

Abstract
(Cancer Sci 2010; 101: 774–781)
Proinflammatory cytokines and growth factors have been thought to play crucial roles in the pathology of acute myelogenous leukemia (AML) by supporting the proliferation and survival of AML cells in an autocrine and paracrine manner, although further elucidation is required. JTE‐607 was originally identified as a multiple cytokine inhibitor that suppresses production of proinflammatory cytokines from lipopolysaccharide (LPS)‐stimulated peripheral blood mononuclear cells. Herein, we report that JTE‐607 exhibits inhibitory activity on the growth of AML cell lines accompanying reduction of the proinflammatory cytokine and growth factor production. In in vitro studies, JTE‐607 suppressed expression and production of cytokines, which are spontaneously up‐regulated in AML cell lines. JTE‐607 also abrogated proliferation of AML cells in a concentration range in which colony formation of normal bone marrow cells was not affected. The growth inhibition by JTE‐607 was characterized by induction of cell‐cycle arrest at the S‐phase and apoptosis, accompanied by a decrease in c‐Myc and increase in p21waf1/cip1. In a leukemia model engrafted with U‐937 cells, JTE‐607 significantly prolonged survival in mice and reduced human cytokine mRNA levels in the bone marrow. These results suggest the usefulness of JTE‐607 in therapeutic applications for patients with hypercytokinemia and aggressive AML cell proliferation.
Although advances in dosage regimens of chemotherapy with conventional cytotoxic agents have improved the prognosis of acute myelogenous leukemia (AML), serious difficulties still remain in the treatment. Approximately 60–80% of young adults with AML achieve complete remission, but due to relapse the 5‐year disease‐free survival rate is only 30–40%.( 1 , 2 ) The outcome in older adult patients is more disappointing, with a lower complete remission induction rate and disease‐free survival, and many cannot tolerate the high burden of chemotherapy and allogenic bone marrow transplantation. Therefore, intensive efforts continue to develop novel therapeutic approaches to prevent recurrence and provide beneficial treatment even after relapse.
The pathogenesis of AML has been implicated in functional and genetic alterations of molecules involved in proliferation, differentiation, survival, and self‐renewal.( 3 , 4 ) Among the malignant phenotypes caused by these heterogeneous alterations, aberrant production of proinflammatory cytokines and growth factors is one of the typical functional alterations of AML cells.( 5 , 6 , 7 ) Several lines of evidence suggest that autocrine and paracrine signaling by these cytokines/growth factors in bone marrow microenvironment potentiate the anomalous growth, anti‐apoptotic phenotype, and resistance to anti‐leukemic agents in AML cells, leading to deterioration of AML.( 8 , 9 , 10 , 11 ) Indeed, the increased production of growth factors from AML blasts in vitro and a strong relationship between levels of proinflammatory cytokines in serum and prognosis of AML have been reported.( 8 , 12 , 13 , 14 ) In addition, increased production of growth factors by leukemic blasts promotes bone marrow neoangiogenesis in a paracrine manner.( 9 , 15 , 16 ) Thus, targeting the cytokine/growth factor network is thought to be a new approach to overcome the limitations of current chemotherapy for AML.
JTE‐607 [(‐)‐Ethyl‐N‐{3,5‐dichloro‐2‐hydroxy‐4‐[2‐(4‐methylpiperazin‐1‐yl)ethoxy]benzoyl}‐L‐phenylalaninate dihydrochloride] (Supplementary Fig. S1) was originally discovered as a cytokine inhibitor that broadly suppresses production of proinflammatory cytokines such as interleukin (IL)‐1β, IL‐6, IL‐8, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), and tumor necrosis factor (TNF)‐α from lipopolysaccharide (LPS)‐stimulated human peripheral blood mononuclear cells (PBMC) at IC50 values on the order of 10−9 m.( 17 ) The inhibitory activity of JTE‐607 is myeloid cell‐specific, and no inhibition was observed in the cytokine production from CD3/CD28‐stimulated T cells.( 17 ) In in vivo studies, JTE‐607 improved the survival rate in a cecal ligation and puncture‐induced mouse septic shock model and showed inhibitory effect on burn insult‐induced mouse lung injury.( 18 , 19 ) Based on these characteristics of JTE‐607 and the presumed importance of cytokines in the pathology of AML, we explored the effect of JTE‐607 on AML cell line U‐937 and found that JTE‐607 had ability to suppress both IL‐8 production and cell proliferation and prolonged the survival of mice in a U‐937‐grafted sever combined immunodeficiency (SCID) mouse acute leukemia model.( 20 ) In the present study, we further examined the inhibitory profile of JTE‐607 on spontaneous and stimulated cytokine/growth factor production from AML cells in vitro and in vivo. We also compared its inhibitory activity on the growth of various AML cell lines in vitro with those of conventional cytotoxic agents, and found unique sequential events including modulation of p21waf1/cip1 protein level, followed by cell‐cycle arrest and apoptosis. These results, together with the result of in vivo experiments, underscore the potential of JTE‐607 to afford a new approach in the therapy of AML.
Materials and Methods
Reagents. JTE‐607 was chemically synthesized by Central Pharmaceutical Research Institute, Japan Tobacco Inc. (Osaka, Japan). Cytosine‐1‐β‐D(+)‐arabinofuranoside (Cytarabine) and Daunorubicin hydrochloride were purchased from Wako Pure Chemical Industries (Osaka, Japan). For in vitro studies, the compounds were dissolved in dimethylsulfoxide (DMSO) and diluted with cell culture medium (final concentration of DMSO, 0.1%). For in vivo studies, JTE‐607 was dissolved in 30% hydroxypropyl β‐cyclodextrin aqueous solution. Antibodies to p15ink4b, p18ink4c, p21waf1/cip1, p27kip1, p53, cyclin‐dependent kinase (CDK) 2, CDK4, CDK6, cyclin D3, cyclin E, c‐Myc, E2F‐1, and α‐tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against total retinoblastoma protein (Rb) and phospho‐Rb were from Cell Signaling Technology (Beverly, MA, USA).
Cell culture. Human myeloid leukemia cell lines U‐937, HL‐60, and THP‐1 were obtained from Dainippon Sumitomo Pharma (Osaka, Japan), and KG‐1, Kasumi‐1, and Kasumi‐3 were from American Type Culture Collection (Rockville, MD, USA). Human myelodysplastic syndrome cell line SKM‐1 was obtained from the Health Science Research Resources Bank (Osaka, Japan). The cells were maintained in RPMI‐1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin, except for KG‐1 cells, which were grown in Iscove’s Modified Dulbecco’s medium with 20% FBS.
Determination of cytokine mRNA and protein levels. U‐937 and HL‐60 cells were exposed to JTE‐607 for 2 h for the determination of cytokine mRNA levels. Total RNA was extracted using the GenElute Mammalian Total RNA Kit (Sigma‐Aldrich, St. Louis, MO, USA), and human IL‐8, IL‐6, and vascular endothelial growth factor (VEGF) mRNA levels were determined by quantitative reverse transcriptase–polymerase chain reaction (RT‐PCR) using an ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and the values were normalized to human glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA levels. All of the specific primers and probes were purchased from Applied Biosystems. For the measurement of cytokine protein levels in the culture supernatant, the cells were exposed to JTE‐607 for 24 h in the absence or presence of phorbol myristate acetate (PMA; 10 ng/mL) or LPS (10 μg/mL). The protein levels of human IL‐6, IL‐8, and VEGF were measured using specific enzyme‐linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA).
Human bone marrow CFU‐GM colony formation assay. Human bone marrow mononuclear cells were purchased from Cambrex Bio Sciences Walkersville (Walkersville, MD, USA). The cells were suspended in semi‐solid culture medium containing recombinant human IL‐3, IL‐6, G‐CSF, GM‐CSF, stem cell factor (Methocult H4535; StemCell Technologies, Vancouver, Canada), and compound to be tested, then cultured for 14 days at 37°C. The colony‐forming unit‐granulocyte/macrophage (CFU‐GM) was counted under a microscope on the last day of culture. All measurements were performed in triplicate, and the IC50 value was calculated.
Cell cycle analysis and detection of apoptosis. U‐937 cells were exposed to various concentrations of JTE‐607 for 1–2 days. For cell cycle analysis, the cells were stained with propidium iodide (PI) using the Cycletest PLUS kit (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) in accordance with the manufacturer’s instructions. The PI fluorescence was measured using a FACSort flow cytometer (Becton Dickinson), and the data were analyzed using FlowJo software (version 4.6.1; Tree Star, Ashland, OR, USA). The proportions of cells in the G0/G1, S, and G2/M phases were determined using the Watson pragmatic algorithm. For the detection of apoptosis, the cells were washed with Annexin V binding buffer and stained with fluorescein isothiocyanate (FITC)‐labeled anti‐Annexin V and PI solution following the manufacturer’s instructions (BD Biosciences Pharmingen, San Diego, CA, USA), then analyzed using the flow cytometer. Annexin V‐positive, PI‐negative cells were determined as the cells undergoing apoptosis.
Preparation of protein extracts and Western blotting. U‐937 cells treated with the indicated concentrations of JTE‐607 were lysed with NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA, USA) containing 0.05 m dithiothreitol. After determination of the protein content with an RCDC protein assay kit (Bio‐Rad Laboratories, Hercules, CA, USA), the lysates were applied on SDS‐PAGE gel (NuPAGE 10% Bis‐Tris gel or 3–8% NuPAGE Tris‐Acetate gel; Invitrogen) and Western blotting was performed using polyvinylidene difluoride membranes (Immobilon‐P; Millipore, Bedford, MA, USA) and antibodies to each cell cycle‐related protein. Horseradish peroxidase‐based chemiluminescence by antimouse or antirabbit second antibodies was developed with enhanced chemiluminescence in accordance with the manufacturer’s recommendations (Amersham Biosciences, Buckinghamshire, UK), and the protein bands were detected using an LAS 3000 Image Analyzer (Fujifilm Corporation, Tokyo, Japan).
U‐937 engrafted SCID mouse AML model. Preparation of the U‐937‐grafted SCID mouse AML model has been described previously.( 20 ) In brief, 8‐week‐old female SCID mice (Fox Chase C.B‐17/Icr‐scidJcl; Clea Japan, Tokyo, Japan) were injected with anti‐asialo GM1 antibody (Wako Pure Chemical Industries) intraperitoneally to deplete natural killer cells. On the next day, the mice were exposed to sub‐lethal total body irradiation at a dose of 3 Gy using a soft X‐ray ionization chamber (M‐150WE; Softex Corporation, Tokyo, Japan) and then inoculated intravenously with U‐937 cells (106 cells/animal) in 200 μL of Hank’s balanced salt solution (day 0). JTE‐607 solution was administered subcutaneously three times daily for 14 days, starting from day 0. General condition, body weight, and survival of the mice were monitored and mean survival time was calculated. In the bolus treatment study, the SCID mice engrafted with U‐937 cells were subcutaneously injected once with JTE‐607 (100 mg/kg) or vehicle solution on day 18. One hour after the injection, bone marrow cells were collected from mouse femurs and tibias, and then total RNA was extracted. The levels of human cytokine mRNA were determined by quantitative RT‐PCR. All procedures for animals were reviewed and approved by the Animal Care and Management Committee of Central Pharmaceutical Research Institute, Japan Tobacco Inc.
Statistical analysis. Statistical significance between the vehicle‐treated control group and drug administration groups was determined using the Kaplan–Meier method and the log‐rank test for the mean survival time (days) using SAS software (version 8.2; SAS Institute Japan, Tokyo, Japan). A P‐value of <0.05 was considered to be statistically significant.
Results
Inhibition of cytokine production from cultured AML cell lines by JTE‐607. Because of the specific activity of JTE‐607 on cytokine production from myeloid cells and the predictive involvement of IL‐6, IL‐8, and VEGF in the pathology of AML,( 6 , 7 , 8 , 9 , 12 , 13 , 14 ) we examined the effect of JTE‐607 on the spontaneous production of these cytokines using AML cell lines. U‐937 and HL‐60 cells were exposed to 0.01–1 μm of JTE‐607 for 2 h in the measurement of cytokine mRNA levels or 24 h to determine cytokine protein levels in the culture supernatant. As shown in Figure 1A, IL‐6 and IL‐8 mRNA levels were reduced by JTE‐607 from a concentration of 0.1 μm in both cell lines. Accordingly, IL‐8 protein levels in the culture supernatant of both cell lines were reduced by JTE‐607 (Fig. 1B). IL‐6 protein levels were below the detection limit of ELISA in the supernatants of 24‐h cultures.
Figure 1.
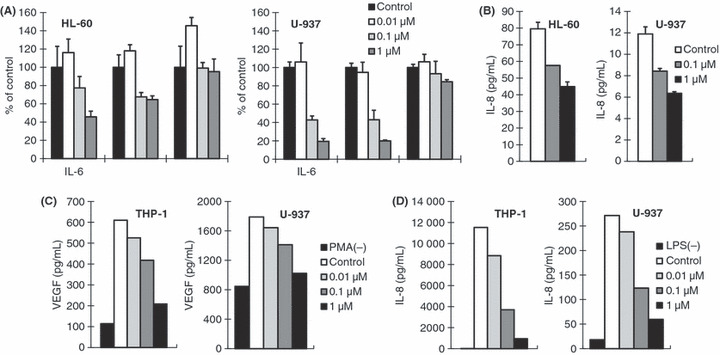
Inhibitory effect of JTE‐607 on spontaneous and lipopolysaccharide (LPS)‐stimulated cytokine production from cultured acute myelogenous leukemia (AML) cells. (A) HL‐60 and U‐937 cells were exposed to JTE‐607 (0.01 to 1 μm) for 2 h. Interleukin (IL)‐6, IL‐8, and vascular endothelial growth factor (VEGF) mRNA levels were determined by quantitative RT‐PCR, and the values normalized to GAPDH mRNA levels are shown. Each bar represents the mean ± SEM of triplicate measurements. (B) HL‐60 and U‐937 cells were exposed to JTE‐607 (0.1 and 1 μm) for 24 h. IL‐8 protein levels in the culture supernatants were measured by ELISA. Each bar represents the mean ± SEM of triplicate measurements. (C,D) THP‐1 and U‐937 cells were stimulated with phorbol myristate acetate (PMA) (C, 10 ng/mL) or LPS (D, 10 μg/mL) in the presence of JTE‐607 at indicated concentrations for 24 h. VEGF (C) and IL‐8 (D) protein levels in the culture supernatants were measured by ELISA. Each bar represents the mean of duplicate measurements.
In the case of VEGF, its spontaneous expression was not affected by JTE‐607 (Fig. 1A). Accordingly, no alteration was observed in VEGF protein levels in the 24‐h culture supernatants of tested cell lines including THP‐1 (data not shown). However, we found that THP‐1 and U‐937 cells produced significant amounts of VEGF in response to PMA stimulation, and that JTE‐607 effectively suppressed this increased production (Fig. 1C). Similar inhibition by JTE‐607 was observed in LPS‐stimulated IL‐8 production from THP‐1 and U‐937 cells (Fig. 1D). These results indicate that JTE‐607 has ability to inhibit both the spontaneous and stimulated production of proinflammatory cytokines and growth factors from AML cell lines, even though the activity is not as efficient as that observed in LPS‐stimulated PBMC.( 17 )
Inhibitory effects of JTE‐607, Cytarabine, and Daunorubicin on proliferation of various AML or MDS cells, and human bone marrow CFU‐GM colony formation. The inhibition of spontaneous cytokine production by JTE‐607 led us to hypothesize that JTE‐607 could interfere with the constitutively activated cellular responses that were ascribed to proliferation and survival of AML cells. To explore this idea, we next examined the inhibitory effect of JTE‐607 on the proliferation of human AML and myelodysplastic syndrome (MDS) cell lines. U‐937, HL‐60, THP‐1, KG‐1, Kasumi‐1, Kasumi‐3, and SKM‐1 cells were cultured for 3 days in the presence of JTE‐607, Cytarabine, or Daunorubicin. In parallel, the effects of these compounds on human CFU‐GM colony formation were examined using normal human bone marrow cells. The results showed significant inhibitory activity of JTE‐607 on the proliferation of AML and MDS cells with IC50 values ranging from 0.043–0.80 μm, except for Kasumi‐3 (Table 1). Cytarabine and Daunorubicin showed potent inhibitory activities on the proliferation of the cell lines with IC50 values of 0.0036–0.19 and 0.0041–0.055 μm, respectively. However, while both of the conventional anti‐leukemic agents also suppressed CFU‐GM at similar concentrations to those in the proliferation assay, the inhibitory effect of JTE‐607 on CFU‐GM was apparently weak. As shown in Table 1, the selectivity ratio of IC50 values in both assays indicates that inhibitory activity of JTE‐607, but neither Cytarabine nor Daunorubicin, is selective for the proliferation of leukemic cells compared to CFU‐GM, except for the case of Kasumi‐3. These data strongly suggest a favorable anti‐AML activity of JTE‐607 not accompanied by bone‐marrow suppression in vivo.
Table 1.
Inhibitory effects of JTE‐607, Cytarabine, and Daunorubicin on proliferation of AML and MDS cell lines, and human bone marrow CFU‐GM colony formation
| JTE‐607 | Cytarabine | Daunorubicin | ||||
|---|---|---|---|---|---|---|
| IC50 (μmol/L) | Ratio | IC50 (μmol/L) | Ratio | IC50 (μmol/L) | Ratio | |
| hCFU‐GM | 2.2 ± 0.1 | — | 0.0059 ± 0.0003 | — | 0.0068 ± 0.0006 | — |
| U‐937 | 0.043 ± 0.002 | 51 | 0.0041 ± 0.0003 | 1.4 | 0.010 ± 0.002 | 0.68 |
| HL‐60 | 0.058 ± 0.01 | 38 | 0.014 ± 0.001 | 0.42 | 0.0041 ± 0.00008 | 1.7 |
| THP‐1 | 0.18 ± 0.02 | 12 | 0.19 ± 0.02 | 0.031 | 0.017 ± 0.0005 | 0.40 |
| KG‐1 | 0.80 ± 0.3 | 2.8 | 0.011 ± 0.002 | 0.54 | 0.019 ± 0.0004 | 0.36 |
| Kasumi‐1 | 0.11 ± 0.02 | 20 | 0.0036 ± 0.0002 | 1.6 | 0.011 ± 0.002 | 0.62 |
| Kasumi‐3 | 4.0 ± 0.6 | 0.6 | 0.0045 ± 0.0004 | 1.3 | 0.055 ± 0.003 | 0.12 |
| SKM‐1 | 0.26 ± 0.01 | 8.5 | 0.024 ± 0.005 | 0.25 | 0.015 ± 0.001 | 0.45 |
Human bone marrow mononuclear cells were incubated for 14 days in semi‐solid culture medium with or without various concentrations of test compound, and formed colony‐forming unit‐granulocyte/macrophage (CFU‐GM) colonies were counted. Acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) cells were cultured for 3 days in the presence of the compound, and were pulsed with [3H]‐thymidine during the last 6 h of the culture. The amount of [3H]‐thymidine incorporated was measured as an indicator of cell proliferation. Each IC50 value represents the mean ± SEM of three independent experiments. Ratio = IC50 from human CFU‐GM assay/IC50 from cell proliferation assay.
Induction of apoptosis, cell‐cycle arrest, and alteration of cell cycle‐related protein levels by JTE‐607. We next characterized the suppressive effect of JTE‐607 on the growth of AML cells in terms of apoptosis and cell‐cycle arrest at the molecular level. To evaluate whether JTE‐607 induces apoptosis, U‐937 cells cultured in the presence or absence of JTE‐607 were stained with FITC‐conjugated anti‐Annexin V and PI. The result of flow cytometry analysis showed that Annexin V‐positive/PI‐negative apoptotic cells and PI‐positive necrotic cells emerged after a 2‐day exposure to JTE‐607, and the proportion of those cells was increased concentration dependently (Fig. 2A). In the cell‐cycle analysis, the proportion of the cells in the S‐phase was increased when U‐937 cells were exposed to over 0.1–0.3 μm of JTE‐607 for 1 day (Fig. 2B).
Figure 2.
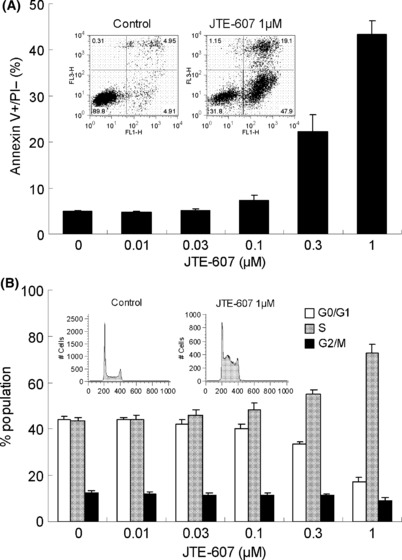
Induction of apoptosis and cell‐cycle arrest by JTE‐607. (A) U‐937 cells were cultured with JTE‐607 for 2 days, and were stained FITC‐labeled anti‐Annexin V and PI, then analyzed using a flow cytometer. The bars represent the mean ± SEM of three independent experiments. (B) U‐937 cells were cultured with JTE‐607 for 1 day, and intracellular DNA levels were measured by flow cytometer after PI staining. Population in each cell‐cycle phase was analyzed by the Watson pragmatic method. The bars represent the mean ± SEM of three independent experiments.
To clarify the molecular basis responsible for the S‐phase arrest by JTE‐607, we analyzed the expression or phosphorylation levels of cell cycle‐related proteins. As shown in Figure 3(C), a significant decrease in c‐Myc and an increase in p21waf1/cip1 occurred after 2–48 h exposure with JTE‐607. The protein levels of CDKs, other INK family proteins, and the phosphorylation level of Rb we examined appeared not to be affected (Fig. 3A,B). We next examined effect of N‐acetyl‐L‐cysteine (LNAC) on the apoptosis in U‐937 cells induced by JTE‐607. The result showed abrogation of apoptosis by LNAC in a concentration‐dependent manner, indicating that reactive oxygen species (ROS) plays a crucial role in the process evoked by JTE‐607 (Fig. 3D). Together, these results suggest that JTE‐607 exerts its antiproliferative effect against AML cells through the sequential process including cell‐cycle arrest at the S‐phase and induction of apoptotic cell death, and that modulation of p21waf1/cip1 and accumulation of ROS are involved in this process.
Figure 3.
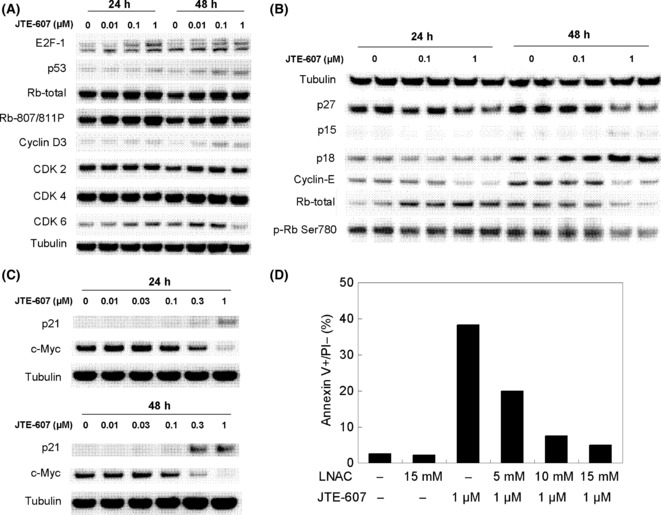
Alteration of cell cycle‐related protein levels induced by JTE‐607 and effect of N‐acetyl‐L‐cysteine (LNAC) on apoptosis in U‐937 cells. (A,B,C) U‐937 cells were cultured with indicated concentrations of JTE‐607 for 1 or 2 days, then cell cycle‐related proteins were detected by Western blotting. Panel (B) shows data from a duplicate experiment, and representative images from multiple experiments are shown in the panels. (D) U‐937 cells were cultured in the presence of JTE‐607 and/or indicated concentrations of LNAC for 2 days, and were stained FITC‐labeled anti‐Annexin V and PI, then analyzed using a flow‐cytometer. Each bar represents the mean of duplicate measurements.
Suppression of in vivo cytokine production by JTE‐607 at the dosage effective in a U‐937‐grafted SCID mouse AML model. Because JTE‐607 exerts its biological activity in a human myeloid cell‐selective manner when compared with other cell types and species, including the mouse,( 17 ) a human AML cell‐grafted SCID mouse model is ideal to evaluate the efficacy of its direct activity on AML cells. We have previously demonstrated that continuous infusion of JTE‐607 significantly prolonged the survival of mice in a U‐937‐grafted SCID mouse AML model in which the survival time of the mice was strictly dependent on the number of AML cells proliferating in vivo.( 20 ) In order to elucidate the inhibition of growth and cytokine production of AML cells by JTE‐607 under the same experimental conditions, we verified the survival‐prolonging effect of bolus injections of JTE‐607, and then examined human cytokine mRNA levels in the bone marrow after one bolus injection of JTE‐607. Based on the pharmacokinetic characteristics of rapid clearance in vivo, JTE‐607 was administrated subcutaneously at doses of 10, 30, and 100 mg/kg three times daily for 14 days, starting on the day of U‐937 engraftment. JTE‐607 prolonged mean survival time (MST) dose‐dependently, and the effect appeared statistically significant at a dose of 100 mg/kg with approximately two‐fold prolongation, compared with that of the vehicle control (Fig. 4A, Table 2). In the next experiment, the mice bearing established leukemia were injected with JTE‐607 at 100 mg/kg on day 18 and sacrificed 1 h after the injection. Human cytokine mRNA levels in the bone marrow cells were then determined by quantitative RT‐PCR and normalized to human GAPDH mRNA levels. The results show that single injection of JTE‐607 dramatically and rapidly reduced human IL‐6 and IL‐8 mRNA (Fig. 4B); in contrast, human GAPDH mRNA levels were not altered (Supplementary Table S1). Moreover, it was observed that human VEGF mRNA levels were also reduced to a lesser extent than the two proinflammatory cytokines, consistent with observations in in vitro studies (Fig. 1). In aggregate, these data indicate that JTE‐607 is able to suppress both the growth and accompanying cytokine/growth factor production from AML cells significantly in an in vivo milieu.
Figure 4.
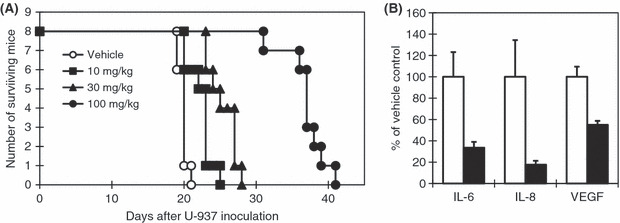
Effect of JTE‐607 on survival and levels of human cytokine mRNA in the bone marrow cells of U‐937‐engrafted sever combined immunodeficiency (SCID) mice. SCID mice were injected with anti‐asialo GM1 antibody intraperitoneally on day (−1). On day 0, the mice were exposed to total body X‐ray irradiation (3 Gy) and injected with U‐937 cells (106 cells/animal) intravenously. (A) JTE‐607 was administered subcutaneously at the indicated dose three times daily for 14 days, starting on day 0. Survival of the mice was monitored every day. (B) U‐937‐engrafted SCID mice were injected with JTE‐607 on day 18, at 100 mg/kg subcutaneously 1 h before collection of bone marrow cells. Human interleukin (IL)‐8, IL‐6, and vascular endothelial growth factor (VEGF) mRNA levels in the bone marrow cells were determined by quantitative RT‐PCR and the values normalized to human GAPDH mRNA levels are shown. Each bar represents the mean ± SEM of five animals.
Table 2.
Effect of JTE‐607 on mean survival time of U‐937 cell‐engrafted SCID mice
| Treatment | Mean survival time (days) | P‐values |
|---|---|---|
| Vehicle | 18.9 ± 0.2 | — |
| 10 mg/kg | 21.4 ± 0.6 | 0.7362 |
| 30 mg/kg | 24.5 ± 0.7 | 0.0781 |
| 100 mg/kg | 36.0 ± 1.0 | <0.0001 |
The data represent mean ± SEM of survival time (days) in Figure 4 (n = 8). P‐values compared to the vehicle control were calculated by the log‐rank test.
Discussion
The importance of autocrine signaling by proinflammatory cytokines and growth factors in AML blasts is well documented. The majority of AML blasts produced IL‐1, IL‐3, IL‐6, IL‐8, GM‐CSF, G‐CSF, VEGF, or TNF‐α at various levels,( 21 , 22 , 23 ) and a large fraction of blasts exhibited autonomous clonogenic proliferation by stimulation with these factors in vitro.( 24 , 25 , 26 , 27 ) In agreement with this, high levels of cytokines were detected in the blood of AML patients, and there is a correlation between the cytokine levels and prognosis of disease.( 12 , 13 , 14 ) In addition, inflammatory cytokines produced from AML cells can also stimulate production of growth factors and angiogenic factors from stromal cells in the bone marrow milieu, which support proliferation of AML cells and induce angiogenesis in a paracrine manner.( 8 , 9 ) Thus, suppression of cytokine production from AML cells is expected to demonstrate significant inhibition of AML cell growth and neo‐angiogenesis in the bone marrow.
JTE‐607 has a potent inhibitory activity on inflammatory cytokine production from PBMC by various stimulations, such as LPS, TNF‐α, and PMA, and as shown in Figure 1 it is also able to suppress the spontaneous production of proinflammatory cytokines. This activity of JTE‐607 cannot be ascribed to direct inhibition of kinases involved in the inflammatory signal transduction (Supplementary Table S2). Interestingly, the activity of JTE‐607 emerges as myeloid cell‐specific, for example selective abrogation of cytokine production was observed with monocyte and polymorphonuclear cells but not with lymphocytes, fibroblasts, or mesangial cells.( 17 ) In accordance, while we observed significant inhibition of the proliferation of AML and MDS cell lines by JTE‐607 (Table 1), sensitivity of lymphoma cells such as T cell and pre‐B cell acute lymphoblastic leukemia cell lines to JTE‐607 emerged to be low, and no leukemic cell‐specificity was observed in comparison with the inhibition of human CFU‐GM (Supplementary Table S3). The fact that Kasumi‐3 is less sensitive against JTE‐607 is also supportive for the myelogenous specificity of JTE‐607 (Table 1), because Kasumi‐3 is an undifferentiated leukemic cell line expressing T‐cell markers and is classified to M0 in the French‐American‐British classification.( 28 ) These data suggest that both the inhibition of cytokine production and proliferation are attained by the same mechanism including a target molecule of JTE‐607, which specifically functions in myeloid cells.
Preferential suppression of growth of AML cells in comparison to that observed in CFU‐GM is one of the unique features of JTE‐607. One possible reason is the mitogenic signal‐dependency of AML cells. In addition to the fact that growth/inflammatory signals such as MAPK and nuclear factor‐kappa B (NF‐kB) are constitutively activated in AML cells,( 29 , 30 ) the dependency of AML cells on mitogenic/cytokine stimulation via these signals is also considered to be higher than those of normal hematopoietic cells.( 8 , 9 , 10 , 11 ) This is supported by other clinical observations that cytokine levels in the blood of AML patients altered depending on the disease condition and therapeutic intervention.( 12 , 13 , 14 ) As CFU‐GM mimics the physiological proliferation and differentiation of bone marrow hematopoietic cells, it is most likely that the difference in the dependency on mitogenic/cytokine signals causes considerable difference in the sensitivity to JTE‐607 between CFU‐GM and growth of AML cell lines. Furthermore, in analysis of cytokine production from human bone marrow cells, we found that JTE‐607 significantly suppressed LPS‐stimulated GM‐CSF production from CD14‐positive cells but not from CD14‐negative stromal cells, which support normal proliferation and differentiation of bone marrow hematopoietic cells in a paracrine manner (Supplementary Fig. S2). Thus, it is conceivable that the myeloid cell‐specificity of JTE‐607 augments selective suppression of growth of AML cells in vivo.
In the analysis of cell cycle‐related proteins in JTE‐607‐exposed AML cells, alterations in the levels of c‐Myc and p21waf1/cip1 were clearly demonstrated (Fig. 3). c‐Myc is well known to be induced by multiple mitogenic signals and to express pleiotropic functions toward cell‐cycle progression,( 31 , 32 ) and it was also reported that c‐Myc negatively regulates p21waf1/cip1 expression.( 33 , 34 ) Our observation of the reciprocal alterations in p21waf1/cip1 and c‐Myc protein levels agreed with this finding, and can be implicated in the depression of mitogenic signals by JTE‐607. The consequent overproduction of p21waf1/cip1 is thought to be a key event leading to growth arrest and apoptosis because of its pivotal roles in cell‐cycle control and cell fate decision.( 35 , 36 , 37 ) Numerous anti‐leukemic agents, such as Daunorubicin, Cytarabine, and histone deacetylase inhibitors, have been reported to exhibit their growth arresting effect with hypophosophorylation of Rb, upregulation of differentiation marker (e.g. CD11b expression), and elevation of p21waf1/cip1 protein.( 38 , 39 , 40 ) However, in these cases, p21waf1/cip1 is thought to behave as an anti‐apoptotic factor, by the fact that dysregulation of p21waf1/cip1 by antisense interference or MAPK inhibition enhanced apoptosis and decreased CD11b‐expressing cells during Cytarabine treatment.( 38 , 40 , 41 , 42 ) In contrast, JTE‐607 induced S‐phase arrest unaccompanied by alteration of phosphorylated RB and CDK protein levels, or increment of CD11b‐expressing cells (data not shown), suggesting a different role of p21waf1/cip1. S‐phase arrest has been thought to be a response to DNA replication impairments, and two triggering mechanisms have been proposed in p21waf1/cip1‐related S‐phase arrest: (1) direct inhibition of CDK2 by p21waf1/cip1; and (2) inhibition of DNA replication through p21waf1/cip1 binding to proliferating cell nuclear antigen (PCNA).( 43 , 44 , 45 ) Because the unchanged pRB levels indicate that the CDK activity was not altered by JTE‐607 (Fig. 3), repression of DNA synthesis via binding to PCNA by p21waf1/cip1 is likely to mediate the defects leading to S‐phase arrest.( 45 , 46 ) Moreover, we observed that the radical scavenger LNAC attenuated apoptosis induced by JTE‐607 in U‐937 cells (Fig. 3D), suggesting that ROS accumulation in AML cells takes place in the course of S‐phase arrest and apoptosis induced by JTE‐607. The findings accumulated recently suggest that mitogenic signals such as MAPK and NF‐kB, which are reported to be activated constitutively in AML cells,( 29 , 30 ) play crucial roles not only in the cell‐cycle progression (e.g. through induction of c‐Myc) but also in the anti‐apoptotic responses. For instance, NF‐kB suppresses apoptosis through several modes of action including induction of Mn‐superoxide dismutase and ROS scavenging.( 47 ) Therefore it is conceivable that inhibition of mitogenic signals by JTE‐607 readily evokes oxidative damages of macromolecules including DNA and impairments of DNA replication. Further experiments should be conducted to illustrate an overall picture of growth inhibition and apoptosis induction by JTE‐607.
In the in vivo study using a U‐937‐grafted SCID mouse AML model, JTE‐607 prolonged the survival of mice in a dose‐dependent fashion (Fig. 4). We also observed reductions in human cytokine mRNA levels in mouse bone marrow cells after a bolus injection of JTE‐607 without affect on human GAPDH mRNA levels, which represent the number of AML cells (Supplementary Table S1). Importantly, JTE‐607 also reduced the levels of VEGF mRNA, of which PMA‐stimulated but not spontaneous expression was suppressed by JTE‐607 in vitro. Similarly, although the spontaneous production level of IL‐8 is low in U‐937 cells, it was drastically up‐regulated by LPS stimulation, and became detectable in the mouse serum after the colonization of human leukemia cells in the host mice (Fig. 1).( 20 ) It is therefore considered that preconditioning of the mice triggers proinflammatory stimuli such as LPS leaked from the intestinal tract and crossreactive inflammatory cytokines released from the mouse tissues, and then accelerated cytokine production of AML cells. The three cytokines/growth factor we observed here were reported in recent studies to be implicated in aggressive malignant phenotype and microvascular formation in the bone marrow of AML patients.( 48 , 49 ) The importance of autocrine and paracrine VEGF signaling pathways in the pathology of AML has also been demonstrated using a mouse AML model.( 50 ) Taking these observations and the findings of the present study together, it is thought that JTE‐607 is a useful agent for elucidating the precise implication of cytokine/growth factor up‐regulation in aggressive growth and other malignant phenotypes of AML cells. In the same line, it is also worthwhile to investigate potential application of this approach to complications after bone marrow transplantation, such as hypercytokinemia caused by the pre‐conditioning and graft‐versus‐host disease.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Structure of JTE‐607.
Fig. S2. Effect of JTE‐607 on granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) production from lipopolysaccharide (LPS)‐stimulated human bone marrow cells.
Table S1. Effect of JTE‐607 on levels of human cytokine mRNA in the bone marrow cells of U‐937‐engrafted sever combined immunodeficiency (SCID) mice.
Table S2. Kinase inhibitory profiling of JTE‐607 against 50 kinases.
Table S3. Inhibitory effects of JTE‐607, Cytarabine, and Daunorubicin on proliferation of T cell and pre‐B cell acute lymphoblastic leukemia cell lines.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank Yukiya Hirata for technical support, Korekiyo Wakitani and Kunio Iwata for their critical comments, and Hiromitsu Watanabe for his research coordination over the course of this study.
References
- 1. Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med 1999; 341: 1051–62. [DOI] [PubMed] [Google Scholar]
- 2. Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005; 106: 1154–63. [DOI] [PubMed] [Google Scholar]
- 3. Licht JD, Sternberg DW. The molecular pathology of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2005; 137–42. [DOI] [PubMed] [Google Scholar]
- 4. Renneville A, Roumier C, Biggio V et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 2008; 22: 915–31. [DOI] [PubMed] [Google Scholar]
- 5. Tobler A, Moser B, Dewald B et al. Constitutive expression of interleukin‐8 and its receptor in human myeloid and lymphoid leukemia. Blood 1993; 82: 2517–25. [PubMed] [Google Scholar]
- 6. Sugiyama H, Inoue K, Ogawa H et al. The expression of IL‐6 and its related genes in acute leukemia. Leuk Lymphoma 1996; 21: 49–52. [DOI] [PubMed] [Google Scholar]
- 7. Hsu HC, Lee YM, Tsai WH et al. Circulating levels of thrombopoietic and inflammatory cytokines in patients with acute myeloblastic leukemia and myelodysplastic syndrome. Oncology 2002; 63: 64–9. [DOI] [PubMed] [Google Scholar]
- 8. Ryningen A, Wergeland L, Glenjen N, Gjertsen BT, Bruserud Ø. In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine networks results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leuk Res 2005; 29: 185–96. [DOI] [PubMed] [Google Scholar]
- 9. Padró T, Bieker R, Ruiz S et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR‐2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002; 16: 1302–10. [DOI] [PubMed] [Google Scholar]
- 10. Hatfield K, Ryningen A, Corbascio M, Bruserud O. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int J Cancer 2006; 119: 2313–21. [DOI] [PubMed] [Google Scholar]
- 11. Kiss C, Benko I, Kovács P. Leukemic cells and the cytokine patchwork. Pediatr Blood Cancer 2004; 42: 113–21. [DOI] [PubMed] [Google Scholar]
- 12. Aguayo A, Kantarjian HM, Estey EH et al. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer 2002; 95: 1923–30. [DOI] [PubMed] [Google Scholar]
- 13. De Bont ES, Vellenga E, Molema G, Van Wering E, De Leij LF, Kamps WA. A possible role for spontaneous interleukin‐8 production by acute myeloid leukemic cells in angiogenesis related processes: work in progress. Med Pediatr Oncol 2001; 37: 511–7. [DOI] [PubMed] [Google Scholar]
- 14. Hatfield KJ, Olsnes AM, Gjertsen BT, Bruserud Ø. Antiangiogenic therapy in acute myelogenous leukemia: targeting of vascular endothelial growth factor and interleukin 8 as possible antileukemic strategies. Curr Cancer Drug Targets 2005; 5: 229–48. [DOI] [PubMed] [Google Scholar]
- 15. Aguayo A, Kantarjian H, Manshouri T et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000; 96: 2240–5. [PubMed] [Google Scholar]
- 16. Padró T, Ruiz S, Bieker R et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 2000; 95: 2637–44. [PubMed] [Google Scholar]
- 17. Kakutani M, Takeuchi K, Waga I, Iwamura H, Wakitani K. JTE‐607, a novel inflammatory cytokine synthesis inhibitor without immunosuppression, protects from endotoxin shock in mice. Inflamm Res 1999; 48: 461–8. [DOI] [PubMed] [Google Scholar]
- 18. Iwamura H, Sato M, Wakitani K. Comparative study of glucocorticoids, cyclosporine A, and JTE‐607[(‐)‐ethyl‐N‐{3,5‐dichloro‐2‐hydroxy‐4‐[2‐(4‐methylpiperazin‐1‐yl)ethoxy]benzoyl}‐L‐phenylalaninate dihydrochloride] in a mouse septic shock model. J Pharmacol Exp Ther 2004; 311: 1256–63. [DOI] [PubMed] [Google Scholar]
- 19. Sasaki J, Fujishima S, Iwamura H, Wakitani K, Aiso S, Aikawa N. Prior burn insult induces lethal acute lung injury in endotoxemic mice: effects of cytokine inhibition. Am J Physiol Lung Cell Mol Physiol 2003; 284: L270–8. [DOI] [PubMed] [Google Scholar]
- 20. Uesato N, Fukui K, Maruhashi J, Tojo A, Tajima N. JTE‐607, a multiple cytokine production inhibitor, ameliorates disease in a SCID mouse xenograft acute myeloid leukemia model. Exp Hematol 2006; 34: 1385–92. [DOI] [PubMed] [Google Scholar]
- 21. Young DC, Griffin JD. Autocrine secretion of GM‐CSF in acute myeloblastic leukemia. Blood 1986; 68: 1178–81. [PubMed] [Google Scholar]
- 22. Murohashi I, Tohda S, Suzuki T, Nagata K, Yamashita Y, Nara N. Autocrine growth mechanisms of the progenitors of blast cells in acute myeloblastic leukemia. Blood 1989; 74: 35–41. [PubMed] [Google Scholar]
- 23. Dunbar CE, Browder TM, Abrams JS, Nienhuis AW. COOH‐terminal‐modified interleukin‐3 is retained intracellularly and stimulates autocrine growth. Science 1989; 245: 1493–6. [DOI] [PubMed] [Google Scholar]
- 24. Miyauchi J, Kelleher CA, Wong GG et al. The effects of combinations of the recombinant growth factors GM‐CSF, G‐CSF, IL‐3, and CSF‐1 on leukemic blast cells in suspension culture. Leukemia 1988; 2: 382–7. [PubMed] [Google Scholar]
- 25. Rodriguez‐Cimadevilla JC, Beauchemin V, Villeneuve L, Letendre F, Shaw A, Hoang T. Coordinate secretion of interleukin‐1β and granulocyte‐macrophage colony‐stimulating factor by the blast cells of acute myeloblastic leukemia: role of interleukin‐1 as endogenous inducer. Blood 1990; 76: 1481–9. [PubMed] [Google Scholar]
- 26. Murohashi I, Tohda S, Imai Y, Hirai Y, Nara N. Growth potentiating activity of endogenous production of interleukin‐1 and tumor necrosis factor alpha in blast cells of acute myeloblastic leukemia. Exp Hematol 1993; 21: 846–51. [PubMed] [Google Scholar]
- 27. Saily M, Koistinen P, Zheng A, Savolainen ER. Signaling through interleukin‐6 receptor supports blast cell proliferation in acute myeloblastic leukemia. Eur J Haematol 1998; 61: 190–6. [DOI] [PubMed] [Google Scholar]
- 28. Asou H, Suzukawa K, Kita K et al. Establishment of an undifferentiated leukemia cell line (Kasumi‐3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn J Cancer Res 1996; 87: 269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Milella M, Precupanu CM, Gregorj C et al. Beyond single pathway inhibition: MEK inhibitors as a platform for the development of pharmacological combinations with synergistic anti‐leukemic effects. Curr Pharm Des 2005; 11: 2779–95. [DOI] [PubMed] [Google Scholar]
- 30. Birkenkamp KU, Geugien M, Schepers H, Westra J, Lemmink HH, Vellenga E. Constitutive NF‐kB DNA‐binding activity in AML is frequently mediated by a Ras/PI3‐K/PKB‐dependent pathway. Leukemia 2004; 18: 103–12. [DOI] [PubMed] [Google Scholar]
- 31. Coller HA, Grandori C, Tamayo P et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A 2000; 97: 3260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoffman B, Amanullah A, Shafarenko M, Liebermann DA. The proto‐oncogene c‐myc in hematopoietic development and leukemogenesis. Oncogene 2002; 21: 3414–21. [DOI] [PubMed] [Google Scholar]
- 33. Seoane J, Le H, Massagué J. Myc suppression of the p21Cip1 cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002; 419: 729–34. [DOI] [PubMed] [Google Scholar]
- 34. Wu S, Cetinkaya C, Munoz‐Alonso MJ et al. Myc represses differentiation‐induced p21CIP1 expression via Miz‐1‐dependent interaction with the p21 core promoter. Oncogene 2003; 22: 351–60. [DOI] [PubMed] [Google Scholar]
- 35. Gartel AL, Tyner AL. The role of the cyclin‐dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1: 639–49. [PubMed] [Google Scholar]
- 36. Wu Q, Kirschmeier P, Hockenberry T et al. Transcriptional regulation during p21WAF1/CIP1‐induced apoptosis in human ovarian cancer cells. J Biol Chem 2002; 277: 36329–37. [DOI] [PubMed] [Google Scholar]
- 37. Liu S, Bishop WR, Liu M. Differential effects of cell cycle regulatory protein p21WAF1/Cip1 on apoptosis and sensitivity to cancer chemotherapy. Drug Resist Updat 2003; 6: 183–95. [DOI] [PubMed] [Google Scholar]
- 38. Wang Z, Wang S, Fisher PB, Dent P, Grant S. Evidence of a functional role for the cyclin‐dependent kinase inhibitor p21CIP1 in leukemic cell (U937) differentiation induced by low concentrations of 1‐beta‐D‐arabinofuranosylcytosine. Differentiation 2000; 66: 1–13. [DOI] [PubMed] [Google Scholar]
- 39. Radosevic N, Delmer A, Tang R, Marie JP, Ajchenbaum‐Cymbalista F. Cell cycle regulatory protein expression in fresh acute myeloid leukemia cells and after drug exposure. Leukemia 2001; 15: 559–66. [DOI] [PubMed] [Google Scholar]
- 40. Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS‐275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 . Cancer Res 2003; 63: 3637–45. [PubMed] [Google Scholar]
- 41. Freemerman AJ, Vrana JA, Tombes RM et al. Effects of antisense p21 (WAF1/CIP1/MDA6) expression on the induction of differentiation and drug‐mediated apoptosis in human myeloid leukemia cells (HL‐60). Leukemia 1997; 11: 504–13. [DOI] [PubMed] [Google Scholar]
- 42. Wang Z, Tuyle GV, Conrad D, Fisher PB, Dent P, Grant S. Dysregulation of the cyclin‐dependent kinase inhibitor p21WAF1/CIP1/MDA6 increases the susceptibility of human leukemia cells (U937) to 1‐β‐D‐arabinofuranosylcytosine‐mediated mitochondrial dysfunction and apoptosis. Cancer Res 1999; 59: 1259–67. [PubMed] [Google Scholar]
- 43. Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 2004; 5: 793–804. [DOI] [PubMed] [Google Scholar]
- 44. Kan Q, Jinno S, Kobayashi K, Yamamoto H, Okayama H. Cdc6 determines utilization of p21WAF1/CIP1‐dependent damage checkpoint in S phase cells. J Biol Chem 2008; 283: 17864–72. [DOI] [PubMed] [Google Scholar]
- 45. Gottifredi V, McKinney K, Poyurovsky MV, Prives C. Decreased p21 levels are required for efficient restart of DNA synthesis after S phase block. J Biol Chem 2004; 279: 5802–10. [DOI] [PubMed] [Google Scholar]
- 46. Ogryzko VV, Wong P, Howard BH. WAF1 retards S‐phase progression primarily by inhibition of cyclin‐dependent kinases. Mol Cell Biol 1997; 17: 4877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bubici C, Papa S, Dean K, Franzoso G. Mutual cross‐talk between reactive oxygen species and nuclear factor‐kappa B: molecular basis and biological significance. Oncogene 2006; 25: 6731–48. [DOI] [PubMed] [Google Scholar]
- 48. Negaard HF, Iversen N, Bowitz‐Lothe IM et al. Increased bone marrow microvascular density in haematological malignancies is associated with differential regulation of angiogenic factors. Leukemia 2009; 23: 162–9. [DOI] [PubMed] [Google Scholar]
- 49. Weigiel B, Ekberg J, Talasila KM, Jalili S, Persson JL. The role of VEGF and a functional link between VEGF and p27Kip1 in acute myeloid leukemia. Leukemia 2009; 23: 251–61. [DOI] [PubMed] [Google Scholar]
- 50. Dias S, Hattori K, Heissig B et al. Inhibition of both paracrine and autocrine VEGF/VEGFR‐2 signaling pathways is essential to induce long‐term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A 2001; 98: 10857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Structure of JTE‐607.
Fig. S2. Effect of JTE‐607 on granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) production from lipopolysaccharide (LPS)‐stimulated human bone marrow cells.
Table S1. Effect of JTE‐607 on levels of human cytokine mRNA in the bone marrow cells of U‐937‐engrafted sever combined immunodeficiency (SCID) mice.
Table S2. Kinase inhibitory profiling of JTE‐607 against 50 kinases.
Table S3. Inhibitory effects of JTE‐607, Cytarabine, and Daunorubicin on proliferation of T cell and pre‐B cell acute lymphoblastic leukemia cell lines.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item