

Abstract
We investigated the ability of TZT‐1027 (Soblidotin), a novel antimicrotubule agent, to induce antivascular effects, because most vascular targeting agents that selectively disrupt tumor vasculature also inhibit tubulin polymerization. Treatment with 10−7 g/mL TZT‐1027 rapidly disrupted the microtubule cytoskeleton in human umbilical vascular endothelial cells (HUVEC), and significantly enhanced vascular permeability in HUVEC monolayers. In addition, single intravenous administration of 2 mg/kg TZT‐1027 to mice bearing Colon26 tumors significantly reduced tumor perfusion and caused extravascular leakage of erythrocytes 1 h after administration. Subsequently, thrombus formation with deposition of fibrin and tumor necrosis was observed 3 and 24 h after administration, respectively. These results strongly suggest that TZT‐1027 possesses antivascular effects. TZT‐1027 induced apoptosis not only in HUVEC but also in C26 cancer cells (cell line of Colon26 solid tumor) in vitro, suggesting it exerts direct cytotoxicity against tumor cells in addition to its antivascular effects. A single intravenous administration of 1, 2 and 4 mg/kg TZT‐1027 significantly prolonged the survival of mice with advanced‐stage Colon26 tumors in a dose‐dependent manner. Furthermore, TZT‐1027 itself less markedly enhanced the permeability of normal vessels, but was additive with vascular endothelial growth factor, indicating the possibility that TZT‐1027 selectively exerts its activity on tumor vessels. In summary, these results suggest that TZT‐1027 exerts both an indirect antivascular effect and a direct cytotoxic effect, resulting in strong antitumor activity against advanced‐stage tumors, and that TZT‐1027 may be useful clinically for treating solid tumors. (Cancer Sci 2006; 97: 1410–1416)
Tumor vasculature has a crucial role in producing the oxygen and nutrient supply necessary for survival and proliferation of solid tumors.( 1 , 2 ) Differences between tumor vessels and normal tissue vessels may frequently be caused by specific vascular barrier dysfunction in tumor vessels, because of abnormal characteristics such as fragility, chaotic arrangements, tortuosity and lack of a vessel wall (smooth muscle cells and pericytes).( 3 , 4 , 5 ) These features may explain why tumor vasculature may be more susceptible than normal vessels to vascular targeting agents (VTA), which selectively disrupt tumor vasculature. Disruption of the tumor vasculature may be an effective strategy for various solid tumors, regardless of cancer type, without being influenced by tumor cell drug resistance mechanisms, suggesting its clinical usefulness. Therefore, a number of VTA have recently been developed, as opposed to anti‐angiogenic agents that inhibit tumor neovascularization. Most VTA, including flavone acetic acid,( 6 , 7 ) 5,6‐dimethylxanthenone‐4‐acetic acid,( 8 ) combretastatin A‐4‐P (CA‐4‐P),( 9 , 10 ) ZD6126,( 11 , 12 ) AVE8062( 13 ) and Oxi4503,( 14 ) have the common feature of inhibiting tubulin polymerization.
TZT‐1027 (Soblidotin) is a novel antimicrotubule agent synthesized to have enhanced antitumor activity and reduced toxicity compared with dolastatin 10.( 15 , 16 ) TZT‐1027 inhibits the growth of various human cancer cells in vitro, with a 50% inhibitory concentration (IC50) value of 1 × 10−12 to 1 × 10−9 g/mL.( 17 , 18 ) Its growth‐inhibitory effect is less affected by P‐glycoprotein overexpression than are other tubulin inhibitors, and it is not affected by overexpression of breast cancer resistance protein or multidrug resistance‐associated protein.( 19 ) Furthermore, TZT‐1027 has a broad spectrum of antitumor activity in vivo against murine syngeneic transplantable tumors, including P388 leukemia, Colon26 adenocarcinoma, B16 melanoma and M5076 sarcoma, with an efficacy superior or comparable to those of several reference agents (dolastatin 10, cisplatin, vincristine and 5‐fluorouracil), and against human tumor xenografts, including LX‐1 lung and MX‐1 breast carcinomas, without causing a serious reduction in bodyweight.( 20 ) In addition, TZT‐1027 arrests the cell cycle at the G2/M phases with Bcl‐2 phosphorylation, followed by apoptosis with DNA fragmentation and Caspase‐3 activation in cancer cells.( 17 ) However, TZT‐1027 caused rapid tumoral hemorrhage and a transient increase in hemoglobin content in advanced‐stage Colon26 tumors in vivo, and it induced membrane blebbing and cell detachment of human umbilical vascular endothelial cells (HUVEC) in vitro,( 21 ) suggesting that TZT‐1027 may have an antivascular effect against tumor vasculature in addition to potent cytotoxicity against cancer cells.
The aim of the present study was to evaluate whether TZT‐1027 is a VTA and to elucidate the mechanism of its antivascular effect. For that purpose, we first investigated the actions of TZT‐1027 on the microtubule cytoskeleton and on vascular barrier function in HUVEC, because we assumed that the antivascular effect of TZT‐1027 involved vascular endothelial cells, as for other VTA.( 22 , 23 , 24 ) Subsequently, we examined the in vivo antivascular effect of TZT‐1027 sequentially on tumor perfusion and histopathology using Colon26 tumors implanted into syngeneic mice. In addition, we investigated its in vitro cytotoxicity against C26 cancer cells (cell line of Colon26 solid tumor) and its in vivo antitumor activity against advanced‐stage Colon26 tumors. Furthermore, we assessed its influence on normal vessels, owing to concerns regarding side‐effects.
Materials and Methods
Experimental animals and cell lines. Female BALB/c and CDF1 mice were purchased from Japan Charles River Co. (Kanagawa, Japan), and male Hartley guinea pigs from Japan SLC (Shizuoka, Japan). A murine adenocarcinoma, Colon26 solid tumor, and its cell line for culture, C26 cancer cells, were kindly supplied by the Cancer Chemotherapy Center, Japanese Foundation for Cancer Research (Tokyo, Japan). The Colon26 solid tumor was maintained in syngeneic BALB/c mice in our laboratory according to the protocol of the National Cancer Institute.( 25 ) All animal experiments were conducted according to the Rules for the Care and Use of Laboratory Animals of ASKA Pharmaceutical Co. The C26 cancer cells were cultured in RPMI‐1640 medium containing 10% heat‐inactivated fetal calf serum, 100 IU/mL of penicillin and 100 µg/mL of streptomycin (standard growth medium). HUVEC were purchased from Cambrex (Walkersville, MD, USA), cultured in EGM‐2 medium (EGM‐2 Bullet Kit; Cambrex) and used for the experiments until passage 5. All cells were maintained at 37°C in the presence of 5% CO2.
Agents. TZT‐1027 was synthesized in our laboratory, and dissolved in dimethylsulfoxide in the skin reaction test and in 0.05 mol/L lactate buffer (LB) (pH 4.5) in the other experiments. The chemical structure of TZT‐1027 is shown in Fig. 1 (molecular weight 701.98). Vascular endothelial growth factor (VEGF), bradykinin and Evans blue dye were purchased from Pepro Tech (London, UK), Cosmo Bio Co. (Tokyo, Japan) and Nacalai Tesque (Kyoto, Japan), respectively. All compounds were dissolved and diluted in saline.
Figure 1.
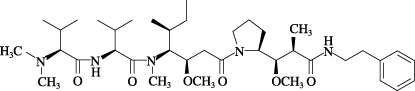
The chemical structure of TZT‐1027.
Immunofluorescent staining of β‐tubulin. Human umbilical vascular endothelial cells were plated on four‐well Laboratory Tek Chamber Slides (Nalge Nunc International, Rochester, NY, USA) at 1 × 104 cells/well. After 24 h, they were washed twice with phosphate‐buffered saline (PBS) and then treated with 10−7 g/mL TZT‐1027 for 15 min at 37°C. After two washes with PBS, they were fixed in 4% paraformaldehyde in PBS for 30 min, washed twice with PBS again, and treated with 0.25% Triton X‐100/1% bovine serum albumin (BSA) in PBS for 5 min. Thereafter, they were washed three times with 0.05% Tween 20 in PBS (Tween/PBS), treated with primary mouse monoclonal anti‐β‐tubulin antibody (1:500, clone TUB2.1; Sigma, St Louis, MO) in 1% BSA in PBS (BSA/PBS) for 1 h at room temperature, washed three times with Tween/PBS, and treated with Alexa Fluor 488‐conjugated secondary goat antimouse IgG antibody (1:500; Invitrogen, Frederick, MD, USA) in BSA/PBS for 1 h at room temperature. After three washes with Tween/PBS and one rinse with PBS, the fluorescent images of β‐tubulin were visualized using a fluorescence microscope (AX80; Olympus, Tokyo, Japan) and photographed.
Human umbilical vascular endothelial cell monolayer permeability study. Diffusion of fluorescein‐isothiocyanate (FITC)–dextran (70 kDa; Sigma) passing through the HUVEC monolayer was determined as described previously.( 26 , 27 ) Briefly, HUVEC were cultured on fibronectin‐coated culture inserts (upper compartment, pore size 3 µm) set on 24‐well companion plates (lower compartment; Becton Dickinson, Bedford, MA, USA) containing EGM‐2 medium at 2 × 105 cells/well. When confluent monolayers were obtained after 2 days of culture, the two compartments were washed twice with PBS, and the agent at 0.15 mL diluted with 1 mg/mL FITC–dextran in EBM medium (Cambrex) and the same concentration of the agent at 0.75 mL diluted with EBM medium was added to the upper and lower compartments, respectively. After 15, 30 and 60 min of treatment, a 50‐µL aliquot was sampled from the lower compartment, and its fluorescence intensity was measured (excitation 490 nm, emission 530 nm) using a 96‐well microplate reader (Corona Electric Co., Ibaragi, Japan).
Tumor perfusion study. Tumor blood volume was measured by the Evans blue dye perfusion technique, as described previously.( 6 , 21 , 28 ) Briefly, tumor fragments (2 mm3) of Colon26 tumors were inoculated subcutaneously into the right flank of female CDF1 mice, and a single dose of 2 mg/kg TZT‐1027 or vehicle (LB) was administered intravenously at 10 mL/kg when the tumor volume reached approximately 400–600 mm3. At 1, 6 and 24 h after drug administration, 1% Evans blue was injected intravenously at 10 mL/kg. After 2 min, the mice were exsanguinated and killed, and the tumor tissues were extirpated, weighed and homogenized in a five‐fold volume of digestive solution (0.5% sodium sulfate‐acetone [2:3]). After incubation for 48 h at room temperature to extract the Evans blue dye, the suspensions were centrifuged at 1700 g for 10 min, and the amount of Evans blue in the supernatant was measured using a 96‐well microplate reader (Nippon InterMed, Tokyo, Japan) with absorbance set at 620 nm.
Histological analysis. A single dose of 2 mg/kg TZT‐1027 or vehicle (LB) was administered as described above when the tumor volume reached approximately 700–800 mm3 after inoculation with fragments of Colon26 solid tumors. At 1, 3 and 6 h after drug administration, tumor tissues were extirpated and fixed in 10% neutral buffered formalin (pH 7.4), and the subsequent preparation of thin sections and hematoxylin and eosin staining were carried out by Hist Science Laboratory Co. (Tokyo, Japan).
DNA fragmentation analysis. Human umbilical vascular endothelial cells and C26 cancer cells were plated on a 90 mm ‐dish at 5 × 106 cells/well. After 24 h, they were washed three times with PBS and then treated with 10−7 g/mL TZT‐1027. At 12, 24 and 48 h after treatment, they were again washed three times with PBS, and intracellular DNA was extracted and purified using Sepa Gene (Sanko Junyaku Co., Tokyo, Japan). The samples were dissolved in TE buffer (10 mM Tris‐HCl and 1 mM ethylenediaminetetracetic acid [pH 8.0]) and treated with 20 µg/mL RNase A for 30 min at 37°C. Additionally, DNA was purified again, and 2 µg DNA was electrophoresed on a 2% agarose gel for 30 min at 50 V. Then, the gel was stained with ethidium bromide, and DNA bands were visualized and photographed under ultraviolet irradiation. A stable 100‐bp DNA ladder (Sigma Genosys Japan, Hokkaido, Japan) served as a weight standard marker.
Antitumor activity against advanced‐stage Colon26 tumors. A single dose of 1, 2 or 4 mg/kg TZT‐1027 or vehicle (LB) was administered as described above when the tumor volume reached approximately 400–600 mm3 after inoculation with Colon26 solid tumor fragments. The median survival time (MST, days) of the treated (T) and control (C) groups was determined, and the increase in life span (ILS, %) was calculated using the following formula:
| ILS (%) = ([MST treated]/[MST control] − 1) × 100. |
Skin reaction test in the dorsal skin of guinea pigs. TZT‐1027 diluted with 5% dimethylsulfoxide in saline, and bradykinin and VEGF diluted with saline were injected intradermally into the dorsal skin of guinea pigs at 50 µL per reaction site. In the case of the combination experiment, a single intravenous dose of 0.5 mg/kg TZT‐1027 or vehicle (LB) was administered to the guinea pigs 15 min prior to the intradermal injection of saline, bradykinin or VEGF. Immediately after intradermal injection, 1% Evans blue in saline was administered intravenously at 2 mL/kg. After 30 min, the guinea pigs were exsanguinated and killed, and the dorsal skin was extirpated using a punch, then minced and homogenized in 3 mL of digestive solution. Thereafter, the amount of Evans blue in the skin was measured using the same method as in the tumor perfusion study.
Statistical analysis. The data obtained from the HUVEC monolayer permeability study, the tumor perfusion study, and the skin reaction test were analyzed using the parametric Student's t‐test. For the analysis of survival prolongation, we used the log‐rank test based on the Kaplan–Meier method. All statistical analyses were carried out using SAS‐system Release 8.2 software (SAS Institute Japan, Tokyo, Japan), and a P‐value of less than 0.05 was considered statistically significant.
Results
TZT‐1027 disrupts the microtubule cytoskeleton in HUVEC. TZT‐1027 targets microtubules of the cytoskeleton and induces their depolymerization, resulting in cell cycle arrest at the G2/M phases and induction of apoptosis in cancer cells.( 16 , 17 ) Though it was thought that vascular endothelial cells were the most important target in the antivascular effect, the action of TZT‐1027 remained unclear. Therefore, we first examined the effect of TZT‐1027 on HUVEC morphology by immunofluorescent staining of β‐tubulin (Fig. 2A). Treatment with 10−7 g/mL TZT‐1027 for 15 min, corresponding to a maximum concentration (Cmax) after administration at an optimal dose (2 mg/kg intravenously), caused the disappearance of radial microtubule filaments extending throughout the cytoplasm compared with untreated HUVEC, and induced extensive fragmentation of microtubule filaments. Disruption of the microtubule cytoskeleton was observed throughout the cytoplasm very acutely (within 15 min), indicating that TZT‐1027 acts not only on spindle microtubules, but also on cytoskeletal microtubules in a cell cycle‐independent manner.
Figure 2.
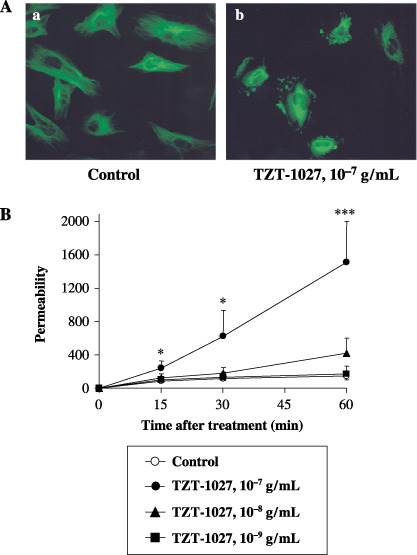
TZT‐1027 disrupts the microtubule cytoskeleton in human umbilical vascular endothelial cells (HUVEC) and induces vascular barrier dysfunction on HUVEC monolayers. (A) HUVEC were treated (a) without or (b) with 10−7 g/mL TZT‐1027 for 15 min, followed by immunostaining with anti‐β‐tubulin antibody. (B) Confluent HUVEC monolayers were treated with vehicle or various concentrations of TZT‐1027 (10−7, 10−8 and 10−9 g/mL) at the indicated time, and then diffusion of fluorescein‐isothiocyanate–dextran passing through the HUVEC monolayer was measured using a 96‐well microplate reader. Each point represents the mean ± SD of three independent experiments. *P < 0.05, ***P < 0.001, significantly different from the vehicle control group (Student's t‐test).
TZT‐1027 induces vascular barrier dysfunction in HUVEC monolayers. To estimate the influence of TZT‐1027 on the barrier function of vascular endothelial cells, we next examined whether TZT‐1027 enhanced vascular permeability, by measuring the diffusion of FITC–dextran passing through the HUVEC monolayer (Fig. 2B). TZT‐1027 at 10−7 g/mL enhanced vascular permeability significantly in the HUVEC monolayer more than 15 min after treatment in a time‐dependent manner, although vehicle or TZT‐1027 at 10−9 g/mL did not, and TZT‐1027 at 10−8 g/mL enhanced it slightly. These results suggested that TZT‐1027 caused barrier dysfunction of vascular endothelial cells when the higher concentration corresponding to the Cmax was used, even with short‐term treatment.
TZT‐1027 causes tumor vascular shutdown. To clarify the effect of TZT‐1027 on tumor vasculature in vivo, sequential changes in Colon26 tumor perfusion after TZT‐1027 administration were assessed in CDF1 mice by measuring the amount of Evans blue in the tumor (Fig. 3). A single intravenous administration of 2 mg/kg TZT‐1027 to mice bearing advanced‐stage Colon26 tumors reduced tumor perfusion dramatically compared to vehicle. The significant reduction in tumor perfusion (P < 0.001) occurred rapidly, at 1 h after administration; the maximum reduction was observed at 6 h, and it persisted for at least 24 h. The reduction rates at 1, 6 and 24 h after treatment with TZT‐1027 compared with the control were 65.9, 86.6 and 61.2%, respectively. These findings suggest that TZT‐1027 induces acute vascular shutdown in tumors at a dose of 2 mg/kg intravenously, the dose at which TZT‐1027 exhibits the most potent antitumor activity in animal models.( 20 )
Figure 3.
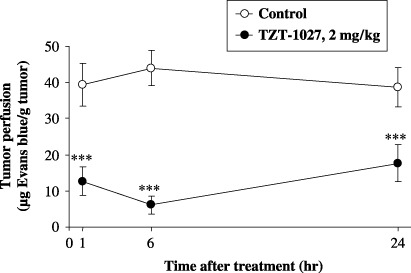
TZT‐1027 causes tumor vasculature shutdown. TZT‐1027 (2 mg/kg) or vehicle (control) was administered intravenously to mice bearing Colon26 tumors. The Evans blue content of tumors was measured at 1, 6 and 24 h after administration. Each point represents the mean ± SD of 10 animals. ***P < 0.001, significantly different from the vehicle treatment group (Student's t‐test).
Gross findings of advanced‐stage Colon26 tumors after administration of TZT‐1027. To observe the antitumor activity of TZT‐1027, advanced‐stage Colon26 tumors extirpated from mice were photographed periodically after a single intravenous administration of 2 mg/kg TZT‐1027 (Fig. 4A). TZT‐1027 induced notable and widespread hemorrhage in advanced‐stage Colon26 tumors 1 h after administration (Fig. 4Ab) compared with the vehicle control (Fig. 4Aa), and hemorrhage increased prominently in a time‐dependent manner (Fig. 4Ac,d). At 24 h after administration, marked hemorrhagic necrosis with scab formation was observed at the peripheral rim of the tumor (Fig. 4Ad).
Figure 4.
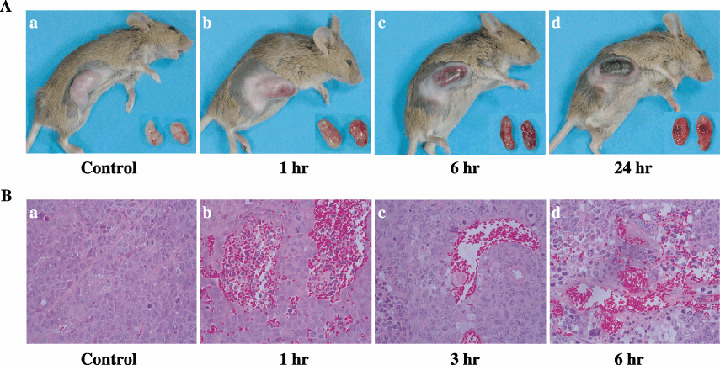
Gross findings and histological appearances of Colon26 tumors after administration of TZT‐1027. TZT‐1027 (2 mg/kg) was administered intravenously to mice bearing Colon26 tumors. (A) Photographs of mice bearing tumors and extirpated tumors are (a) vehicle control and (b) 1 h, (c) 6 h and (d) 24 h after administration of TZT‐1027. (B) After treatment with TZT‐1027, tumor tissues were extirpated, fixed in 10% neutral buffered formalin (pH 7.4), and stained with hematoxylin and eosin. Specimens are (a) vehicle control and (b) 1 h, (c) 3 h and (d) 6 h after administration with TZT‐1027.
Histological appearance of advanced‐stage Colon26 tumors after administration of TZT‐1027. After a single intravenous administration of 2 mg/kg TZT‐1027 to mice bearing advanced‐stage Colon26 tumors, extirpated tumors were obtained at successive time points and stained with hematoxylin and eosin (Fig. 4B). TZT‐1027 caused marked extravascular leakage of erythrocytes 1 h after administration (Fig. 4Bb) compared with the vehicle control (Fig. 4Ba), suggesting that vascular permeability was enhanced in the tumor tissue. Subsequently, thrombus formation with pink‐colored deposition of fibrin was observed 3 h after administration (Fig. 4Bc), and multiple areas of erythrocyte leakage and thrombus formation existed simultaneously 6 h after administration (Fig. 4Bd). In addition, the vascular network in the tumor tissue had become unclear, indicating that TZT‐1027 induced an extensive vascular shutdown. These results strongly suggested that TZT‐1027 possesses characteristic antivascular effects.
Induction of apoptosis. We next evaluated whether TZT‐1027 induces apoptosis in C26 cancer cells comprising a tumor mass and HUVEC comprising blood vessels (Fig. 5). A characteristic of typical apoptosis, a 180‐bp DNA ladder, was observed after more than 12 and 48 h of treatment with 10−7 g/mL TZT‐1027 in HUVEC (Fig. 5A) and C26 cancer cells (Fig. 5B), respectively. Although there was a difference between the two cellular types in the interval required for induction of apoptosis, we confirmed that TZT‐1027 induced apoptosis not only in HUVEC but also in C26 cancer cells in vitro, and the results suggested that TZT‐1027 exerts a direct cytotoxic effect against tumor cells in addition to its antivascular effects.
Figure 5.
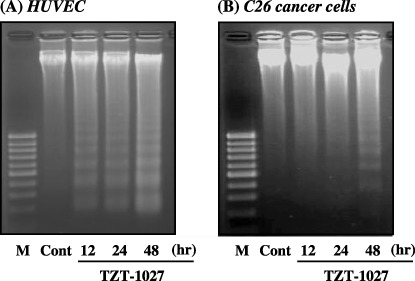
TZT‐1027 induced apoptosis not only in human umbilical vascular endothelial cells (HUVEC) but also in C26 cancer cells. (A) HUVEC and (B) C26 cancer cells were treated with 10−7 g/mL TZT‐1027. At 12, 24 and 48 h after treatment with TZT‐1027, intracellular DNA was extracted, purified and electrophoresed on a 2% agarose gel. Then, the gel was stained with ethidium bromide, and DNA bands were visualized and photographed under ultraviolet irradiation. Cont, untreated control; M, molecular size marker (100 bp).
Antitumor activity of TZT‐1027 against advanced‐stage Colon26 tumors. Next, we evaluated the antitumor activity of TZT‐1027 against advanced‐stage Colon26 tumors (Table 1). A single intravenous administration of 1, 2 and 4 mg/kg TZT‐1027 significantly prolonged survival (P < 0.05) compared with control in a dose‐dependent manner, with ILS of 39.6, 58.3 and 108.3%, respectively. However, no toxic death was observed at an optimal dose of 2 mg/kg TZT‐1027, although one of eight mice was dead only at the higher dose of 4 mg/kg TZT‐1027. Thus, we confirmed that a single administration of TZT‐1027 exhibited potent antitumor activity against advanced‐stage Colon26 tumors.
Table 1.
Antitumor activity of TZT‐1027 against advanced‐stage Colon26 tumors
| Drug | Dose (mg/kg) | MST (days) | ILS (%) | P‐value | Toxic death |
|---|---|---|---|---|---|
| Control | – | 12.0 | – | – | 0/8 |
| TZT‐1027 | 1 | 16.8 | 39.6 | 0.033 | 0/8 |
| TZT‐1027 | 2 | 19.0 | 58.3 | 0.0019 | 0/8 |
| TZT‐1027 | 4 | 16.8 | 108.3 | 0.0013 | 1/8 |
A single dose of 1, 2 or 4 mg/kg TZT‐1027 or vehicle (LB) was administered when the tumor volume reached approximately 400–600 mm3. Antitumor activity was evaluated in terms of the increase in life span (ILS), defined as ([Median survival time {MST} treated]/[MST control] − 1) × 100. Statistical analysis was carried out using the log‐rank test based on the Kaplan–Meier method. A significant difference from the survival of the control group is represented. Toxic death means the number of mice that died/number of mice that were used.
Bradykinin, VEGF and TZT‐1027 enhance vascular permeability in the dorsal skin of the guinea pig. Furthermore, we examined the influence of TZT‐1027 on normal vessels in a skin reaction test using guinea pigs, because we were concerned whether the antivascular action caused side‐effects (Fig. 6). Intradermal administration of bradykinin or VEGF caused marked extravasation of Evans blue at concentrations ranging from 10 to 100 ng/site, suggesting that they enhanced the permeability of normal vessels, as reported previously (Fig. 6A,B). In contrast, intradermal administration of TZT‐1027 enhanced vascular permeability only at concentrations ranging from 10 000 to 50 000 ng/site (Fig. 6C). It was confirmed that TZT‐1027 less markedly enhanced the permeability of normal vessels; therefore, the concentrations of TZT‐1027 inducing vascular permeability were not physiological, and corresponded to approximately 1/1000 of those of bradykinin or VEGF.
Figure 6.
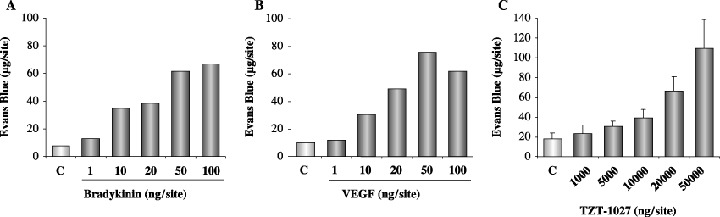
Bradykinin (BK), vascular endothelial growth factor (VEGF) and TZT‐1027 induce enhanced vascular permeability in the dorsal skin of the guinea pig. (A) BK (n = 2), (B) VEGF (n = 2) and (C) TZT‐1027 (n = 6) were injected intradermally into the dorsal skin of guinea pigs at 50 µL per reaction site, followed by an intravenous injection of 1% Evans blue at 2 mL/kg. After 30 min, the dorsal skins were extirpated and amounts of Evans blue in the skin were measured.
TZT‐1027 potentiates the enhanced vascular permeability induced by VEGF, but not by bradykinin. Finally, we investigated the interaction of TZT‐1027 with bradykinin or VEGF (Fig. 7). After intravenous pretreatment with vehicle or 0.5 mg/kg of TZT‐1027, bradykinin or VEGF was administered intradermally in the dorsal skin of the guinea pig. It is interesting to note that TZT‐1027 potentiated the enhanced vascular permeability induced by 10 or 20 ng/site of VEGF, but not that induced by bradykinin. In fact, these results indicated that the effect of TZT‐1027 was additive with that of VEGF.
Figure 7.
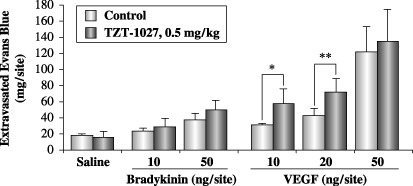
TZT‐1027 potentiates enhanced vascular permeability induced by vascular endothelial growth factor (VEGF), but not by bradykinin. A single dose of 0.5 mg/kg TZT‐1027 or vehicle (saline) was administered to the guinea pigs intravenously 15 min prior to intradermal injection of saline, bradykinin or VEGF. Immediately after the intradermal injection, 1% Evans blue in saline was administered intravenously at 2 mL/kg. After 30 min, the dorsal skins were extirpated and amounts of Evans blue were measured. Each point represents the mean ± SD of six animals. *P < 0.05, **P < 0.01, significantly different from the vehicle treatment group (Student's t‐test).
Discussion
Several studies have reported that VTA such as CA4P( 22 ) and ZD6126( 23 , 24 ) disrupt the microtubule cytoskeleton in HUVEC 30–40 min after treatment, inducing morphological changes, membrane blebbing and cell detachment. In the present study, TZT‐1027 also completely disrupted the microtubule cytoskeleton in HUVEC 15 min after treatment (Fig. 2A), suggesting that TZT‐1027 targets microtubules in vascular endothelial cells as well as in cancer cells, as demonstrated for other VTA. In the vascular permeability study using HUVEC monolayers, TZT‐1027 enhanced vascular permeability significantly more than 15 min after treatment (Fig. 2B). The onset of both microtubule disruption (Fig. 2A) and enhanced vascular permeability (Fig. 2B) was observed at nearly the same time (within 15 min), suggesting that enhanced vascular permeability would be due to disruption of the microtubule cytoskeletal tight junctions in vascular endothelial cells. Furthermore, TZT‐1027 increased hemoglobin levels transiently,( 21 ) and induced marked extravascular leakage of erythrocytes (Fig. 4Bb) 1 h after treatment in tumor tissue, and enhanced vascular permeability in vitro. Taken together, these results demonstrated that TZT‐1027 could cause enhanced vascular permeability in tumor tissues. In histological analysis, thrombus formation (Fig. 4Bc) was confirmed after enhancement of tumor vascular permeability (Fig. 4Bb), suggesting that the contact of plasma components with the exposed basement membrane induces thrombosis formation. Subsequently, elicited vessel occlusion might cause vascular shutdown (Fig. 3) and secondary ischemic tumor necrosis (Fig. 4Ad).
We reported previously that TZT‐1027 arrests the cell cycle at the G2/M phases 12 h after treatment and induces apoptosis 24–48 h after treatment, indicating that the cell‐kill kinetics of TZT‐1027 against cancer cells occurs in a cell cycle‐ or time‐dependent manner.( 17 ) In the present study, TZT‐1027 induced apoptosis against HUVEC at 12 h (Fig. 5A), which was more rapid than against C26 cancer cells at 48 h (Fig. 5B), and disrupted the microtubules throughout the cytoplasm in HUVEC within 15 min (Fig. 2). Taken together, these data indicate that TZT‐1027 may exert its action against vascular endothelial cells in a cell cycle‐independent manner, not but in a cell cycle‐dependent manner, as against cancer cells.
Because it was very important to ascertain whether the antivascular action of TZT‐1027 caused side‐effects, we next examined the influence of TZT‐1027 on normal vessels in a skin reaction test using guinea pigs. Despite the markedly enhanced permeability induced by bradykinin (Fig. 6A) and VEGF (Fig. 6B), TZT‐1027 (Fig. 6C) did not enhance vascular permeability at concentrations other than non‐physiological concentrations (incidentally, the Cmax of TZT‐1027 in mice was approximately 200 ng/mL, data not shown). These results indicated that TZT‐1027 itself did not affect normal vessels, which are well‐matured and surrounded by a vessel wall (smooth muscle cells and pericytes), differing from tumor vessels. TZT‐1027 did enhance vascular permeability in the VEGF‐coexistent situation (Fig. 7), indicating that it acts additively with VEGF to enhance vascular permeability. However, a previous report has shown that the effect of combretastatin A4, one of the VTA, was larger in tumor tissue than in normal tissues, including brain, heart and kidney.( 10 ) In addition, we previously compared the antitumor activity of TZT‐1027 with those of other anticancer agents, including CA‐4‐P, vincristine, docetaxel and cisplatin, against two sublines of human small‐cell lung cancer cells (SBC‐3): mock transfectant SBC‐3/Neo and VEGF transfectant SBC‐3/VEGF.( 29 ) As the SBC‐3/VEGF transfectant was highly malignant, with enhanced proliferation, the antitumor activities of the above anticancer agents against it were less marked than those against SBC‐3/Neo. However, TZT‐1027 exhibited potent antitumor activities against not only SBC‐3/Neo, but also SBC‐3/VEGF. This result supports the synergistic effects of TZT‐1027 and VEGF, and suggests that TZT‐1027 is useful for treating highly malignant solid tumors with strong VEGF expression.
In the present report, the in vivo study used the optimal dose of TZT‐1027 (2 mg/kg, intravenously) at which it exhibits the most potent antitumor effects, whereas the in vitro study used TZT‐1027 at the Cmax value, as antivascular effects were rapid after administration of TZT‐1027. Consequently, we obtained the results of characteristic action closely involved in antivascular effects; disruption of the microtubule cytoskeleton in vascular endothelial cells and enhanced vascular permeability in HUVEC monolayers in the in vitro study, and enhanced tumor vascular permeability, thrombus formation, vascular perfusion shutdown and tumor necrosis in the in vivo study. Therefore, these results strongly suggest that TZT‐1027 possesses antivascular effects.
Whereas classical antimicrotubule agents, such as colchicines, vincristine and vinblastine,( 30 , 31 ) disrupt tumor vasculature at a maximum tolerated dose (MTD), VTA such as CA‐4‐P( 10 ) and ZD6126( 12 ) exert their antivascular effects at doses below the MTD with fewer side‐effects. However, these VTA have serious problems in common( 10 , 12 , 32 ) in that they cause necrosis only in the central region of tumor, not at the peripheral region, as the oxygen and nutrients from normal blood vessels surrounding the tumor could be provided to the rim of tumor cells. Consequently, the surviving rim of tumor cells rapidly reassembles the tumor mass, resulting in weak antitumor activity when VTA are given as monotherapy.( 12 , 32 ) Therefore, several studies have investigated combination therapy with VTA and irradiation( 33 , 34 ) or chemotherapeutic agents( 12 , 32 , 35 , 36 ) in xenograft models, and recently, combination therapy with CA‐4‐P and carboplatin was conducted in a phase I clinical trial,( 37 ) and combination therapy with ZD6126 and 5‐fluorouracil or oxaliplatin was considered.( 38 ) In contrast, TZT‐1027, differing from other VTA, has not only antivascular effects against tumor vasculature but also potent cytotoxicity against cancer cells, suggesting the benefit of a potential synergistic effect. In particular, TZT‐1027 may efficiently invade tumor tissue owing to enhanced vascular permeability early after administration, and then, diffusion of TZT‐1027 from the tumor tissue may be prevented, owing to disruption of the tumor vasculature. Consequently, keeping TZT‐1027 in the tumor tissue for a long time may be useful to exert its cell cycle‐dependent cytotoxicity against tumor cells, including those at the peripheral region. Thus, TZT‐1027 monotherapy may prolong survival significantly against advanced‐stage Colon26 tumors.
In summary, in the present study we have confirmed that TZT‐1027 has antivascular activity against tumor vasculature from our results of the disruption of the microtubule cytoskeleton, the enhanced vascular permeability in HUVEC monolayers and in tumor tissue, the induction of marked extravascular leakage of erythrocytes, and the reduction of tumor blood perfusion, in addition to the results in our previous study of the induction of membrane blebbing and cell detachment in HUVEC, a transient increase in hemoglobin in tumor tissue, and rapid tumoral hemorrhage.( 21 ) Therefore, we suggest that TZT‐1027 exerts both an indirect antivascular effect and a direct cytotoxic effect, resulting in strong antitumor activity in advance‐stage tumors, and that TZT‐1027 may be useful for treating solid tumors in the clinical setting.
Acknowledgments
The authors wish to thank Sayuri Sakano for her excellent technical assistance.
References
- 1. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors. Cancer Res 1989; 49: 6449–65. [PubMed] [Google Scholar]
- 2. Denekamp J. Angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol 1993; 66: 181–96. [DOI] [PubMed] [Google Scholar]
- 3. Denekamp J. Vascular attack as a therapeutic strategy for cancer. Cancer Metastasis Rev 1990; 9: 267–82. [DOI] [PubMed] [Google Scholar]
- 4. Ruoslahti E. Targeting tumor vasculature with homing peptides from phage display. Semin Cancer Biol 2000; 10: 435–42. [DOI] [PubMed] [Google Scholar]
- 5. St Croix B, Rago C, Velculescu V et al. Genes expressed in human tumor endothelium. Science 2000; 289: 1197–202. [DOI] [PubMed] [Google Scholar]
- 6. Bibby MC, Double JA, Loadman PM, Duke CV. Reduction of tumor blood flow by flavone acetic acid: a possible component of therapy. J Natl Cancer Inst 1989; 81: 216–20. [DOI] [PubMed] [Google Scholar]
- 7. Zwi LJ, Baguley BC, Gavin JB, Wilson WR. Blood flow failure as a major determinant in the antitumor action of flavone acetic acid. J Natl Cancer Inst 1989; 81: 1005–13. [DOI] [PubMed] [Google Scholar]
- 8. Philpott M, Baguley BC, Ching LM. Induction of tumour necrosis factor‐α by single and repeated doses of the antitumour agent 5,6‐dimethylxanthenone‐4‐acetic acid. Cancer Chemother Pharmacol 1995; 36: 143–8. [DOI] [PubMed] [Google Scholar]
- 9. Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Combretastatin A‐4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res 1997; 57: 1829–34. [PubMed] [Google Scholar]
- 10. Tozer GM, Prise VE, Wilson J et al. Combretastatin A‐4 phosphate as a tumor vascular‐targeting agent: early effects in tumors and normal tissues. Cancer Res 1999; 59: 1626–34. [PubMed] [Google Scholar]
- 11. Blakey DC, Westwood FR, Walker M et al. Antitumor activity of the novel vascular targeting agent ZD6126 in a panel of tumor models. Clin Cancer Res 2002; 8: 1974–83. [PubMed] [Google Scholar]
- 12. Davis PD, Dougherty GJ, Blakey DC et al. ZD6126: a novel vascular‐targeting agent that causes selective destruction of tumor vasculature. Cancer Res 2002; 62: 7247–53. [PubMed] [Google Scholar]
- 13. Nihei Y, Suga Y, Morinaga Y et al. A novel combretastatin A‐4 derivative, AC‐7700, shows marked antitumor activity against advanced solid tumors and orthotopically transplanted tumors. Jpn J Cancer Res 1999; 90: 1016–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hill SA, Toze GM, Pettit GR, Chaplin DJ. Preclinical evaluation of the antitumour activity of the novel vascular targeting agent Oxi 4503. Anticancer Res 2002; 22: 1453–8. [PubMed] [Google Scholar]
- 15. Miyazaki K, Kobayashi M, Natsume T et al. Synthesis and antitumor activity of novel dolastatin 10 analogs. Chem Pharm Bull 1995; 43: 1706–18. [DOI] [PubMed] [Google Scholar]
- 16. Natsume T, Watanabe J, Fujio N, Miyasaka K, Kobayashi M. Characterization of the interaction of TZT‐1027a potent antitumor agent with tubulin. Jpn J Cancer Res 2000; 91: 737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watanabe J, Natsume T, Fujio N, Miyasaka K, Kobayashi M. Induction of apoptosis in human cancer cells by TZT‐1027, an antimicrotubule agent. Apoptosis 2000; 5: 345–53. [DOI] [PubMed] [Google Scholar]
- 18. Natsume T, Koh Y, Kobayashi M et al. Enhanced antitumor activities of TZT‐1027 against TNF‐α or IL‐6 secreting Lewis lung carcinoma in vivo . Cancer Chemother Pharmacol 2002; 49: 35–47. [DOI] [PubMed] [Google Scholar]
- 19. Watanabe J, Minami M, Kobayashi M. Antitumor activity of TZT‐1027 (Soblidotin). Anticancer Res 2006; 26: 1973–82. [PubMed] [Google Scholar]
- 20. Kobayashi M, Natsume T, Tamaoki S et al. Antitumor activity of TZT‐1027a novel dolastatin 10 derivative. Jpn J Cancer Res 1997; 88: 316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otani M, Natsume T, Watanabe J et al. TZT‐1027, an antimicrotubule agent, attacks tumor vasculature and induces tumor cell death. Jpn J Cancer Res 2000; 91: 837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galbraith SM, Chaplin DJ, Lee F et al. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo . Anticancer Res 2001; 21: 93–102. [PubMed] [Google Scholar]
- 23. Micheletti G, Poli M, Borsotti P et al. Vascular‐targeting activity of ZD6126, a novel tubulin‐binding agent. Cancer Res 2003; 63: 1534–7. [PubMed] [Google Scholar]
- 24. Blakey DC, Ashton SE, Westwood FR, Walker M, Ryan AJ. ZD6126: a novel small molecule vascular targeting agent. Int J Radiat Oncol Biol Phys 2002; 54: 1497–502. [DOI] [PubMed] [Google Scholar]
- 25. Geran RI, Greenberg NH, Macdonald MM, Schumacher AM, Abott BJ. Protocols for Screening Chemical Agents and Natural Products Against Animal Tumor and Other Biochemical Systems. Cancer Chemother Rep , 1972; 3: 1–103. [Google Scholar]
- 26. Lal BK, Varma S, Pappas PJ, Hobson RW 2nd, Duran WN. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric‐oxide synthase, and MAP kinase pathways. Microvasc Res 2001; 62: 252–62. [DOI] [PubMed] [Google Scholar]
- 27. Kanthou C, Tozer GM. The tumor vascular targeting agent combretastatin A‐4‐phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood 2002; 99: 2060–9. [DOI] [PubMed] [Google Scholar]
- 28. Laws AL, Matthew AM, Double JA, Bibby MC. Preclinical in vitro and in vivo activity of 5,6‐dimethylxanthenone‐4‐acetic acid. Br J Cancer 1995; 71: 1204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Natsume T, Watanabe J, Koh Y et al. Antitumor activity of TZT‐1027 (Soblidotin) against vascular endothelial growth factor‐secreting human lung cancer in vivo . Cancer Sci 2003; 94: 826–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baguley BC, Holdaway KM, Thomsen LL, Zhuang L, Zwi LJ. Inhibition of growth of colon 38 adenocarcinoma by vinblastine and colchicine: evidence for a vascular mechanism. Eur J Cancer 1991; 27: 482–7. [DOI] [PubMed] [Google Scholar]
- 31. Hill SA, Lonergan SJ, Denekamp J, Chaplin DJ. Vinca alkaloids: anti‐vascular effects in a murine tumour. Eur J Cancer 1993; 29: 1320–4. [DOI] [PubMed] [Google Scholar]
- 32. Chaplin DJ, Pettit GR, Hill SA. Anti‐vascular approaches to solid tumour therapy: evaluation of combretastatin A4 phosphate. Anticancer Res 1999; 19: 189–95. [PubMed] [Google Scholar]
- 33. Siemann DW, Rojiani AM. Enhancement of radiation therapy by the novel vascular targeting agent ZD6126. Int J Radiat Oncol Biol Phys 2002; 53: 164–71. [DOI] [PubMed] [Google Scholar]
- 34. Siemann DW, Rojiani AM. The vascular disrupting agent ZD6126 shows increased antitumor efficacy and enhanced radiation response in large, advanced tumors. Int J Radiat Oncol Biol Phys 2005; 62: 846–53. [DOI] [PubMed] [Google Scholar]
- 35. Siemann DW, Mercer E, Lepler S, Rojiani AM. Vascular targeting agents enhance chemotherapeutic agent activities in solid tumor therapy. Int J Cancer 2002; 99: 1–6. [DOI] [PubMed] [Google Scholar]
- 36. Goto H, Yano S, Matsumori Y, Ogawa H, Blakey DC, Sone S. Sensitization of tumor‐associated endothelial cell apoptosis by the novel vascular‐targeting agent ZD6126 in combination with cisplatin. Clin Cancer Res 2004; 10: 7671–6. [DOI] [PubMed] [Google Scholar]
- 37. Bilenker JH, Flaherty KT, Rosen M et al. Phase I trial of combretastatin A‐4 phosphate with carboplatin. Clin Cancer Res 2005; 11: 1527–33. [DOI] [PubMed] [Google Scholar]
- 38. Siemann DW, Chaplin DJ, Horsman MR. Vascular‐targeting therapies for treatment of malignant disease. Cancer 2004; 100: 2491–9. [DOI] [PubMed] [Google Scholar]