP7, indicating that this cell line represents a highly aggressive state and might be a good candidate for investigating EMT and metastasis. Furthermore, our previous study showed that zebrafish embryo proteins at the 50%‐epiboly stage could affect WM‐266‐4 cell growth (36) so we selected BMP7, which is one of the highly expressed morphogens at this developmental stage, as a potential communicating molecule for this cell line.
The growth inhibitory effects of BMP7 are unclear (Fig. 1A). BMP7 inhibited growth of WM‐266‐4 cells only at the highest concentration tested (400 ng/mL), but did not inhibit growth of HEK293T cells. However, morphological changes were evident in BMP7‐treated WM‐266‐4 cells at concentrations as low as 50 ng/mL. MET induced by BMP7 treatment was further supported by semiquantitative RT‐PCR results for Alk2, Alk6, noggin, Slug, SMA, TWIST, and snail (Fig. 2C) as well as by western blotting results of TWIST and p‐Smad1/5/8 (Fig. 3B). We could not obtain exact dose–response results for these genes in the semiquantitative RT‐PCR, but patterns of increased levels of Alk2, Alk6, and noggin caused by BMP7 did exist, which shows direct BMP7‐mediated effects, as did decreased levels of Slug, SMA, TWIST, and snail, which shows MET. Biphasic effects in Figure 2(C) could be explained because BMP7 is one kind of endogenous molecule; thus, it might not be fit to apply dose–response curves in this case. On the other hand, no morphological changes were observed in cisplatin‐treated melanoma cells (Fig. 2A) or in HEK293T cells treated with either BMP7 or cisplatin (data not shown). Our results are consistent with the original concept that the more aggressive melanoma cells are, the more responsive they are to the embryonic microenvironment.( 26 ) As HEK293T is not a cancer cell line, it would be interesting to include a less aggressive melanoma cell line as an experimental group. The observation that BMP7 affects MET, not to the level of cell proliferation, is in total contrast to the effects of cisplatin. The cisplatin concentration used in the present study was 4 μmol/L, which caused 50% growth inhibition in HEK293T and approximately 60% inhibition in WM‐266‐4. Even after treatment with this toxic concentration of cisplatin, cells did not lose their capacity to invade through the basement membrane. This might give clues to us because the high rates of tumor recurrence and metastasis after chemotherapy are well documented.
BMP7 has previously been investigated in several cell lines including human prostate, breast cancer, kidney, colon, and melanoma cell lines.( 18 , 21 , 22 , 23 , 37 , 38 , 39 ) Although the mechanism of action of BMP7 is involved with Smad1/5/8 activation,( 28 ) the biological effects of BMP7 seem to vary. Evidence from a variety of tumors suggests that the effects of BMP are cell specific and can be either protumorigenic or antitumorigenic.( 40 ) For example, BMP7 has been shown to induce EMT, to counteract EMT by inducing MET, or to have no effect.( 22 , 28 , 41 , 42 , 43 ) Moreover, BMP7 inhibited prostate cancer bone metastasis and colonization in a mouse model, but did not inhibit growth of an orthotopically implanted tumor in the prostate.( 23 ) Based on this finding, the authors proposed the importance of the bone microenvironment, in particular, specific factors such as TGF‐β that act as inhibitors for BMP7. In contrast to these in vivo experiments, our in vitro data clearly indicate that BMP7 alone induces MET and inhibits metastatic potential in WM‐266‐4 melanoma cells in the absence of any bone‐specific microenvironment. It seems that BMP7‐induced Smad1/5/8 phosphorylation counteracts TWIST expression. Because TWIST represses E‐cadherin, which is a cell–cell adhesion molecule that is consistently observed at sites of EMT during development and cancer,( 4 ) we infer about BMP7‐mediated metastasis inhibition through TWIST inhibition. Because of the complexity of BMP signaling in cancer, this study has certain limitations including the need to test various kinds of melanoma cell lines and the lack of an in vivo animal model. However, our observations that metastatic potential was inhibited by BMP7, but not by cisplatin, do provide valuable insights into the need for alternative combination therapies that can inhibit tumor metastasis. New theories are emerging that we must focus on carcinogenesis centered at the tissue level, specifically, the tissue microenvironmental conditions.( 44 , 45 ) Our results fully support current aspects regarding the interaction of tissue microenvironment and carcinogenesis.
In summary, our results suggest that BMP7 may play a significant role in WM‐266‐4 melanoma cell metastasis. BMP7 inhibits cell invasion and migration and reduces drug resistance to cisplatin; these actions might be caused by the induction of a MET state, which is a key step in reversal of the cellular metastatic process. Furthermore, the inability of cisplatin to induce MET or inhibit metastatic potential emphasizes the significance of BMP7 as an alternative therapeutic candidate for metastatic melanoma cells.
Disclosure Statement
All the authors declare that they have no financial or personal relationships with other people or organizations that could inappropriately influence (bias) their work.
Acknowledgments
We gratefully acknowledge financial support from a Korea Research Foundation Grant (KRF‐005‐E00077) and additional financial support from the BK21 Program for Veterinary Science.
References
- 1. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up‐regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007; 67: 1979–87. [DOI] [PubMed] [Google Scholar]
- 2. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2: 563–72. [DOI] [PubMed] [Google Scholar]
- 3. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer 2004; 4: 448–56. [DOI] [PubMed] [Google Scholar]
- 4. Wu Y, Zhou BP. New insights of epithelial–mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin (Shanghai) 2008; 40: 643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacDonald IC, Groom AC, Chambers AF. Cancer spread and micrometastasis development: quantitative approaches for in vivo models. BioEssays 2002; 24: 885–93. [DOI] [PubMed] [Google Scholar]
- 6. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005; 17: 548–58. [DOI] [PubMed] [Google Scholar]
- 7. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 8. Dudley AT, Lyons KM, Robertson EJ. A requirement for bone morphogenetic protein‐7 during development of the mammalian kidney and eye. Genes Dev 1995; 9: 2795–807. [DOI] [PubMed] [Google Scholar]
- 9. Vukicevic S, Kopp JB, Luyten FP, Sampath TK. Induction of nephrogenic mesenchyme by osteogenic protein 1 (bone morphogenetic protein 7). Proc Natl Acad Sci USA 1996; 93: 9021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res 2006; 66: 4549–52. [DOI] [PubMed] [Google Scholar]
- 11. Cheung M, Chaboissier MC, Mynett A, Hirst E, Schedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell 2005; 8: 179–92. [DOI] [PubMed] [Google Scholar]
- 12. Martin SS, Ridgeway AG, Pinkas J et al. A cytoskeleton‐based functional genetic screen identifies Bcl‐xL as an enhancer of metastasis, but not primary tumor growth. Oncogene 2004; 23: 4641–5. [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Belguise K, Kersual N et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol 2007; 9: 470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ozkaynak E, Rueger DC, Drier EA et al. OP‐1 cDNA encodes an osteogenic protein in the TGF‐beta family. EMBO J 1990; 9: 2085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ducy P, Karsenty G. The family of bone morphogenetic proteins. Kidney Int 2000; 57: 2207–14. [DOI] [PubMed] [Google Scholar]
- 16. Itoh S, Itoh F, Goumans MJ, Ten Dijke P. Signaling of transforming growth factor‐beta family members through Smad proteins. Eur J Biochem 2000; 267: 6954–67. [DOI] [PubMed] [Google Scholar]
- 17. Simic P, Vukicevic S. Bone morphogenetic proteins in development and homeostasis of kidney. Cytokine Growth Factor Rev 2005; 16: 299–308. [DOI] [PubMed] [Google Scholar]
- 18. Zeisberg M, Hanai J, Sugimoto H et al. BMP‐7 counteracts TGF‐beta1‐induced epithelial‐to‐mesenchymal transition and reverses chronic renal injury. Nat Med 2003; 9: 964–8. [DOI] [PubMed] [Google Scholar]
- 19. Howe JR, Sayed MG, Ahmed AF et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet 2004; 41: 484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waite KA, Eng C. From developmental disorder to heritable cancer: it’s all in the BMP/TGF‐beta family. Nat Rev Genet 2003; 4: 763–73. [DOI] [PubMed] [Google Scholar]
- 21. Hsu MY, Rovinsky SA, Lai CY et al. Aggressive melanoma cells escape from BMP7‐mediated autocrine growth inhibition through coordinated Noggin upregulation. Lab Invest 2008; 88: 842–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buijs JT, Henriquez NV, Van Overveld PG et al. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res 2007; 67: 8742–51. [DOI] [PubMed] [Google Scholar]
- 23. Buijs JT, Rentsch CA, Van Der Horst G et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo . Am J Pathol 2007; 171: 1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Notting I, Buijs J, Mintardjo R et al. Bone morphogenetic protein 7 inhibits tumor growth of human uveal melanoma in vivo . Invest Ophthalmol Vis Sci 2007; 48: 4882–9. [DOI] [PubMed] [Google Scholar]
- 25. Alonso SR, Tracey L, Ortiz P et al. A high‐throughput study in melanoma identifies epithelial–mesenchymal transition as a major determinant of metastasis. Cancer Res 2007; 67: 3450–60. [DOI] [PubMed] [Google Scholar]
- 26. Kulesa PM, Kasemeier‐Kulesa JC, Teddy JM et al. Reprogramming metastatic melanoma cells to assume a neural crest cell‐like phenotype in an embryonic microenvironment. Proc Natl Acad Sci USA 2006; 103: 3752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 28. Yang S, Zhong C, Frenkel B, Reddi AH, Roy‐Burman P. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res 2005; 65: 5769–77. [DOI] [PubMed] [Google Scholar]
- 29. Hsu MY, Rovinsky S, Penmatcha S, Herlyn M, Muirhead D. Bone morphogenetic proteins in melanoma: angel or devil? Cancer Metastasis Rev 2005; 24: 251–63. [DOI] [PubMed] [Google Scholar]
- 30. Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor‐1 (HIF‐1): implications in metastasis and development. Cell Cycle 2008; 7: 2090–6. [DOI] [PubMed] [Google Scholar]
- 31. Yang MH, Wu MZ, Chiou SH et al. Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol 2008; 10: 295–305. [DOI] [PubMed] [Google Scholar]
- 32. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier‐Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer 2007; 7: 246–55. [DOI] [PubMed] [Google Scholar]
- 33. Kuphal S, Palm HG, Poser I, Bosserhoff AK. Snail‐regulated genes in malignant melanoma. Melanoma Res 2005; 15: 305–13. [DOI] [PubMed] [Google Scholar]
- 34. Poser I, Dominguez D, De Herreros AG, Varnai A, Buettner R, Bosserhoff AK. Loss of E‐cadherin expression in melanoma cells involves up‐regulation of the transcriptional repressor Snail. J Biol Chem 2001; 276: 24661–6. [DOI] [PubMed] [Google Scholar]
- 35. Tsutsumida A, Hamada J, Tada M et al. Epigenetic silencing of E‐ and P‐cadherin gene expression in human melanoma cell lines. Int J Oncol 2004; 25: 1415–21. [PubMed] [Google Scholar]
- 36. Na YR, Seok SH, Kim DJ et al. Zebrafish embryo extracts promote sphene‐forming abilities of human melanoma cell line. Cancer Sci. 2009; 100: 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buijs JT, Henriquez NV, Van Overveld PG, Van Der Horst G, Ten Dijke P, Van Der Pluijm G. TGF‐beta and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis 2007; 24: 609–17. [DOI] [PubMed] [Google Scholar]
- 38. Grijelmo C, Rodrigue C, Svrcek M et al. Proinvasive activity of BMP‐7 through SMAD4/src‐independent and ERK/Rac/JNK‐dependent signaling pathways in colon cancer cells. Cell Signal 2007; 19: 1722–32. [DOI] [PubMed] [Google Scholar]
- 39. Ro TB, Holt RU, Brenne AT et al. Bone morphogenetic protein‐5, ‐6 and ‐7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene 2004; 23: 3024–32. [DOI] [PubMed] [Google Scholar]
- 40. Tu WH, Thomas TZ, Masumori N et al. The loss of TGF‐beta signaling promotes prostate cancer metastasis. Neoplasia 2003; 5: 267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Piek E, Moustakas A, Kurisaki A, Heldin CH, Ten Dijke P. TGF‐(beta) type I receptor/ALK‐5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 1999; 112(Pt 24): 4557–68. [DOI] [PubMed] [Google Scholar]
- 42. Prindull G. Hypothesis: cell plasticity, linking embryonal stem cells to adult stem cell reservoirs and metastatic cancer cells? Exp Hematol 2005; 33: 738–46. [DOI] [PubMed] [Google Scholar]
- 43. Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF‐beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial–mesenchymal cell transition. Mol Biol Cell 2005; 16: 1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ingber DE. Can cancer be reversed by engineering the tumor microenvironment? Semin Cancer Biol 2008; 18: 356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sonnenschein C, Soto AM. Theories of carcinogenesis: an emerging perspective. Semin Cancer Biol 2008; 18: 372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]