

Abstract
Nek2 (NIMA‐related kinase 2) is involved in cell division and mitotic regulation by centrosome splitting. We previously reported that Nek2 depletion causes growth suppression and cell death in cholangiocarcinoma and breast cancer cells. In this report, we examine the effect of a combination treatment using Nek2 siRNA with the cytotoxic chemotherapeutic agent cisplatin (CDDP) on colorectal cancer. Nek2 was overexpressed in all colorectal cancer cell lines examined (HCT‐15, DLD‐1, Colo205, and Colo320). Nek2 short‐interfering RNA (siRNA) resulted in the inhibition of cell proliferation and the induction of apoptosis in vitro. Nek2 siRNA suppressed tumor growth compared to control siRNA in a xenograft mouse model. To investigate the potential utility of Nek2 siRNA for clinical cancer therapy, we examine the effect of a combination treatment using Nek2 siRNA with CDDP on colorectal cancer. The combined administration of both Nek2 siRNA and CDDP inhibited cell proliferation and induced apoptotic cell death in vitro. Furthermore, the combined administration of both Nek2 siRNA and CDDP suppressed tumor growth compared to either the single administration of Nek2 siRNA or the combined administration of control siRNA and CDDP. Our results suggest that combination treatment using Nek2 siRNA and chemotherapeutic agents may be an effective therapeutic option for colorectal cancer.
(Cancer Sci 2010; 101: 1163–1169)
Colorectal cancer is the second most common malignancy and the third most frequent cause of cancer‐related death in Japan. Significant progress has been made in the chemotherapy of colorectal cancer over the past decade. Chemotherapy has improved the outcome of early stage colorectal cancer. However, the outcome of advanced colorectal cancer has not been satisfactory. Novel therapeutic agents need to be developed to further improve the outcome of chemotherapy for advanced colorectal cancer.
Nek2 is a serine/threonine kinase of the NIMA–related kinase family, which consists of 11 different members (Nek1–11).( 1 ) In humans, Nek2 has three splice variants: Nek2A, Nek2B, and Nek2A‐T. Nek2A and Nek2B are identical up to residue 370 and differ in their extreme C‐termini, and Nek2A‐T lacks eight amino acids in the C‐terminal region of Nek2A.( 2 , 3 ) Nek2 is localized to the centrosome and involved in centrosome separation during mitosis.( 4 , 5 )
We previously reported that Nek2 is highly expressed in cholangiocarcinoma and breast cancer cell lines.( 6 , 7 ) Furthermore, we reported that Nek2 depletion causes growth suppression and cell death in cholangiocarcinoma and breast cancer cells, and that the administration of short‐interfering RNA (siRNA) against Nek2 in tumor‐bearing mice substantially prolonged survival.( 6 , 7 ) These results suggested that the regulation of Nek2 may also affect colorectal cancer growth. We hypothesized that a combination treatment using Nek2 suppression with conventional anticancer drugs would further inhibit tumor growth.
In this report, we show that Nek2 is overexpressed in colorectal cancer cell lines, and that silencing of Nek2 results in the inhibition of cell proliferation and the induction of apoptosis. In addition, we show that subcutaneous injection of Nek2 siRNA around tumor nodules suppresses tumor growth compared to control siRNA in a xenograft nude mouse model. Moreover, the combined administration of both Nek2 siRNA and cisplatin (CDDP) results in the suppression of tumor growth compared to the single administration of Nek2 siRNA or the combined administration of control siRNA and CDDP. These results indicate that combination treatment using Nek2 siRNA and chemotherapeutic agents may be effective and can serve as a therapeutic option for the treatment of colorectal cancer.
Materials and Methods
Cell culture, antibodies, and reagents. The cell lines (HCT‐15, DLD‐1, Colo205, Colo320) derived from human colorectal cancers were obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan), and Riken Cell Bank (Tsukuba, Japan). Human foreskin fibroblast (HFF) cells were kindly supplied by Dr T. Tsurumi (Aichi Cancer Center, Nagoya, Japan). Cells were cultured according to the supplier’s instructions. Antibodies and reagents were purchased from the following manufacturers: mouse monoclonal anti‐Nek2 (BD Biosciences, San Jose, CA, USA), rabbit polyclonal anti‐Nek2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and mouse monoclonal anti β‐actin (Sigma–Aldrich, St. Louis, MO, USA). Cis‐diamminedichloroplatinum (II) (Cisplatin or CDDP, Briplatin) was purchased from Bristol‐Myers Squibb (Tokyo, Japan).
Transfection of siRNA. siRNAs targeting Nek2, synthesized by Sigma‐Aldrich, are referred to as Nek2‐1 siRNA, Nek2‐2, Nek2‐3, and Nek2‐4.
Nek2‐1 siRNA, 623–645, sense: CUUUGGGCUAGCUAG‐AAUAUU, antisense: UAUUCUAGCUAGCCCAAAGUC;
Nek2‐2 siRNA, 1951–1973, sense: GUGAUUAAUACCAUGACAUCU, antisense: AUGUCAUGGUAUUAAUGACCA;
Nek2‐3 siRNA, 1940–1962, sense: GAUGCAAUUUGGU‐CAUUAAUA, antisense: UUAAUGACCAAAUUGCAUCUA;
and Nek2‐4 siRNA, 1853–1875, sense: CUGAGUGGUAU‐GCUUACAAUU, antisense: UUGUAAGCAUACCACUCA‐GUC.
Control siRNA was used as a control (Qiagen, Valencia, CA, USA).
Cells were transfected with siRNA using HiperFect Transfection Reagent (Qiagen), according to the manufacturer’s instructions.
Western blot analysis. Total proteins were separated by 10% SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE), transferred to a polyvinylidene difluoride membrane (Immobilon‐P; Millipore, Billerica, MA, USA), and immunoblotted with the appropriate antibodies. Signals were detected using the ECL system (Nakalai, Kyoto, Japan).
Cell viability and proliferation assays. Cell death was determined using the Trypan blue dye exclusion test. Viable cells that have intact cell membranes exclude the dye, whereas dead cells do not and thus are stained. Floating and attached cells were collected 72 h after transfection and stained with 1% Trypan blue. Dead cells were counted under microscope. Cell proliferation was determined using a colorimetric assay of cell viability that is based on the cleavage of tetrazolium salt by mitochondrial dehydrogenases (Tetra Color One; Seikagaku, Tokyo, Japan). Absorbance of the formazan dye was measured at 450 and 630 nm 1.5 h after adding the reagent.
Apoptosis detection assay. Apoptosis was quantified by terminal deoxyribonucleotidyl transferase–mediated dUTP nicked labeling (TUNEL) analysis (Dead End TUNEL system; Promega, Madison, WI, USA) according to the manufacturer’s protocol.
Animal efficacy studies. BALB/c nude mice (aged 7–8 weeks, 20–25 g in weight) were purchased from Japan SLC (Shizuoka, Japan). DLD‐1 cells (1 × 106) were suspended in 100 μL of Hank’s balanced salt solution (Gibco, Invitrogen, Carlsbad, CA, USA) and injected into the right femoral area of each mouse. After 4 days of tumor inoculation, PBS, control siRNA (50 μm), or Nek2 siRNA (50 μm) diluted in 100 μL of cell matrix was administered directly around the tumor twice a week for 3 weeks. PBS or control siRNA were used as controls. Tumor dimensions were measured by a digital caliper twice a week and the tumor volume was calculated using the following formula: [length (cm) × width (cm) × width (cm) × 0.523]. Animal experiments were performed in compliance with the guidelines of the Institute for Laboratory Animal Research, Nagoya University Graduate School of Medicine.
Combination treatment using siRNA and cisplatin. For in vitro combined chemotherapeutic treatments, DLD‐1 cells were first transfected with siRNA, and then treated with CDDP after 24 h. For in vivo studies using nude mice, DLD‐1 cells (1 × 106) were injected into the right femoral area of each mouse. siRNAs were administered around the tumor, and then CDDP was injected intraperitoneally at a dose of 4 mg/kg body weight for each mouse 4 days after tumor inoculation. Both siRNA and CDDP were administered twice a week for 2 weeks.
Combination‐Index (CI) isobologram. The Combination‐Index isobologram by Chou and Talalay( 8 ) was used to analyze the drug combination assays. Nek2 siRNA were added to DLD‐1 cells. After 24‐h incubation, CDDP was added, and the plates were harvested 24 h later. Each CI was calculated from the mean affected fraction at each drug ratio concentration (triplicate). CI > 1.1, 1.1 > CI > 0.9, and CI < 0.8 indicate antagonism, additive effect, or synergy, respectively.
PCR array. The PCR array (Cancer Pathway Finder RT2Profiler; SABioscience, Frederick, MD, USA) was used to analyze the main molecule (pathway), which is activated (or inhibited) by the combination of Nek2 siRNA and CDDP. Nek2 siRNA were added to DLD‐1 cells. After 24‐h incubation, CDDP was added and the plates were harvested 24 h later. Genes related to cancer development were detected according to the manufacturer’s protocol.
Statistical analysis. Results are expressed as mean ± SE. The statistical differences were analyzed by Student’s t‐test or repeated measures of anova. P‐values <0.05 were considered statistically significant.
Results
Nek2 expression in colorectal cancer cell lines and inhibition with Nek2 siRNA. We first investigated the expression of Nek2 in colorectal cancer cell lines (HCT‐15, DLD‐1, Colo205, and Colo320) by Western blot analysis (Fig. 1A, left). We observed higher expression of Nek2 in four colorectal cancer cell lines compared to normal HFF cells. Furthermore, we performed Western blot analysis to confirm Nek2 expression in human colorectal cancer tissue and normal colonic epithelium. The expression of Nek2 in colorectal cancer tissue was greater than that of normal colonic epithelium (Fig. 1A, right). We then designed four siRNAs targeting Nek2 (Nek2‐1, Nek2‐2, Nek2‐3, and Nek2‐4), and transfected them into DLD‐1 cells and HCT‐15 cells. The effect of Nek2 silencing on DLD‐1 was stronger than that of HCT‐15 cells (Fig. 1B). At a concentration of 30 nm, all four siRNAs efficiently suppressed the expression of both Nek2A and Nek2B in DLD‐1 cells. Nek2‐1 siRNA showed the strongest suppression of Nek2 protein expression. In contrast, control siRNA and empty liposomes did not alter Nek2 expression. We next transfected different concentrations of Nek2‐1 siRNA (10–50 nm) into DLD‐1 cells and Colo320 cells, to determine the minimum concentration required for the suppression of Nek2 expression. As shown in Figure 1(C), 20 nm of siRNA was sufficient for the suppression of Nek2 expression in DLD‐1 cells and 30 nm for suppression in Colo320 cells. Nek2‐1 siRNA suppressed the expression of Nek2 in a dose‐dependent manner.
Figure 1.
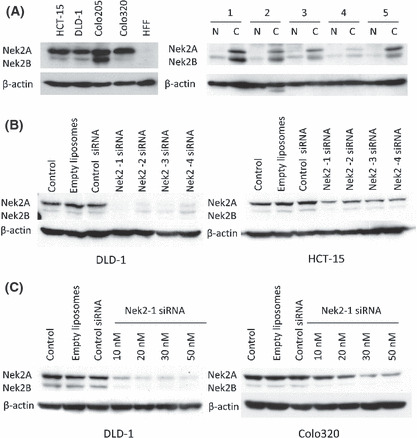
(A) Nek2 expression was examined in four different colon cancer cell lines (HCT‐15, DLD‐1, Colo205, Colo320) and human foreskin fibroblast (HFF) cells by Western blot analysis (left). The expression of Nek2 in five colorectal cancer patients (C, cancer tissue; N, normal colonic epithelium; right). β‐Actin was used as an internal loading control. (B) Nek2 suppression by siRNA was analyzed by Western blot analysis in DLD‐1 cells (left) and HCT‐15 cells (right). (C) Nek2 suppression by different concentrations of siRNA was observed by Western blot analysis in DLD‐1 (left) and Colo320 (right) cells.
Effect of Nek2 silencing on cell proliferation in DLD‐1 and colo320 cells. We next examined the effect of Nek2 suppression on proliferation in DLD‐1 cells and Colo320 cells. Cell proliferation was assessed by MTT assay 72 h after transfection with either control siRNA or Nek2 siRNA. Nek2 siRNA substantially decreased the proliferative rate of both DLD‐1and Colo320 cells compared with control siRNA (Fig. 2A). The antiproliferative effect of Nek2 siRNA was greater in DLD‐1 cells than in Colo320 cells. We also performed a Trypan blue dye exclusion test to estimate the cell death after siRNA transfection. As shown in Figure 2(B), Nek2 siRNA induced cell death in both DLD‐1 cells and Colo320 cells. To confirm whether the suppression of Nek2 expression induces apoptosis of colorectal cancer cells, we performed TUNEL assay, which detects fragmented DNA. We observed more TUNEL‐positive cells in Nek2 siRNA‐transfected cells compared to control siRNA‐transfected cells. These results indicate that the suppression of cell proliferation by Nek2 siRNA was at least partly due to the induction of apoptosis (Fig. 2C).
Figure 2.
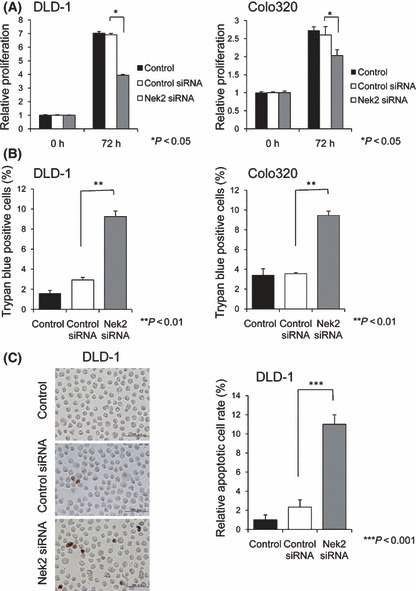
(A) Effect of Nek2 siRNA on the proliferation of DLD‐1 (left) and Colo320 (right) cells was examined by MTT assay. Graphs show the relative proliferation ratio of cells 72 h after Nek2 siRNA transfection. (B) Effect of Nek2 siRNA on cell viability was analyzed by Trypan blue dye exclusion test. Graphs indicate the level of cell death in DLD‐1 (left) and Colo320 (right) cells at 72 h after Nek2 siRNA treatment. (C) Detection of apoptotic cells after Nek2 siRNA treatment was observed by TUNEL assay (left). Graphs show the relative numbers of apoptotic cell after Nek2 siRNA treatment using Nek2 siRNA for 72 h (right). The number of TUNEL‐positive cells in Nek2 siRNA‐treated cells is significantly larger than that of control siRNA‐treated cells.
Effect of Nek2 silencing on tumor formation in a xenograft nude mouse model. We next examined the effect of Nek2 siRNA on the growth of colorectal cancer tumors in a xenograft nude mouse model. Mice were subcutaneously inoculated with DLD‐1 cells. Four days following the tumor inoculation, the tumors in all mice grew up to an average diameter of 4 mm. Nek2 siRNA, control siRNA, or PBS dissolved in cell matrix was then injected around the tumor nodules twice a week for 3 weeks. The growth rate and final volume of tumors were significantly reduced in Nek2 siRNA‐treated mice compared with those in control siRNA‐treated mice and PBS‐treated mice (Fig. 3A,B). The Nek2 expression in Nek2 siRNA‐treated tumors was significantly reduced compared to control siRNA‐treated tumors (Fig. 3C).
Figure 3.
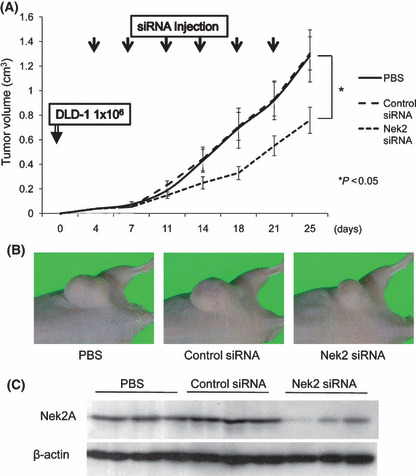
(A) The effect of Nek2 siRNA treatment was examined on tumor growth in a xenograft nude mouse model. Graphs show the tumor volume of DLD‐1 xenograft tumors in nude mice. Four days after DLD‐1 cell (1 × 106) inoculation, animals were randomly divided into three groups (PBS, control siRNA, and Nek2 siRNA). siRNA was injected around the tumor twice weekly for 3 weeks, as indicated by arrows. Tumor volume was measured twice every week. Each bar in the graph represents the average of tumor volume in each experimental group ± SE. *Statistically significant (P < 0.05 vs control siRNA by one‐way anova). (B) DLD‐1 xenograft tumors 1week after the final siRNA injection. A representative mouse per treatment group shows substantial reduction in tumors that received Nek2 siRNA treatment. (C) Nek2 expression was examined in the tumor tissues by Western blot analysis after Nek2 siRNA treatment. Nek2 expression in the tumor tissues was significantly suppressed in Nek2 siRNA‐treated tumors as compared with control siRNA‐treated tumors and PBS‐treated tumors.
Effect of combination treatment using Nek2 siRNA and CDDP on colorectal cancer cell proliferation and in vivo tumor formation. To further enhance the effect of Nek2 siRNA, we performed a combination treatment using Nek2 siRNA and the chemotherapeutic drug, CDDP. We chose CDDP because it is one of the most widely used anticancer drug in combination with multiple drugs such as 5‐fluorouracil (5‐FU), gemcitabine, irrinotecan, and others. CDDP enhances the antitumor effect via the cell death signaling pathway in cancer cells.( 9 ) CDDP induces DNA‐damage, whereas Nek2 siRNA may induce cell cycle arrest and apoptosis. By combining these drugs with different actions, we expected to observe additive or synergistic effects on the growth of colon cancer cells.
CDDP alone inhibited the proliferation of DLD‐1 cells in dose‐dependent manner. Combined administration of Nek2 siRNA (30 nm) with CDDP (0–30 μm) significantly suppressed proliferation of DLD‐1 cells compared to control or control siRNA (Fig. 4A). The number of TUNEL‐positive cells treated with Nek2 siRNA and CDDP was significantly higher than that of cells treated with control siRNA and CDDP (Fig. 4B). Furthermore, Nek2 siRNA and CDDP induced apoptosis strongly compared with the number of TUNEL‐positive cells by Nek2 siRNA alone in Figure 2(C). We next examined whether the combination of Nek2 siRNA and CDDP is more efficient for the suppression of tumor formation in a xenograft nude mouse model. To confirm the amount of CDDP that could inhibit tumor growth, we administered different concentrations of CDDP (2 mg/kg, 4 mg/kg, or 6 mg/kg) intraperitoneally in colorectal cancer tumor twice a week for 2 weeks. We found that the efficacy of tumor suppression reached at plateau at a dose of 4 mg/kg CDDP; therefore, we decided to use 4 mg/kg of CDDP for subsequent experiments. Four days following inoculation with DLD‐1 cells, we injected Nek2 siRNA (50 μm, 50 μL) or control siRNA (50 μm, 50 μL) with cell matrix (50 μL) to the area surrounding tumors and administered CDDP (4 mg/kg) intraperitoneally. As shown in Figure 5(A,B), combination treatment using Nek2 siRNA and CDDP suppressed tumor growth more efficiently than control siRNA and CDDP, and was also more effective than the treatment with Nek2 siRNA alone. Furthermore, we performed Western blot analysis to confirm Nek2 expression in xenograft tumors after combination of Nek2 siRNA and CDDP (Fig. 5C). There is no difference in Nek2 expression between Nek2 siRNA alone and combination of Nek2 siRNA and CDDP. The data demonstrated that Nek2 suppression was independent of CDDP treatment.
Figure 4.
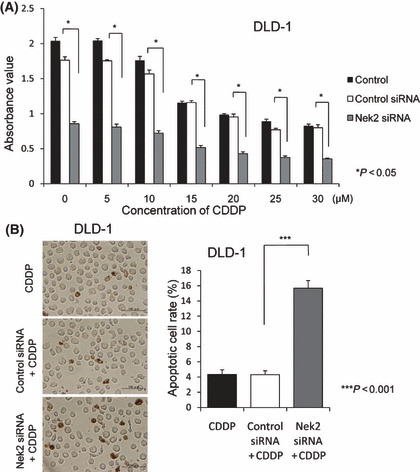
(A) Effect of combination treatment using Nek2 siRNA and cisplatin (CDDP) on the proliferation of DLD‐1 cells was analyzed by MTT assay. Various concentrations of CDDP additively suppressed the proliferation of DLD‐1 cells together with Nek2 siRNA. (B) Detection of apoptotic cells after combination treatment using Nek2 siRNA and CDDP was observed by TUNEL assay (left). Graphs show the relative numbers of apoptotic cell after combination treatment using Nek2 siRNA and CDDP for 72 h (right). The number of TUNEL‐positive cells in Nek2 siRNA and CDDP‐treated cells is significantly larger than that of control siRNA and CDDP‐treated cells.
Figure 5.
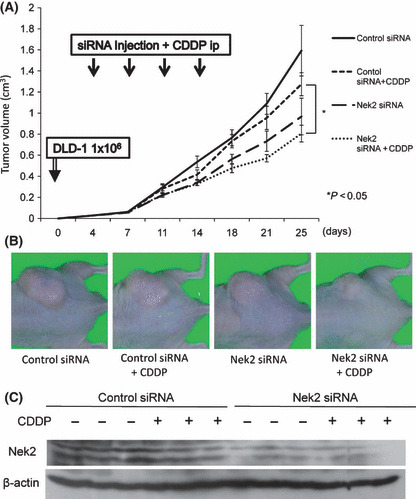
(A) The effect of combination treatment using Nek2 siRNA and cisplatin (CDDP) on the growth of DLD‐1 xenograft tumor volume in nude mice was examined. Four days after DLD‐1 cell (1 × 106) inoculation, animals were randomly divided into four groups (control siRNA, Nek2 siRNA, control siRNA plus CDDP, and Nek2 siRNA plus CDDP). siRNA was administered around the tumor, along with intraperitoneal injection of CDDP at a dose of 4 mg/kg body weight for each mouse twice a week for 2 weeks. Tumor volume was measured twice every week. Each represents the average of tumor volume in each group ± SE. *Statistically significant (P < 0.05 vs control siRNA by one‐way anova). (B) DLD‐1 xenograft tumors 11 days after the final siRNA and CDDP injection. A representative mouse per treatment group shows significant reduction of the tumors that received a combination of Nek2 siRNA and CDDP compared to control siRNA and CDDP. (C)The expression of Nek2 in xenograft tumors after combination of Nek2 siRNA and CDDP by Western blot analysis. β‐Actin was used as an internal loading control.
Growth inhibitory effect of combination treatment using Nek2 siRNA and CDDP on colorectal cancer cell proliferation. To examine whether combination of Nek2 siRNA with CDDP might be an effective strategy for the treatment of colorectal cancer, we evaluated the growth inhibitory effects of combination of Nek2 siRNA with CDDP by CI isobologram. To calculate CI, we determined the IC50 value for both Nek2 siRNA and CDDP. We found that the IC50 values for Nek2 siRNA and CDDP were 26.6 nm and 78.4 μm, respectively (Fig. 6A). The interaction between Nek2 siRNA and CDDP was further examined and is shown in Figure 6(B). The isobologram showed that the combination index was 1.0 at 25 nm of Nek2 siRNA plus 10 μm CDDP, indicating that combination of Nek2 siRNA and CDDP at these concentrations exerted additive growth inhibition (Fig. 6C). To clarify the detailed mechanisms of the efficiency with a combination of Nek2 siRNA plus CDDP, we examined the main molecule (pathway) using a PCR array in DLD‐1 cells. We identified the suppression of 14 genes and the overexpression of 20 genes among a total of 84 cancer‐related genes (Table 1). The data demonstrated a suppression of oncogenes, such as FOS and JUN, which is related to transformation of cancer. In addition, we identified an overexpression of genes such as APAF‐1 and BCL2L1, which are related to apoptosis.
Figure 6.
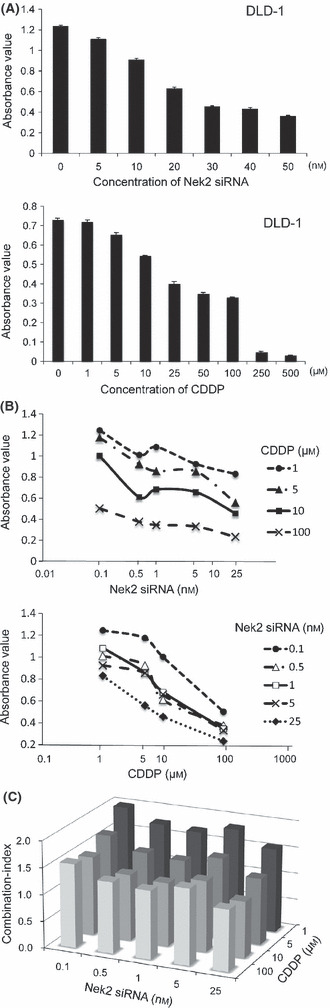
(A) Inhibition of cell growth by Nek2 siRNA alone, cisplatin (CDDP) alone with various concentrations in DLD‐1 cells. (B) The interaction between Nek2 siRNA and CDDP with various concentrations in DLD‐1 cells. (C) Combination effects of Nek2 siRNA with CDDP on DLD‐1 cells. CI > 1.1, 1.1 > CI > 0.9, and CI < 0.8 indicate antagonism, additive effect, or synergy, respectively.
Table 1.
Suppression or overexpression of genes, which are related to cancer development, by Nek2 siRNA alone or Nek2 siRNA plus cisplatin (CDDP) in DLD‐1 cells
| Suppressed genes |
| TNF, CDKN2A, MMP1, GZMA, FOS, IFNA1, ITGB3, JUN, TNFRSF25, SERPINE5, SERPINE1, MCAM, PLAUR, S100A4 |
| Overexpressed genes |
| FGFR, BRCA1, ITGAV, PDGFA, ITGA2, MYC, NFKB1, PIK3R1, BCL2L1, VEGFA, MET, ITGB5, ITGA3, APAF1, RB1, PDGFB, RAF1, MTSS1, CHEK2, FAS |
Discussion
As we previously reported that Nek2 was highly expressed in cholangiocarcinoma and breast cancer cells, in this report we investigated the expression of Nek2 in another tumor type, that is colorectal cancer. As expected, we found that Nek2 was overexpressed in four colorectal cancer cell lines, and that silencing of Nek2 expression induced cell apoptosis and growth suppression. To investigate the potential utility of Nek2 siRNA for clinical cancer therapy, we examined the effect of Nek2 siRNA administration in areas surrounding tumors in nude mice. Injection of Nek2 siRNA reduced Nek2 expression in tumor cells, as confirmed by Western blot analysis, and significantly suppressed tumor growth. Furthermore, treatment of tumor‐bearing mice with a combination of Nek2 siRNA and CDDP was more effective than administration of Nek2 siRNA alone. Thus, combination treatment using Nek2 siRNA and CDDP may be a useful therapeutic option for colorectal cancer.
In recent years, several new drugs such as oxaliplatin, irinotecan, and capecitabine have been developed and added to conventional chemotherapy regimens for colorectal cancer.( 10 , 11 , 12 , 13 , 14 ) In addition, other biological agents have also been proven to be efficient colorectal cancer therapeutics. Bevacizumab and cetuximab are monoclonal antibodies for vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR).( 15 , 16 , 17 , 18 , 19 ) The combination of these molecular targeting drugs with chemotherapy such as FOLFOX (oxaliplatin plus 5‐fluorouracil and leucovorin) or FOLFIRI (irinotecan plus 5‐fluorouracil and leucovorin) have led to significant improvements in colorectal cancer survival.( 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 ) However, these treatments are frequently terminated due to intolerance, resistance, or toxicity. Therefore, new therapeutic agents need to be developed to further improve the outcome of chemotherapy for colorectal cancer.
RNA interference technology has been developed as a potential therapeutic agent for several diseases, including cancer.( 27 , 28 , 29 ) siRNAs may be the best candidates for cancer therapy because of their exquisite specificity, efficiency, and endurance in gene‐specific silencing. In the present study, we showed that Nek2 siRNA induces cell apoptosis and growth suppression of colorectal cancer cells in vitro and significantly inhibits tumor growth in a xenograft nude mouse model. Thus, molecular target therapy using Nek2 siRNA is effective for colorectal cancer as well as in cholangiocarcinoma and breast cancer. Based on these data, we believe that Nek2 can be a strong candidate in molecular target therapy for wide variety of cancers.
Furthermore, to enhance the effect of Nek2 siRNA on tumor treatment, we developed a therapeutic strategy using Nek2 siRNA combined with the cytotoxic chemotherapy drug CDDP. The combination of Nek2 siRNA and CDDP achieved better tumor suppression in DLD‐1 cells than single administration of either Nek2 siRNA or CDDP. CDDP, or cisplatin, (cis‐dichlorodiammineplatinum [II]), is a platinum‐based cytotoxic compound that is widely used for chemotherapy of solid tumors, such as head and neck tumors, and ovarian, lung, and gastrointestinal cancers.( 30 ) However, the clinical use of this drug is limited due to the emergence of intrinsic and acquired resistance and severe side effects, such as acute nephrotoxicity and chronic neurotoxicity.( 31 , 32 ) The combination treatment using Nek2 siRNA and CDDP can reduce the total amount of CDDP needed in order to minimize side effects.
The mechanism of increased efficiency with a combination therapy using Nek2 siRNA and CDDP is still unclear. Platinum‐based agents are thought to produce intra‐ and interstrand DNA crosslinks, which induce DNA damage and activate a number of signal transduction pathways that eventually cause apoptosis.( 30 , 33 , 34 , 35 ) Nek2 is involved in centrosome separation in mitosis.( 1 , 5 ) Recent data also implicate a functional role for Nek2 in chromatin condensation, mitotic checkpoint, and cytokinesis.( 4 , 36 , 37 ) Depletion of Nek2 causes premature chromosome segregation and abrogation of the checkpoint pathway.( 38 , 39 ) PCR array data demonstrated that several genes, such as FOS and JUN, were suppressed, whereas APAF‐1 (apoptotic protease activating factor 1) and BCL2L1, were overexpressed by combination therapy with Nek2 siRNA plus CDDP. FOS and JUN, as a JUN‐JUN homodimer or a JUN‐FOS heterodimer, regulate cell proliferation in colorectal cancer.( 40 , 41 ) APAF‐1 is required to reconstitute caspase activity.( 42 ) BCL2L1 constitutes a critical intracellular checkpoint in the intrinsic pathway of apoptosis.( 43 ) Therefore, these data suggested that the combination of Nek2 siRNA plus CDDP is associated with suppression of cell growth and induction of apoptosis. However, we also observed an overexpression of MYC and MET, which are related to cell proliferation. The mechanisms of cell death and inhibition of cell growth induced by a combination of Nek2 siRNA and CDDP may be complicated. Further study is necessary to clarify the detailed mechanism regulating cell growth by a combination of Nek2 siRNA and CDDP.
Several reports previously demonstrated therapeutic usage of siRNAs combined with cytotoxic chemotherapy drugs for colorectal cancer in vitro. Karasawa reported that cellular inhibitor of apoptosis 2 (cLAP2) siRNA efficiently enhanced tumor cell sensitivity to 5‐FU.( 44 ) Guo reported that nuclear factor‐kappa B (NF‐κB) siRNA inhibited chemotherapy resistance.( 45 ) The targets of these siRNAs are modulators, enhancers, or suppressors of chemotherapeutic drugs. The therapeutic strategy used in these studies is direct activation of a single intrinsic pathway, which leads to improvement in the response to chemotherapy. Our therapeutic strategy, on the other hand, targets on multiple pathways involved in cell proliferation and cell death. Further studies are needed to elucidate how Nek2 siRNA modulate the effects of CDDP in cancer cells.
In summary, this study demonstrates for the first time the utility of a novel combination treatment using Nek2 siRNA and CDDP for colorectal cancer tumors. Our previous and present data indicate that Nek2 siRNA can be used in combination with conventional chemotherapeutic agents for cancer therapy, and has the potential to improve regression of human malignancies in the clinic.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgment
This work was supported by a grant from the Ministry of Education, Science, Sports and Culture of Japan.
References
- 1. Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J 1998; 17: 470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uto K, Nakajo N, Sagata N. Two structural variants of Nek2 kinase, termed Nek2A and Nek2B, are differentially expressed in Xenopus tissues and development. Dev Biol 1999; 208: 456–64. [DOI] [PubMed] [Google Scholar]
- 3. Fardilha M, WW U, Sa R et al. Alternatively spliced protein variants as potential therapeutic targets for male infertility and contraception. Ann N Y Acad Sci 2004; 1030: 468–78. [DOI] [PubMed] [Google Scholar]
- 4. Fletcher L, Cerniglia GJ, Nigg EA, Yend TJ, Muschel RJ. Inhibition of centrosome separation after DNA damage: a role for Nek2. Radiat Res 2004; 162: 128–35. [DOI] [PubMed] [Google Scholar]
- 5. Fry AM. The Nek2 protein kinase: a novel regulator of centrosome structure. Oncogene 2002; 21: 6184–94. [DOI] [PubMed] [Google Scholar]
- 6. Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res 2007; 67: 9637–42. [DOI] [PubMed] [Google Scholar]
- 7. Tsunoda N, Kokuryo T, Oda K et al. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci 2009; 100: 111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chou T‐C, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol Sci 1983; 4: 450–4. [Google Scholar]
- 9. Shamimi‐Noori S, Yeow WS, Ziauddin MF et al. Cisplatin enhances the antitumor effect of tumor necrosis factor‐related apoptosis‐inducing ligand gene therapy via recruitment of the mitochondria‐dependent death signaling pathway. Cancer Gene Ther 2008; 15: 356–70. [DOI] [PubMed] [Google Scholar]
- 10. Grivicich I, Mans DRA, Peters GJ, Schwartsmann G. Irinotecan and oxaliplatin: an overview of the novel chemotherapeutic options for the treatment of advanced colorectal cancer. Braz J Med Biol Res 2001; 34: 1087–103. [DOI] [PubMed] [Google Scholar]
- 11. Andre T, Boni C, Mounedji‐Boudiaf L et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004; 350: 2343–51. [DOI] [PubMed] [Google Scholar]
- 12. Kuebler JP, Wieand HS, O’Connell MJ et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C‐07. J Clin Oncol 2007; 25: 2198–204. [DOI] [PubMed] [Google Scholar]
- 13. Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology 2008; 134: 1296–310.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Omura K. Advances in chemotherapy against advanced or metastatic colorectal cancer. Digestion 2008; 77: 13–22. [DOI] [PubMed] [Google Scholar]
- 15. Wong S‐F. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther 2005; 27: 684–94. [DOI] [PubMed] [Google Scholar]
- 16. Ranieri G, Patruno R, Ruggieri E, Montemurro S, Valerio P, Ribatti D. Vascular endothelial growth factor (VEGF) as a target of bevacizumab in cancer: from the biology to the clinic. Curr Med Chem 2006; 13: 1845–57. [DOI] [PubMed] [Google Scholar]
- 17. Diaz‐Rubio E, Schmoll HJ. The future development of bevacizumab in colorectal cancer. Oncology 2005; 69 (Suppl 3): 34–45. [DOI] [PubMed] [Google Scholar]
- 18. Jean GW, Shah SR. Epidermal growth factor receptor monoclonal antibodies for the treatment of metastatic colorectal cancer. Pharmacotherapy 2008; 28: 742–54. [DOI] [PubMed] [Google Scholar]
- 19. Tol J, Koopman M, Cats A et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 2009; 360: 563–72. [DOI] [PubMed] [Google Scholar]
- 20. Kohne C‐H, Lenz H‐J. Chemotherapy with targeted agents for the treatment of metastatic colorectal cancer. Oncologist 2009; 14: 478–88. [DOI] [PubMed] [Google Scholar]
- 21. Samantas E, Dervenis C, Rigatos SK. Adjuvant chemotherapy for colon cancer: evidence on improvement in survival. Dig Dis 2007; 25: 67–75. [DOI] [PubMed] [Google Scholar]
- 22. Giantonio BJ, Catalano PJ, Meropol NJ et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 2007; 25: 1539–44. [DOI] [PubMed] [Google Scholar]
- 23. Moosmann N, Heinemann V. Cetuximab plus oxaliplatin‐based chemotherapy in the treatment of colorectal cancer. Expert Rev Anticancer Ther 2008; 8: 319–29. [DOI] [PubMed] [Google Scholar]
- 24. O’Neil BH, Goldberg RM. Innovations in chemotherapy for metastatic colorectal cancer: an update of recent clinical trials. Oncologist 2008; 13: 1074–83. [DOI] [PubMed] [Google Scholar]
- 25. Hegde SR, Sun W, Lynch JP. Systemic and targeted therapy for advanced colon cancer. Expert Rev Gastroenterol Hepatol 2008; 2: 135–49. [DOI] [PubMed] [Google Scholar]
- 26. Van Cutsem E, Kohne C‐H, Hitre E et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360: 1408–17. [DOI] [PubMed] [Google Scholar]
- 27. Karagiannis TC, El‐Osta A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther 2005; 12: 787–95. [DOI] [PubMed] [Google Scholar]
- 28. Lu PY, Xie F, Woodle MC. In vivo application of RNA interference: from functional genomics to therapeutics. Advances in Genetics 2005; 54; 117–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ameyar‐Zazoua M, Guasconi V, Ait‐Si‐Ali S. siRNA as a route to new cancer therapies. Expert Opin Biol Ther 2005; 5: 221–4. [DOI] [PubMed] [Google Scholar]
- 30. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22: 7265–79. [DOI] [PubMed] [Google Scholar]
- 31. McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther 2009; 8: 10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci 2007; 334: 115–24. [DOI] [PubMed] [Google Scholar]
- 33. Chu G. Cellular responses to cisplatin. The roles of DNA‐binding proteins and DNA repair. J Biol Chem 1994; 269: 787–90. [PubMed] [Google Scholar]
- 34. Faivre S, Chan D, Salinas R, Woynarowska B, Woynarowski JM. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem Pharmacol 2003; 66: 225–37. [DOI] [PubMed] [Google Scholar]
- 35. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005; 4: 307–20. [DOI] [PubMed] [Google Scholar]
- 36. Fu G, Ding X, Yuan K et al. Phosphorylation of human Sgo1 by NEK2A is essential for chromosome congression in mitosis. Cell Res 2007; 17: 608–18. [DOI] [PubMed] [Google Scholar]
- 37. Du J, Cai X, Yao J et al. The mitotic checkpoint kinase NEK2A regulates kinetochore microtubule attachment stability. Oncogene 2008; 27: 4107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li JJ, Li SA. Mitotic kinases: the key to duplication, segregation, and cytokinesis errors, chromosomal instability, and oncogenesis. Pharmacol Ther 2006; 111: 974–84. [DOI] [PubMed] [Google Scholar]
- 39. Qiu X‐L, Li G, Wu G et al. Synthesis and biological evaluation of a series of novel inhibitor of Nek2/Hec1 analogues. J Med Chem 2009; 52: 1757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Milde‐Langosch K. The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer 2005; 41: 2449–61. [DOI] [PubMed] [Google Scholar]
- 41. Shaulian E, Karin M. AP‐1 in cell proliferation and survival. Oncogene 2001; 20: 2390–400. [DOI] [PubMed] [Google Scholar]
- 42. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116: 205–19. [DOI] [PubMed] [Google Scholar]
- 43. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 2009; 9: 501–7. [DOI] [PubMed] [Google Scholar]
- 44. Karasawa H, Miura K, Fujibuchi W et al. Down‐regulation of cIAP2 enhances 5‐FU sensitivity through the apoptotic pathway in human colon cancer cells. Cancer Sci 2009; 100: 903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo J, Verma UN, Gaynor RB, Frenkel EP, Becerra CR. Enhanced chemosensitivity to irinotecan by RNA interference‐mediated down‐regulation of the nuclear factor‐{kappa}B p65 subunit. Clin Cancer Res 2004; 10: 3333–41. [DOI] [PubMed] [Google Scholar]