

Abstract
3‐Phosphoinositide–dependent protein kinase‐1 (PDK1) is a key regulator of cell proliferation and survival signal transduction. PDK1 is known to be constitutively active and is further activated by Src‐mediated phosphorylation at the tyrosine‐9, ‐373, and ‐376 residues. To identify novel regulators of PDK1, we performed E. coli‐based two‐hybrid screening and revealed that tumor suppressor candidate 4 (TUSC4), also known as nitrogen permease regulator‐like 2 (NPRL2), formed a complex with PDK1 and suppressed Src‐dependent tyrosine phosphorylation and activation of PDK1 in vitro and in cells. The NH2‐terminal 133 amino acid residues of TUSC4 were involved in binding to PDK1. The deletion mutant of TUSC4 that lacked the NH2‐terminal domain showed no inhibitory effects on PDK1 tyrosine phosphorylation or activation. Thus, complex formation is indispensable for TUSC4‐mediated PDK1 inactivation. The siRNA‐mediated down‐regulation of TUSC4 induced cell proliferation, while ectopic TUSC4 expression inactivated the PDK1 downstream signaling pathway, including Akt and p70 ribosomal protein S6 kinase, and increased cancer cell sensitivity to several anticancer drugs. Our results suggest that TUSC4/NPRL2, a novel PDK1‐interacting protein, plays a role in regulating the Src/PDK1 signaling pathway and cell sensitivity to multiple cancer chemotherapeutic drugs. (Cancer Sci 2008; 99: 1827–1834)
Abbreviations:
- BSA
bovine serum albumin
- cDDP
cisplatin
- EGF
epidermal growth factor
- NPRL2
Nitrogen permease regulator‐like 2
- PARP
poly(ADP‐ribose) polymerase
- PDK1
3‐phosphoinositide‐dependent protein kinase‐1
- PH
Pleckstrin homology
- PI3K
phosphoinositide‐3‐kinase
- PtdIns(3,4,5)P3
phosphatidilinositol‐3,4,5‐trisphosphate
- S6K
p70 ribosomal protein S6 kinase
- SGKs
serum‐ and glucocorticoid‐inducible kinases
- siRNA
small interfering RNA
- TUSC4
tumor suppressor candidate 4
- VP‐16
etoposide
3‐Phosphoinositide‐dependent protein kinase‐1 (PDK1) was originally identified as a protein serine/threonine kinase that could phosphorylate Akt at the Thr308 residue in its activation loop.( 1 , 2 ) Later studies have shown that PDK1 is not only an Akt kinase but also a master regulator of a group of protein kinases known as the AGC (cAMP‐dependent, cGMP‐dependent, and protein kinase C) family, including p70 ribosomal protein S6 kinase (S6K), serum‐ and glucocorticoid‐inducible kinases (SGKs), protein kinase A (PKA), protein kinase C (PKC) isoforms, and p90 ribosomal protein S6 kinases (RSKs) at the equivalent residues of Thr308 in Akt (T‐loop).( 3 ) Therefore, PDK1 functions as a pivotal molecule for activation of a number of signaling pathways involved in proliferation and cell survival. Many components involved in the downstream of PDK1 have been elucidated, though the regulatory mechanism to control PDK1 activity is still controversial.
It has long been thought that auto‐phosphorylation at the Ser241 residue in the activation loop is sufficient for PDK1 activation.( 4 ) Thus, PDK1 is thought to be constitutively active in resting cells and not further activated by growth factor stimulation.( 5 ) However, recent reports suggest that PDK1 activity and stability are regulated by interaction with other proteins.( 6 , 7 , 8 , 9 , 10 , 11 ) For example, PDK1 binds to Hsp90, and its binding protects PDK1 from proteasome‐dependent degradation.( 6 ) PDK1 also binds to 14‐3‐3 through the residues surrounding the auto‐phosphorylation site Ser241 and its binding decreases PDK1 kinase activity.( 7 ) Moreover, Src kinase directly phosphorylates PDK1 at Tyr9 Tyr373, and Tyr376 leading to an increase in PDK1 kinase activity.( 8 , 9 , 10 , 11 ) It has also been proposed that Abl, RET/PTC, Pyk2, insulin receptor, and Hsp90 were candidates for PDK1 activation.( 11 ) To identify novel regulators of PDK1, we performed E. coli‐based two‐hybrid screening using the Pleckstrin homology (PH) domain of human PDK1 as bait. We were fortunate to identify the tumor suppressor candidate 4 (TUSC4), also known as nitrogen permease regulator‐like 2 (NPRL2), as a novel PDK1‐interacting protein.
TUSC4/NPRL2 is located at chromosome 3p21.3 and expresses in many normal tissues, including those of the heart, liver, skeletal muscle, kidney, and pancreas.( 12 ) Chromosomal deletions of the region are frequently observed in lung, breast, kidney, and other cancers.( 12 ) Allelic losses and overlapping homozygous deletions in lung and breast tumor cell lines have defined a 120‐kb region in 3p21.3, including TUSC4.( 12 , 13 ) Thus, tumor suppressor candidate(s) would be harbored in this region. It has been reported that overexpression of TUSC4 inhibited proliferation and induced apoptosis in a variety of tumor cell lines.( 14 ) Further studies show that TUSC4 induced susceptibility to anticancer drugs and apoptosis.( 15 , 16 ) How TUSC4 suppresses tumor proliferation has yet to be clarified.
In this study, we identified TUSC4 as a novel PDK1‐interacting protein. We revealed that TUSC4 inhibits Src‐mediated PDK1 tyrosine phosphorylation, leading to inactivation of Akt and S6K after stimulation. Moreover, aberrant expression of TUSC4 enhances sensitivity of tumor cells to several chemotherapeutic drugs.
Materials and Methods
Cell culture conditions. Human embryonic kidney 293 (HEK293) cells and its SV40 large T antigen–expressing 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Nissui, Tokyo, Japan) supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO, USA) (DMEM complete medium). Human fibrosarcoma HT1080 cells and human breast adenocarcinoma MCF‐7 cells were cultured in RPMI‐1640 medium (Nissui) supplemented with 10% FBS (RPMI complete medium). Stable HT1080 transfectants were cultured in RPMI complete medium supplemented with 200 µg/mL of geneticin (Sigma).
E. coli two‐hybrid screening. The BacterioMatch II two‐hybrid system (Stratagene, La Jolla, CA, USA) was used for the screening of PDK1‐interacting protein, according to the manufacturers’ instructions. The cDNA encoding Pleckstrin homology (PH) domain of PDK1 and additional 14 amino acids (445–556 a.a. of human PDK1) were ligated into a pBT vector for use as bait. A human fetal brain cDNA library was used as the prey for screening.
Reagents. Cisplatin (cDDP), taxol, and etoposide (VP‐16) were kindly provided by Bristol‐Myers Squibb (Tokyo, Japan). The geldanamycin derivative, 17‐(allylamino)‐17‐demethoxygeldanamycin (17‐AAG), was purchased from Alomone (Jerusalem, Israel). The recombinant Src and human active PDK1 were purchased from Millipore (Bedford, MA, USA). The 3XFLAG peptide was purchased from Sigma. The recombinant human epidermal growth factor (EGF) was purchased from Peprotech (London, UK).
Plasmid construction. Human full‐length TUSC4‐WT cDNA in a pc5FLAG vector was generated by polymerase chain reaction (PCR) with an I.M.A.G.E clone (clone ID 6169151; Open Biosystems, Huntsville, AL, USA) as the template. The p60 c‐src cDNA in a pHM6 vector was generated by PCR with an I.M.A.G.E clone (clone ID 4548399; Open Biosystems) as the template. Deletion mutants of TUSC4 were generated by PCR with pc5FLAG‐TUSC4‐WT as the template, using a QuikChange Mutagenesis Kit (Stratagene). The HA‐tagged human full‐length PDK1‐WT cDNA in a pHM6 vector was established in our laboratory. The Myc‐tagged human full‐length PDK1‐WT cDNA in a pCMV3 vector was kindly provided by Drs P. Hawkins and K. Anderson (The Babraham Institute, Cambridge, UK). Several HA‐tagged or Myc‐tagged PDK1 mutants were generated using the QuikChange Mutagenesis Kit (Stratagene). A constitutively active form of src cDNA in a pUSEamp vector was purchased from Millipore.
Antibodies. For Western blot analysis, we used the following: an antibody to TUSC4 (Proteintech Group, Chicago, IL, USA); antibodies to Src (clone GD11), S6K, and phospho‐Thr412 S6K (Millipore); an antibody to phospho‐Tyr (clone pY20; BD Transduction Laboratories, Lexington, KY, USA); an antibody to poly (ADP‐ribose) polymerase (PARP) (clone C2‐10; BD Pharmingen, San Diego, CA, USA); antibodies to Caspase‐3 (clone H‐277) and β‐actin (clone C‐2) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); antibodies to PDK1, phospho‐Ser241 PDK1, Akt, phospho‐Thr308 Akt, and cleaved PARP (Cell Signaling Technology, Danvers, MA, USA); antibodies to FLAG tag (clone M2) and tubulin‐α (Sigma); or antibodies to an HA tag (clone 3A10) and Myc tag (clone 9E10) (Roche, Mannhein, Germany).
Transient transfections. Cells were transfected with appropriate plasmids or siRNA using Superfect transfection reagent (Qiagen, Valencia, CA, USA), Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), or Lipofectamine RNAiMAX (Invitrogen), according to the manufacturers’ instructions. Negative control siRNA (medium GC duplex), TUSC4‐3 siRNA, and TUSC4‐4 siRNA were purchased from Invitrogen. These oligonucleotides were stealth siRNAs. The siRNA coding strands were ACUUCCAGCAGAUGAUGAUGUUGGG (TUSC4‐3; directed to nucleotides 1117–1141) and AUAAGCCCGAACUGGAUCAGCUUCC (TUSC4‐4; directed to nucleotides 932–956).
Immunoprecipitations and Western blot analysis. Cells were harvested and solubilized in lysis buffer.( 7 ) Tagged proteins were immunoprecipitated with an anti‐HA antibody–conjugated agarose (clone HA‐7; Sigma), an anti‐FLAG antibody–conjugated agarose (clone M2; Sigma), an anti‐Myc antibody–conjugated agarose (Sigma), or an anti‐PDK1 antibody–conjugated agarose (Millipore or Santa Cruz Biotechnology). In some experiments, cell lysates were preincubated with protein G sepharose (Zymed Laboratories, San Francisco, CA, USA) before immunoprecipitation to remove the nonspecific binding proteins. Then, the immunoprecipitated proteins or the cell lysates were electrophoresed and immunoblotted. Blots were scanned using an Image Reader LAS‐3000 mini (Fujifilm, Tokyo, Japan) and were quantified using Multi Gauge software.
In vitro estimation for PDK1 tyrosine phosphorylation. After transfection of mock or FLAG‐tagged TUSC4 plasmids into 293T cells, cells were lyzed in SDS lysis buffer (20 mM Tris‐HCl [pH 7.5], 1% SDS, and 10% glycerol). FLAG‐tagged TUSC4 in the lysed was immunopurified with an anti‐FLAG antibody–conjugated agarose. The immunopurified proteins were then washed in a PDK1 assay dilution buffer (Millipore, Cat. #20–151). The proteins were then incubated in 30 µL of PDK1 assay dilution buffer containing a 10‐fold diluted magnesium/ATP cocktail (Millipore, Cat. #20–113), recombinant Src, and recombinant PDK1. The mixture was incubated at 37°C for 30 min. Then, the samples were electrophoresed and immunoblotted with the appropriate antibodies.
MTT assay. To assess the change in cell viability, we used the MTT assay. In brief, cells were incubated with 2 mg/mL of 3‐(4, 5‐methylthiazol‐2‐yl)‐2, 5‐diphenyl‐tetrazolium bromide (MTT) for 2.5 h. Formazon products were solubilized with DMSO, and the optical density was measured at 525 nm, with a reference at 650 nm, using a microplate spectrophotometer (Benchmark Plus, Bio‐Rad, Tokyo, Japan). The rate of viability was calculated by dividing the absorbance of each sample by that of the corresponding control.
Results
Identification of TUSC4 as a novel PDK1‐interacting protein. To identify novel regulators of PDK1, we employed an E. coli‐based two‐hybrid screening system with a COOH‐terminal domain of human PDK1 containing a PH domain (445–556 a.a.) as bait and human fetal brain cDNA library as the prey. We successively isolated nine positive clones (BP1–BP9) and analyzed the partial sequence of each. Among the isolated clones, the COOH terminal of BP9 contained 266 amino acids (115–380 a.a.) corresponding to human TUSC4/NPRL2 cDNA. After cloning each full‐length cDNA, we tested their binding ability to PDK1 in mammalian cells by examining the presence of PDK1 in each clone's immunoprecipitant. As shown in Fig. 1a, each isolated full‐length clone, except BP2 and BP7, could form a complex with HA‐tagged full‐length human PDK1‐WT in 293T cells, as was the case in our previously identified PDK1 binding protein 14‐3‐3θ.( 7 ) Among the clones, the full‐length TUSC4 (= BP9) showed the highest PDK1 binding ability (Fig. 1a, lane 11). To confirm the interaction specificity between TUSC4 and PDK1, we transfected FLAG‐tagged full‐length human TUSC4 with HA‐tagged full‐length human PDK1 into 293T cells, following immunoprecipitation of PDK1 with an anti‐HA antibody. We clearly detected FLAG‐tagged TUSC4 in the HA‐PDK1 immunoprecipitant (Fig. 1b). The binding seemed to be specific because the FLAG‐tagged TUSC4 could not be coimmunoprecipitated without HA‐tagged PDK1. In addition, endogenous TUSC4 could be detected in the endogenous PDK1 immunoprecipitant but not in control immunoprecipitant, indicating endogenous interaction between TUSC4 and PDK1 (Fig. 1c).
Figure 1.
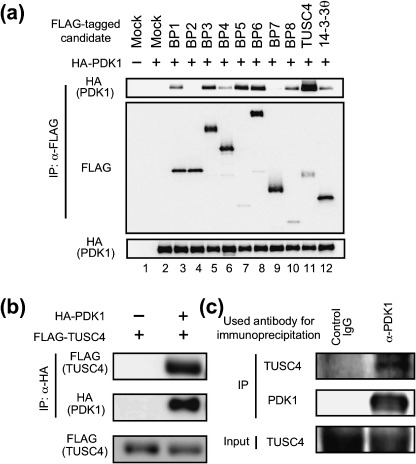
Identification of TUSC4/NPRL2 as a PDK1‐interacting protein. (a) After cloning the full‐length cDNA of each PDK1 binding protein (BP), we transfected a pc5FLAG vector encoding none (mock, lanes 1 and 2), full‐length candidate proteins (BP1‐BP8 and TUSC4, lanes 3–11), or 14‐3‐3θ (lane 12), together with a pHM6 vector encoding none (–, lane 1) or human full‐length PDK1‐WT (+, lanes 2–12) into 293T cells. After transfection, the FLAG‐tagged proteins were immunoprecipitated. The immunoprecipitated proteins (top and middle) and cell lysates (bottom) were immunoblotted with the indicated antibodies. (b) The FLAG‐tagged full‐length TUSC4‐WT was transfected into 293T cells together with (+) or without (–) HA‐tagged full‐length PDK1‐WT. After transfection, the HA‐tagged protein was immunoprecipitated. The immunoprecipitated proteins (top and middle) and cell lysates (bottom) were immunoblotted with the indicated antibodies. (c) Protein G Sepharose was incubated with normal sheep IgG (control IgG) or an anti‐PDK1 antibody (α‐PDK1; Millipore). Then, precleared MCF‐7 cell lysates were incubated with the antibody–conjugated Sepharose. After washing, the immunoprecipitated proteins (top and middle) and cell lysates (input, bottom) were immunoblotted with antibodies to TUSC4 or PDK1 (Cell Signaling Technology). IP, immunoprecipitation.
To identify the binding site in TUSC4, we prepared COOH‐terminal (ΔC103, ΔC199, and ΔC295) and NH2‐terminal (ΔN103, ΔN133, and ΔN173) deletion mutants of TUSC4 (Fig. 2a). Then, 293T cells were cotransfected with the FLAG‐tagged TUSC4 deletion mutants together with HA‐tagged full‐length PDK1‐WT. As shown in Fig. 2b, PDK1 formed a complex with TUSC4‐WT, ΔC103, ΔC199, ΔC295, and ΔN103 but not with TUSC4‐ΔN133 and ΔN173, indicating that TUSC4 amino acid residues 1–133 were associated with the binding to PDK1 (Fig. 2a, hatched area). Because PDK1 bound to both TUSC4‐ΔC103 and TUSC4‐ΔN103, 1–102 a.a. residue and 103–133 a.a. residues both seemed to be associated with binding to PDK1. In subsequent experiments, we used TUSC4‐ΔN133 as a negative control of full‐length TUSC4 because it could not form a complex with PDK1 in cells.
Figure 2.
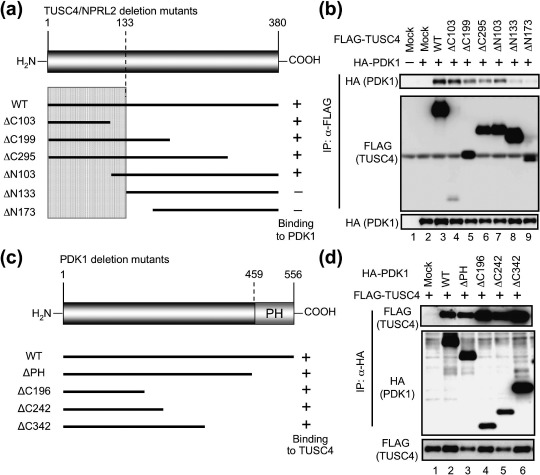
TUSC4/NPRL2 interacts with PDK1 via its NH2‐terminal 133 amino acids. (a) TUSC4 deletion mutants used in the experiments are represented by black bars. The predicted sites involved in the binding to PDK1 are shown schematically (hatched). (b) 293T cells were transfected with a pHM6 vector encoding none (–) or full‐length PDK1‐WT (+) together with a pc5FLAG vector encoding none (mock, lanes 1 and 2), TUSC4‐WT (lane 3), or the indicated TUSC4 deletion mutants (lanes 4–9). After transfection, the FLAG‐tagged proteins were immunoprecipitated. The immunoprecipitants (top and middle) and cell lysates (bottom) were immunoblotted with the indicated antibodies. (c) Structural domains of PDK1 and PDK1 deletion mutants used in the experiments are represented by black bars. (d) 293T cells were transfected with a pHM6 vector encoding none (mock, lane 1), PDK1‐WT (lane 2), or the indicated PDK1 mutants (lanes 3–6) together with FLAG‐tagged TUSC4‐WT. After transfection, the HA‐tagged proteins were immunoprecipitated. The immunoprecipitants (top and middle) and cell lysates (bottom) were immunoblotted with the indicated antibodies. IP, immunoprecipitation.
Using two‐hybrid screening, we isolated TUSC4 as the protein bound near the PH domain of PDK1 (445–556 a.a.) (see ‘Materials and Methods’). To verify the binding domain in PDK1, we first transfected FLAG‐tagged full‐length TUSC4‐WT together with HA‐tagged PDK1 deletion mutants (Fig. 2c), then the FLAG‐tagged TUSC4 was immunoprecipitated with an anti‐FLAG antibody–conjugated agarose. An unexpected finding was that TUSC4 could bind to PDK1 mutants that lacked the PH domain (ΔPH, Fig. 2d). Further deletion of the COOH‐terminal domains slightly increased the amount of the coimmunoprecipitated TUSC4, indicating that TUSC4 formed a complex with PDK1 via the PH and NH2‐terminal domains in PDK1. The PDK1 PH domain is not the only residue associated with the binding to TUSC4.
TUSC4 attenuated PDK1 kinase activity and inhibited its tyrosine phosphorylation by Src. TUSC4 was reported to suppress cell proliferation and metastasis,( 14 , 16 ) but its mechanism remains unknown. Because TUSC4 interacted with PDK1 (1, 2), we hypothesized that it suppressed tumor growth by negatively affecting PDK1 kinase activity. To verify our hypothesis, we performed in vitro PDK1 kinase assay using recombinant Akt as a substrate in the presence of PtdIns(3,4,5)P3‐containing vesicle. The kinase activity of PDK1 that was immunoprecipitated from TUSC4‐WT‐expressing cells was about 40% of that immunoprecipitated from mock transfectants (data not shown). This result suggests that TUSC4 has the ability to attenuate PDK1 kinase activity. We also examined the change in phospho‐Thr308 Akt and phospho‐Thr412 S6K levels after TUSC4 expression because these sites were reported to be phosphorylated by PDK1 in cells.( 1 , 2 , 3 , 17 ) Actually, ectopic expression of PDK1‐WT, but not of the PDK1 mutant with weak kinase activity (3F; Y9/373/376F), increased phospho‐Thr308 Akt and phospho‐Thr412 S6K levels in cells (Fig. 3a). TUSC4‐WT expression inhibited PDK1‐mediated Akt and S6K phosphorylation (Fig. 3a). These results suggest that TUSC4 down‐regulates PDK1 kinase activity. To clarify the mechanisms of PDK1 inhibition by TUSC4, we first examined phosphorylation at the Ser241 residue in the activation loop of PDK1, which is essential for PDK1 activation.( 4 ) Although TUSC4‐WT expression suppressed PDK1‐mediated Akt and S6K phosphorylation, it had no effect on the phospho‐Ser241 PDK1 level (Fig. 3a).
Figure 3.
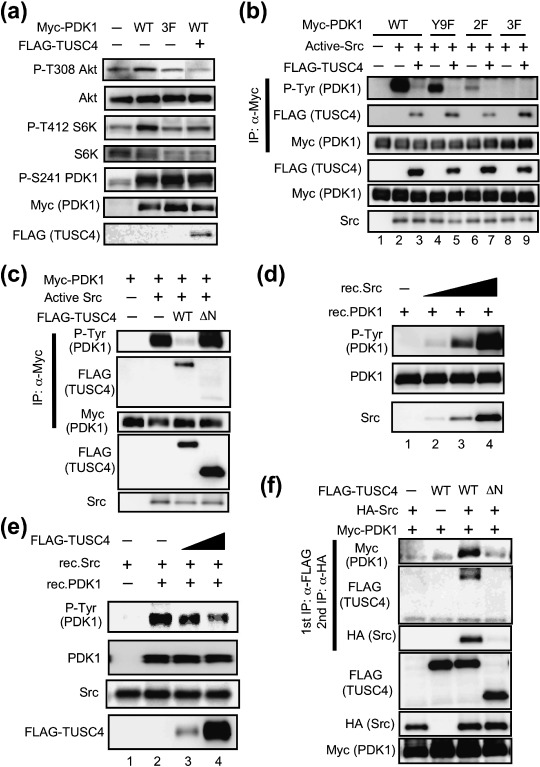
TUSC4/NPRL2 suppresses PDK1 tyrosine phosphorylation mediated by Src. (a) 293T cells were transfected with a pCMV3 vector encoding none (–), full‐length PDK1‐WT (WT), PDK1‐Y9/373/376F mutant (3F) together with (+) or without (–) FLAG‐tagged TUSC4‐WT. After transfection, the cell lysates were immunoblotted with the indicated antibodies. (b) 293T cells were transfected with a pCMV3 vector encoding full‐length PDK1‐WT (WT, lanes 1–3), PDK1‐Y9F mutant (lanes 4 and 5), PDK1‐Y373/376F mutant (2F, lanes 6 and 7), or PDK1‐Y9/373/376F mutant (3F, lanes 8 and 9). The cells were cotransfected with a pUSEamp vector encoding none (–, lane 1) or active Src (+, lanes 2–9) together with a pc5FLAG vector encoding none (–) or TUSC4‐WT (+). After transfection, the Myc‐tagged PDK1‐WT and its mutants were immunoprecipitated. The immunoprecipitants and cell lysates were immunoblotted with the indicated antibodies. (c) 293T cells were transfected with a pCMV3 vector encoding full‐length PDK1‐WT. The cells were cotransfected with a pUSEamp vector encoding none (–) or active Src (+) together with a pc5FLAG vector encoding none (–), TUSC4‐WT (WT), or TUSC4‐ΔN133 (ΔN). After transfection, the Myc‐tagged PDK1‐WT were immunoprecipitated. The immunoprecipitants and cell lysates were immunoblotted with the indicated antibodies. (d) The recombinant Src (rec.Src; 0.15, 1.5, and 15 U; lanes 2–4, respectively) or vehicle control (lane 1) was reacted with 500 ng of recombinant PDK1 (rec.PDK1) for 10 min at 37°C. Then the samples were immunoblotted with the indicated antibodies. Anti‐PDK1 antibody was obtained from Cell Signaling Technology. (e) The increasing amount of the immunoprecipitated FLAG‐tagged TUSC4‐WT proteins (lanes 3 and 4) or the vehicle control (–; lanes 1 and 2) were incubated with 0.6 U of recombinant Src and 200 ng of PDK1 in 30 µL of reaction buffer. The mixture was incubated for 30 min at 37°C. Then the samples were immunoblotted with the indicated antibodies. Anti‐PDK1 antibody was purchased from Cell Signaling Technology. (f) 293T cells were transfected with a pHM6 vector encoding none (–) or human full‐length c‐Src‐WT (+) and a pc5FLAG vector encoding none (–), TUSC4‐WT (WT), or TUSC4‐ΔN133 (ΔN) together with Myc‐tagged PDK1‐WT. After transfection, the FLAG‐tagged proteins were immunoprecipitated. The bound proteins were eluted using 3XFLAG peptide. The supernatant was subsequently incubated with an anti‐HA antibody–conjugated agarose. The sequentially immunoprecipitated proteins (top to third) and the cell lysates (fourth to bottom) were immunoblotted with the indicated antibodies. IP, immunoprecipitation.
It has been reported that several kinases, including Src, phosphorylate PDK1 at Tyr9, Tyr373, and Tyr376 residues, leading to the up‐regulation of PDK1 kinase activity.( 8 , 9 , 10 , 11 , 18 , 19 , 20 ) We examined the change in phospho‐Tyr9, Tyr373, and Tyr376 levels after TUSC4 expression. Consistent with the previous reports, ectopic expression of Src increased the tyrosine‐phosphorylated PDK1 (Fig. 3b, lane 2). TUSC4‐WT expression almost completely suppressed PDK1 phosphorylation at the tyrosine residues (Fig. 3b, lane 3). To identify the phosphorylation sites, we generated PDK1 mutants in which Tyr9 was converted to phenylalanine (Y9F), both Tyr373 and Tyr376 were replaced with phenylalanines (2F; Y373/376F), and all Tyr9, Tyr373, and Tyr376 were substituted with phenylalanines (3F; Y9/373/376F). Consistent with the previous reports,( 8 , 9 , 10 , 11 , 18 , 19 , 20 ) Tyr9, Tyr373, and Tyr376 mutations in PDK1 decreased the reactivity to an antiphospho‐tyrosine antibody, indicating that TUSC4 down‐regulates PDK1 kinase activity by interfering with tyrosine phosphorylation at the Tyr9 Tyr373 and Tyr376 residues. It has previously been shown that phosphorylation at Tyr9 of PDK1 is necessary for subsequent phosphorylation at Tyr373 and Tyr376 in HEK293 cells.( 10 , 19 ) In 293T cells, Src tended to phosphorylate the PDK1‐Y9F mutant rather than the PDK1–2F mutant (Fig. 3b). Thus, PDK1 tyrosine phosphorylation might not be fully sequential, and Src preferentially phosphorylates PDK1 at Tyr373 and Tyr376 over Tyr9 in 293T cells. Then, we investigated whether the TUSC4‐ΔN133 mutant, lacking the PDK1 binding ability, could also inhibit PDK1 tyrosine phosphorylation mediated by Src. As a result, TUSC4‐ΔN133 mutant was unable to suppress Src‐mediated PDK1 tyrosine phosphorylation (Fig. 3c). These results suggested that TUSC4 suppressed PDK1 tyrosine phosphorylation by complex formation.
It has been shown that Src directly phosphorylates PDK1 in vitro.( 8 ) Consistent with those findings, we found that recombinant Src phosphorylated recombinant PDK1 in vitro in a dose‐dependent manner (Fig. 3d). The Src‐mediated in vitro PDK1 tyrosine phosphorylation was suppressed by adding immunopurified TUSC4 in a dose‐dependent fashion (Fig. 3e). These results clearly indicate that TUSC4 inhibits Src‐mediated PDK1 tyrosine phosphorylation. To confirm that TUSC4 formed a ternary complex with PDK1 and Src, we transfected Myc‐tagged PDK1, HA‐tagged Src, and FLAG‐tagged TUSC4 into 293T cells and performed sequential immunoprecipitation assays. The cell lysates of the transfectants were applied first to an anti‐FLAG antibody–conjugated agarose. Then, the bound proteins were gently eluted from the agarose using a 3XFLAG peptide. The eluates were further applied to an anti‐HA antibody–conjugated agarose. Then the bound proteins were analyzed by immunoblot analysis. The sequential immunoprecipitation analysis revealed that TUSC4‐WT, PDK1, and Src were present in the same protein complex, while TUSC4‐ΔN133 (lacking the PDK1 binding ability) could not form a ternary complex with PDK1 and Src (Fig. 3f).
TUSC4 interfered with EGF‐mediated PDK1 tyrosine phosphorylation. We next examined whether TUSC4 expression suppressed PDK1 downstream signaling after growth factor stimulation. Cells were treated with a vehicle control (BSA) or 50 ng/mL of EGF for 10 min at 37°C. We found that EGF addition increased the level of PDK1 tyrosine phosphorylation (Fig. 4a). To estimate the effects of TUSC4 on EGF signaling, we established stable HT1080 cells expressing none (Mock), TUSC4‐WT, or TUSC4‐ΔN133. We used polyclonal HT1080 stable transfectants for the assay to avoid the problem of clonal heterogeneity. As shown in Fig. 4b, EGF treatment induced PDK1 tyrosine phosphorylation in mock and TUSC4‐ΔN133 transfectants but not in TUSC4‐WT transfectants. Consistent with PDK1 phosphorylation and activation, EGF treatment induced Akt phosphorylation at the Thr308 residue in mock and TUSC4‐ΔN133 transfectants but not in polyclonal TUSC4‐WT transfectants (Fig. 4c). EGF‐induced PDK1 activation and its inhibition by TUSC4‐WT were also confirmed on immunoblot analysis of the phospho‐Thr412 S6K level (Fig. 4c). We saw no difference between the levels of phospho‐EGF receptor in mock transfectants and that in TUSC4‐WT transfectants (data not shown). Thus, TUSC4 suppressed EGF‐induced PDK1 downstream signaling by interfering with the Src‐mediated tyrosine phosphorylation and activation of PDK1.
Figure 4.
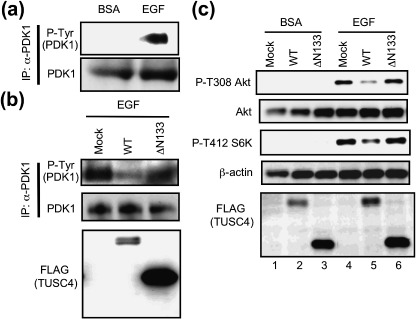
TUSC4/NPRL2 attenuates tyrosine phosphorylation and epidermal growth factor (EGF)–induced activation of PDK1. (a) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none were seeded onto culture plates in RPMI complete medium containing 200 µg/mL of geneticin. After incubation for 24 h, medium was replaced with medium containing 0.1% fetal bovine serum (FBS). After incubation for an additional 24 h, medium was replaced again with medium containing vehicle control (bovine serum albumin; BSA) or 50 ng/mL of EGF and incubated for 10 min. Endogenous PDK1 proteins were immunoprecipitated and immunoblotted with the indicated antibodies. Anti‐PDK1 antibody for immunoblotting was obtained from Cell Signaling Technology. (b) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none (mock), TUSC4‐WT (WT) or TUSC4‐ΔN133 (ΔN133) were seeded and treated with 50 ng/mL of EGF for 10 min as described in (a). The immunoprecipitated endogenous PDK1 proteins or cell lysates were immunoblotted with the indicated antibodies. Anti‐PDK1 antibody for immunoblotting was purchased from Cell Signaling Technology. (c) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none (mock), TUSC4‐WT (WT), or TUSC4‐ΔN133 (ΔN133) were seeded and treated as described in (a). The cell lysates were immunoblotted with the indicated antibodies. IP, immunoprecipitation.
TUSC4 suppressed cell growth and enhanced sensitivity to various anticancer drugs. It has already been reported that the forced expression of TUSC4 in tumor cells could attenuate tumor cell growth.( 14 ) However, it has not been reported whether or not TUSC4 expression suppressed by specific siRNAs accelerates proliferation. Therefore, we designed four siRNAs from the human TUSC4 sequence to study this. In our experiments to suppress human TUSC4 expression, we used two effective siRNAs (TUSC4‐3 and TUSC4‐4) from among the four. Because chromosomal deletions of the region, including TUSC4, are frequently observed in lung, breast, kidney, and other cancers,( 12 ) we introduced TUSC4 siRNAs into normal HEK293 cells. We saw that the transfection of TUSC4‐3 and TUSC4‐4 siRNAs resulted in about 30% elevation of cell growth after 96 h of transfection (Fig. 5a, open symbols). Down‐regulation of TUSC4 expression in TUSC4 siRNA‐transfected HEK293 cells was confirmed by immunoblot analysis of the cell lysates (Fig. 5b). We also found that the TUSC4 siRNA‐mediated down‐regulation of TUSC4‐promoted phosphorylation of downstream Akt substrates, S6K and SGK (data not shown). Thus, we supposed that endogenous TUSC4 regulated endogenous PDK1 signaling pathway.
Figure 5.
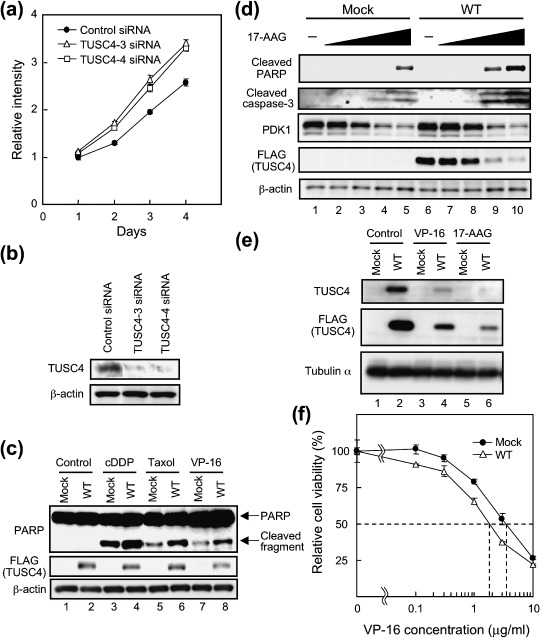
TUSC4/NPRL2 down‐regulates tumor cell growth and enhances the sensitivity to anticancer drugs. (a) HEK293 cells were transfected with non‐silencing control siRNA (closed circles), TUSC4‐3 siRNA (open triangles), or TUSC4‐4 siRNA (open squares). After transfection, the viability of the transfectants was estimated. The error bars represent standard deviations of quadruplicate experiments. (b) The cell lysates of HEK293 cells that had been transfected with the indicated siRNAs for 72 h were immunoblotted with the indicated antibodies. (c) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none (mock) or TUSC4‐WT (WT) were treated with DMSO (control), 5 µg/mL of cDDP, 0.05 µg/mL of taxol, or 1 µg/mL of VP‐16. After incubation for 24 h, cells were harvested. The cell lysates were immunoblotted with the indicated antibodies. (d) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none (mock) or TUSC4‐WT (WT) were treated with DMSO control (–; lanes 1 and 6), 0.03 µM (lanes 2 and 7), 0.1 µM (lanes 3 and 8), 0.3 µM (lanes 4 and 9), or 1 µM (lanes 5 and 10) 17‐AAG for 24 h. The cell lysates were immunoblotted with the indicated antibodies. Anti‐PDK1 antibody for immunoblotting was purchased from Cell Signaling Technology. (e) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none (mock) or TUSC4‐WT (WT) were treated with DMSO (control), 1 µg/mL of VP‐16 or 1 µM 17‐AAG. After incubation for 24 h, cells were harvested. The cell lysates were immunoblotted with the indicated antibodies. (f) Polyclonal HT1080 cells that had been stably transfected with a pc5FLAG vector encoding none (mock) or TUSC4‐WT (WT) were treated with DMSO control, 0.1, 0.3, 1, 3, and 10 µg/mL of VP‐16 for 24 h. After incubation, cell viability was estimated. The error bars represent standard deviations of triplicate experiments. The viability of each polyclonal cell treated with DMSO was set at 100%.
TUSC4 was reported to enhance the sensitivity to the anticancer drugs cDDP and doxorubicin.( 15 , 16 ) Using stable polyclonal HT1080 cells expressing none (mock) or TUSC4‐WT, we compared their sensitivity to cDDP, taxol, and VP‐16. PARP was one of the substrates of caspase‐3 and was cleaved to produce an 85‐kDa fragment. PARP cleavage was not observed in untreated control cells. However, PARP was cleaved to produce the p85 fragment in cells treated with chemotherapeutic drugs (Fig. 5c). PARP cleavage was more clearly observed in stable polyclonal TUSC4‐WT transfectants than in mock transfectants, suggesting that TUSC4 expression enhanced sensitivity to all of the tested anticancer drugs. We also examined the effect of TUSC4 expression on the cells’ susceptibility to an Hsp90 inhibitor, 17‐AAG, because Hsp90 stabilizes PDK1 and enhances Src binding to PDK1.( 6 , 11 ) Consistent with our previous report,( 6 ) inhibition of Hsp90 function induced PDK1 destabilization (Fig. 5d). Treating cells with 17‐AAG produced the 85‐kDa PARP fragment and cleaved a caspase‐3 fragment in a dose‐dependent manner (Fig. 5d). The cleaved fragments were more clearly observed in stable polyclonal TUSC4‐WT transfectants than in mock transfectants. Treating cells with 17‐AAG also decreased exogenous TUSC4 expression (Fig. 5d,e). Because VP‐16 treatment repressed exogenous TUSC4 expression (Fig. 5e), TUSC4 down‐regulation did not specifically occur because of Hsp90 inhibition. Thus, TUSC4 might be destabilized during apoptosis. However, we could not exclude the possibility that Hsp90 (or PDK1) was associated with the TUSC4's stability in cells because 17‐AAG drastically suppressed exogenous TUSC4 expression (Fig. 5d). We are now investigating the role of TUSC4 binding to Hsp90 or to PDK1 in TUSC4's stability. We also estimated the IC50 values of stable polyclonal mock and TUSC4‐WT transfectants in VP‐16‐mediated cytotoxicity. As shown in Fig. 5f, the IC50 value of mock transfectants was approximately two‐fold that of TUSC4‐WT transfectants (3.46 versus 1.77 µg/mL) when the cells were treated with VP‐16. Moreover, the IC50 value of mock transfected cells was approximately 1.3‐fold that of TUSC4‐WT transfected cells (0.88 versus 0.67 µM) when the cells were treated with 17‐AAG for 40 h (data not shown). These data suggest that TUSC4 expression enhanced sensitivity not only to cDDP but also to a variety of other anticancer drugs, including taxol, VP‐16, and 17‐AAG.
Discussion
PDK1 is a Ser/Thr kinase that regulates the activity of at least 23 protein kinases belonging to the AGC protein kinase family, which includes Akt, S6K, SGKs, PKA, PKC isoforms, and RSKs. Activated kinases, then, mediate cell survival, growth, and cell‐cycle progression by phosphorylating downstream key regulatory proteins.( 3 ) All of these kinases require phosphorylation by PDK1 on a conserved Ser/Thr residue (T‐loop). PDK1 has been reported as being able to phosphorylate S6K at Thr412 in its hydrophobic motif, in addition to Thr252 in the T‐loop of S6K.( 17 ) PDK1 itself is also a member of the AGC subfamily of protein kinases and is phosphorylated at the Ser241 residue in the activation T‐loop.( 4 ) Because PDK1 expressed in bacteria is active and phosphorylated at Ser241, it is thought that PDK1 phosphorylates by itself at this site.( 4 ) Although PDK1 kinase activity is thought to be constitutively active and not further activated by growth factor stimulation, several reports have suggested that it is controlled by several PDK1‐associating proteins, such as PKC‐related kinase‐1 (PRK1)/PRK2, PDK1‐interacting fragment of PRK2 (PIF), RSK2, Ft1, 14‐3‐3 protein, and Hsp90.( 3 , 6 , 7 , 21 , 22 ) Moreover, recent studies have proposed that Src‐mediated PDK1 tyrosine phosphorylation increases PDK1 kinase activity.( 8 , 9 , 10 ) Consistent with these reports, we found that tyrosine phosphorylation of PDK1 was important for its kinase activity because mutation of the Tyr9, Tyr373, and Tyr376 residues to phenylalanine (3F; Y9/373/376F) showed reduced phosphorylating activity toward the downstream PDK1 substrates Akt and S6K (Fig. 3a). Additionally, it has been reported that Pyk2 acts as a scaffold protein for Src‐dependent phosphorylation of PDK1 on Tyr9, and Hsp90 acts as a scaffold for Tyr373 and Tyr376 phosphorylation.( 11 , 19 )
In this study, we identified TUSC4 as a novel PDK1‐interacting protein using an E. coli two‐hybrid screening. An endogenous interaction was also found in mammalian cells (Fig. 1c). Because we could not obtain recombinant full‐length TUSC4 expressed in E. coli, we did not confirm the direct binding between full‐length TUSC4 and full‐length PDK1 in vitro. However, we identified TUSC4 as a PDK1‐interacting protein by the E. coli two‐hybrid screening system. In addition, we have already shown that the Src‐mediated PDK1 tyrosine phosphorylation in vitro was suppressed by adding the immunopurified TUSC4 (Fig. 3e). Thus, TUSC4 seemed to bind directly to PDK1. TUSC4 locates at chromosome 3p21.3 and expresses in many normal tissues.( 12 ) Initial studies reported that the 3p21.3 region undergoes allelic loss in about 80% of primary lung cancers.( 14 ) Because forced expression of TUSC4 effectively inhibited the growth of tumor cell lines without affecting the growth of normal cells,( 14 ) the TUSC4 gene was thought to be the tumor suppressor gene, but the mechanism for regulating cell growth remains unknown. We hypothesized that TUSC4 negatively influences PDK1 function by complex formation. We found that TUSC4 bound to PDK1 in mammalian cells and acted as a negative regulator of PDK1 in cells and in vitro (2, 3). We discovered that TUSC4 expression suppressed PDK1 phosphorylation at Tyr9, Tyr373, and Tyr376 (Fig. 3b) and suppressed PDK1 downstream signaling (Fig. 3a) without affecting PDK1 auto‐phosphorylation at Ser241 (Fig. 3a). Src was previously identified as a kinase that could phosphorylate PDK1 at Tyr9, Tyr373, and Tyr376.( 8 , 9 , 10 ) Thus, TUSC4 might exert its effects largely by inhibiting Src‐induced PDK1 tyrosine phosphorylation. Sequential immunoprecipitation analysis revealed that TUSC4 formed a ternary complex with PDK1 and Src (Fig. 3f). Because TUSC4 didn't interfere with PDK1‐Src binding (data not shown), we speculated that TUSC4 would down‐regulate the levels of PDK1 tyrosine phosphorylation by suppressing Src‐mediated phosphorylation or by promoting protein tyrosine phosphatase–mediated dephosphorylation. TUSC4 inhibited Src‐induced PDK1 tyrosine phosphorylation in vitro (Fig. 3e), suggesting that TUSC4 attenuated PDK1 signaling mainly by inhibiting Src‐mediated PDK1 tyrosine phosphorylation.
It is well known that the EGF receptor (EGFR) and Src are overexpressed in a variety of human cancers. In addition, EGFR activation results in recruitment and activation of Src.( 23 , 24 ) As shown in Fig. 4a, EGF treatment induced PDK1 tyrosine phosphorylation. Ectopic expression of TUSC4 suppressed PDK1 tyrosine phosphorylation and downstream Akt and S6K phosphorylations (Fig. 4b,c). Therefore, TUSC4 would suppress PDK1 signaling, mainly by interfering with Src‐mediated PDK1 tyrosine phosphorylation. Because TUSC4‐ΔN133, which lacks PDK1 binding ability, showed no effects on PDK1 signaling (Fig. 4b,c), TUSC4 binding to PDK1 would be essential for TUSC4's inhibitory effects on PDK1 signaling. PDK1 has been reported to be associated with cellular transformation and tumor growth.( 25 , 26 , 27 , 28 , 29 ) Thus, TUSC4 might act as a tumor suppressor, in part, by suppressing the tumorigenic function of PDK1.
Previous studies have shown that TUSC4 enhanced sensitivity to the DNA‐damaging anticancer drugs cDDP and doxorubicin.( 15 , 16 ) In this study, we found that TUSC4 was a sensitizer not only of cDDP and doxorubicin but also of a variety of other anticancer drugs, including taxol, VP‐16, and 17‐AAG (Fig. 5b,c). As we have reported previously, Akt inactivation is upstream and is a prerequisite event for anticancer drugs to induce apoptosis by caspase activation.( 29 ) Further, it has also been reported that Akt1 activity is essential for cDDP resistance in lung cancer cell lines.( 30 ) TUSC4 functions as a negative regulator of PDK1‐Akt signaling; thus, TUSC4‐expressing cells might be vulnerable to apoptosis after chemotherapy. Additionally, recent studies have demonstrated that increased PDK1 activity is associated with tumor cell growth and motility control in tumor cells.( 25 , 26 , 31 , 32 ) These data suggest that the EGFR/Src/PDK1 signaling pathway is an attractive target for developing new anticancer drugs. The development of specific inhibitors of PDK1 is an especially attractive and vital concern which will enable the medication of patients who are deficient in TUSC4 expression.
Acknowledgments
We thank Dr Emi Tokuda for technical advice on two‐hybrid screening. We also thank to Drs Hiroshi Tanaka and Akito Nakamura for helpful discussions. This study was supported in part by special grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan 17016012 and 18015008 (to TT and NF). NF was also supported by the Novartis Foundation (Japan) for the Promotion of Science, by the Kobayashi Institute for Innovative Cancer Chemotherapy, and by the Vehicle Racing Commemorative Foundation.
References
- 1. Alessi DR, James SR, Downes CP et al . Characterization of a 3‐phosphoinositide‐dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol 1997; 7: 261–9. [DOI] [PubMed] [Google Scholar]
- 2. Stephens L, Anderson K, Stokoe D et al . Protein kinase B kinases that mediate phosphatidylinositol 3–5‐trisphosphate‐dependent activation of protein kinase B. Science 1998; 279: 710–4. [DOI] [PubMed] [Google Scholar]
- 3. Vanhaesebroeck B, Alessi DR. The PI3K‐PDK1 connection: more than just a road to PKB. Biochem J 2000; 346: 561–76. [PMC free article] [PubMed] [Google Scholar]
- 4. Casamayor A, Morrice NA, Alessi DR. Phosphorylation of Ser‐241 is essential for the activity of 3‐phosphoinositide‐dependent protein kinase‐1: identification of five sites of phosphorylation in vivo . Biochem J 1999; 342: 287–92. [PMC free article] [PubMed] [Google Scholar]
- 5. Alessi DR, Deak M, Casamayor A et al . 3‐Phosphoinositide‐dependent protein kinase‐1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 1997; 7: 776–89. [DOI] [PubMed] [Google Scholar]
- 6. Fujita N, Sato S, Ishida A, Tsuruo T. Involvement of Hsp90 in signaling and stability of 3‐phosphoinositide‐dependent kinase‐1. J Biol Chem 2002; 277: 10346–53. [DOI] [PubMed] [Google Scholar]
- 7. Sato S, Fujita N, Tsuruo T. Regulation of kinase activity of 3‐phosphoinositide‐dependent protein kinase‐1 by binding to 14‐3‐3. J Biol Chem 2002; 277: 39360–7. [DOI] [PubMed] [Google Scholar]
- 8. Prasad N, Topping RS, Zhou D, Decker SJ. Oxidative stress and vanadate induce tyrosine phosphorylation of phosphoinositide‐dependent kinase 1 (PDK1). Biochemistry 2000; 39: 6929–35. [DOI] [PubMed] [Google Scholar]
- 9. Grillo S, Gremeaux T, Casamayor A, Alessi DR, Le Marchand‐Brustel Y, Tanti JF. Peroxovanadate induces tyrosine phosphorylation of phosphoinositide‐dependent protein kinase‐1 potential involvement of src kinase. Eur J Biochem 2000; 267: 6642–9. [DOI] [PubMed] [Google Scholar]
- 10. Park J, Hill MM, Hess D, Brazil DP, Hofsteenge J, Hemmings BA. Identification of tyrosine phosphorylation sites on 3‐phosphoinositide‐dependent protein kinase‐1 and their role in regulating kinase activity. J Biol Chem 2001; 276: 37459–71. [DOI] [PubMed] [Google Scholar]
- 11. Yang KJ, Shin S, Piao L et al . Regulation of 3‐phosphoinositide‐dependent protein kinase‐1 (PDK1) by Src involves tyrosine phosphorylation of PDK1 and Src homology 2 domain binding. J Biol Chem 2008; 283: 1480–91. [DOI] [PubMed] [Google Scholar]
- 12. Lerman MI, Minna JD. The 630‐kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res 2000; 60: 6116–33. [PubMed] [Google Scholar]
- 13. Sekido Y, Ahmadian M, Wistuba II et al . Cloning of a breast cancer homozygous deletion junction narrows the region of search for a 3p21.3 tumor suppressor gene. Oncogene 1998; 16: 3151–7. [DOI] [PubMed] [Google Scholar]
- 14. Ji L, Nishizaki M, Gao B et al . Expression of several genes in the human chromosome 3p21.3 homozygous deletion region by an adenovirus vector results in tumor suppressor activities in vitro and in vivo . Cancer Res 2002; 62: 2715–20. [PMC free article] [PubMed] [Google Scholar]
- 15. Schenk PW, Brok M, Boersma AW et al . Anticancer drug resistance induced by disruption of the Saccharomyces cerevisiae NPR2 gene: a novel component involved in cisplatin‐ and doxorubicin‐provoked cell kill. Mol Pharmacol 2003; 64: 259–68. [DOI] [PubMed] [Google Scholar]
- 16. Ueda K, Kawashima H, Ohtani S et al . The 3p21.3 tumor suppressor NPRL2 plays an important role in cisplatin‐induced resistance in human non‐small‐cell lung cancer cells. Cancer Res 2006; 66: 9682–90. [DOI] [PubMed] [Google Scholar]
- 17. Balendran A, Currie R, Armstrong CG, Avruch J, Alessi DR. Evidence that 3‐phosphoinositide‐dependent protein kinase‐1 mediates phosphorylation of p70, S6 kinase in vivo at Thr‐412 as well as Thr‐252. J Biol Chem 1999; 274: 37400–6. [DOI] [PubMed] [Google Scholar]
- 18. Kim DW, Hwang JH, Suh JM et al . RET/PTC (rearranged in transformation/papillary thyroid carcinomas) tyrosine kinase phosphorylates and activates phosphoinositide‐dependent kinase 1 (PDK1): an alternative phosphatidylinositol 3‐kinase‐independent pathway to activate PDK1. Mol Endocrinol 2003; 17: 1382–94. [DOI] [PubMed] [Google Scholar]
- 19. Taniyama Y, Weber DS, Rocic P et al . Pyk2‐ and Src‐dependent tyrosine phosphorylation of PDK1 regulates focal adhesions. Mol Cell Biol 2003; 23: 8019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fiory F, Alberobello AT, Miele C et al . Tyrosine phosphorylation of phosphoinositide‐dependent kinase 1 by the insulin receptor is necessary for insulin metabolic signaling. Mol Cell Biol 2005; 25: 10803–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Remy I, Michnick SW. Regulation of apoptosis by the Ft1 protein, a new modulator of protein kinase B/Akt. Mol Cell Biol 2004; 24: 1493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balendran A, Casamayor A, Deak M et al . PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr Biol 1999; 9: 393–404. [DOI] [PubMed] [Google Scholar]
- 23. Mao W, Irby R, Coppola D et al . Activation of c‐Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 1997; 15: 3083–90. [DOI] [PubMed] [Google Scholar]
- 24. Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c‐Src‐mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem 1999; 274: 8335–43. [DOI] [PubMed] [Google Scholar]
- 25. Zeng X, Xu H, Glazer RI. Transformation of mammary epithelial cells by 3‐phosphoinositide‐dependent protein kinase‐1 (PDK1) is associated with the induction of protein kinase Calpha. Cancer Res 2002; 62: 3583–43. [PubMed] [Google Scholar]
- 26. Xie Z, Zeng X, Waldman T, Glazer RI. Transformation of mammary epithelial cells by 3‐phosphoinositide‐dependent protein kinase‐1 activates beta‐catenin and c‐Myc, and down‐regulates caveolin‐1. Cancer Res 2003; 63: 5370–5. [PubMed] [Google Scholar]
- 27. Sato S, Fujita N, Tsuruo T. Interference with PDK1‐Akt survival signaling pathway by UCN‐01 (7‐hydroxystaurosporine). Oncogene 2002; 21: 1727–38. [DOI] [PubMed] [Google Scholar]
- 28. Tsuruo T, Naito M, Tomida A et al . Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci 2003; 94: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rokudai S, Fujita N, Kitahara O, Nakamura Y, Tsuruo T. Involvement of FKHR‐dependent TRADD expression in chemotherapeutic drug‐induced apoptosis. Mol Cell Biol 2002; 22: 8695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu LZ, Zhou XD, Qian G, Shi X, Fang J, Jiang BH. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res 2007; 67: 6325–32. [DOI] [PubMed] [Google Scholar]
- 31. Xie Z, Yuan H, Yin Y, Zeng X, Bai R, Glazer RI. 3‐phosphoinositide‐dependent protein kinase‐1 (PDK1) promotes invasion and activation of matrix metalloproteinases. BMC Cancer 2006; 6: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 2008; 10: 127–37. [DOI] [PubMed] [Google Scholar]