

Abstract
Recent studies have shown that genetically engineered stem cells (GESTECs) to produce suicide enzymes that convert non‐toxic prodrugs to toxic metabolites selectively migrate toward tumor sites and reduce tumor growth. In the present study, we evaluated whether these GESTECs were capable of migrating to human ovarian cancer cells and examined the potential therapeutic efficacy of the gene‐directed enzyme prodrug therapy against ovarian cancer cells in vitro. The expression of cytosine deaminase (CD) or carboxyl esterase (CE) mRNA of GESTECs was confirmed by RT‐PCR. A modified transwell migration assay was performed to determine the migratory capacity of GESTECs to ovarian cancer cells. GESTECs (HB1.F3.CD or HB1.F3.CE cells) engineered to express a suicide gene (CD or CE) selectively migrated toward ovarian cancer cells. A [3H] thymidine incorporation assay was conducted to measure the proliferative index. Treatment of human epithelial ovarian cancer cell line (SKOV‐3, an ovarian adenocarcinoma derived from the ascites of an ovarian cancer patient) with the prodrugs 5‐fluorocytosine (5‐FC) or camptothecin‐11 (CPT‐11) in the presence of HB1.F3.CD or HB1.F3.CE cells resulted in the inhibition of ovarian cancer cell growth. Based on the data presented herein, we suggest that GESTECs expressing CD/CE may have a potent advantage to selectively treat ovarian cancers.
(Cancer Sci 2010; 101: 955–962)
Ovarian malignancies have a significant impact upon women’s health, but the mechanism(s) of the transformation of health cells into cancerous cells and the development of these lethal cancers remain unclear. In order to enhance the effectiveness of therapeutics for treating ovarian cancers, novel strategies are required.( 1 , 2 )
Since gene/prodrug systems can be designed to more selectively target tumor cells than normal cells,( 3 , 4 ) the application of enzyme/prodrug systems to minimize side‐effects has received much attention. The cytosine deaminase (CD)/5‐fluorocytosine (5‐FC) system,( 5 , 6 , 7 , 8 , 9 ) one of the gene‐directed enzyme/prodrug therapies (GEPT), metabolically converts nontoxic 5‐FC into the toxic metabolite 5‐fluorouracil (5‐FU)( 10 , 11 ) and inhibits DNA synthesis in cancer cells.( 12 , 13 ) The CD/5‐FC GEPT system has been applied to several types of cancers, including clinical trials for colorectal and prostate cancers.( 14 , 15 , 16 ) In addition, CPT‐11, which is hydrolyzed to a topoisomerase 1 inhibitor (SN‐38) by carboxyl esterase (CE), has been administered to cancer patients, including colorectal cancer patients, for decades.( 17 , 18 ) The application of the prodrug seems to reduce the toxicity in normal tissues, but there are potential problems with exogenous enzyme delivery in targeting tumor cells selectively.
Stem cells have recently received a great deal of attention for their clinical and therapeutic potential to treat human cancers. For instance, neural stem cells (NSCs) may have great tropic and therapeutic potential for human malignant tumors such as medulloblastomas and gliomas,( 19 , 20 , 21 ) suggesting that enzyme/prodrug therapy using NSCs can be a potent delivery system to target and eradicate tumor cells specifically following systemic prodrug administration. HB1.F3, a new cell line of human NSCs immortalized using a retroviral vector carrying v‐myc, has been generated from fetal telencephalon cells.( 22 ) This clonally isolated, multipotent human NSC line has the ability to self‐renew, and differentiate into cells of neuronal and glial lineages both in vivo and in vitro.( 23 ) In addition to the therapeutic potential of these NSCs to treat brain disorders, their inherent migratory and tumor‐tropic properties represent a novel and potentially powerful approach for the treatment of invasive tumors. As a delivery vehicle to target tumor cells and disseminate therapeutic gene products throughout tumor sites, these therapeutic NSCs may solve major obstacles facing current gene therapy strategies by selectively infiltrating tumor masses. Human NSCs generated from a single clone can be engineered to stably express a therapeutic suicide gene, i.e., CD or CE, to activate the prodrugs 5‐FC or CPT‐11, respectively, because they are homogeneous and can be expanded to large numbers in vitro. HB1.F3 cells, the parental cell line of HB1.F3.CD cells, migrate to subcutaneous xenografts of diverse solid tumors, including prostate, breast, melanoma, glioma, and neuroblastoma, indicating that these cell lines do not possess a tissue‐specific homing tendency, but could be useful therapeutically due to their tendency to migrate to tumor tissues in general.( 20 ) In this study, we investigated whether GESTECs have significant migrating capacity to selectively target human ovarian cancers, as well as the therapeutic value of a suicide/prodrug system in ovarian cancer therapy.
Materials and Methods
Cell culture. The human epithelial ovarian cancer cell line SKOV‐3, the mouse embryonic fibroblast cell line NIH 3T3, the breast cancer cell line MCF‐7, and the human endometrial carcinoma cell line Hec 1A were purchased from ATCC (American Type Cell Culture, Manassas, VA, USA) and cultured in DMEM/F12 (Sigma‐Aldrich Corp., St. Louis, MO, USA) supplemented with 10% FBS (Hyclone, Logan, UT, USA), 100 U/mL penicillin G, and 100 μg/mL streptomycin (Life Technologies, Inc., Rockville, MD, USA) at 37°C in a humidified atmosphere of 5% CO2‐95% air. Non‐tumorigenic SV40 Tag‐immortalized ovarian surface epithelium‐derived cell line (IOSE‐80) was cultured in the same condition as described above. Cells were trypsinized with 0.06% trypsin (1:250)/0.01% EDTA (Life Technologies, Inc.) in Mg2+/Ca2+‐free HBSS.
HB1.F3, immortalized human NSCs derived from human fetal telencephalon at 15 weeks of gestation by introducing a retroviral vector encoding v‐myc,( 22 , 24 ) HB1.F3.CD cells producing Escherichia coli CD, and HB1.F3.CE cells producing rabbit CE were cultured as described above and used in the present study.( 20 , 21 )
RNA extraction and reverse transcription‐polymerase chain reaction. Total RNA extracts were prepared using the TriZol Reagent (Invitrogen Life Technologies Carlsbad, CA, USA) and reverse transcribed from 2.5 ng of total RNA into cDNA using the First‐Strand cDNA Synthesis Kit (Amersham Pharmacia Biotech Inc., Oakville, ON, USA) following the manufacturer’s protocols. PCR was conducted to amplify the bacterial CD gene by touchdown PCR with the following primer sequences based on the published sequence of the gene.( 21 ) The rabbit CE and the human CE genes were amplified by touchdown PCR using primer sequences described in Table 1. Human glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and human actin was used to confirm equal loading. The PCR amplification for chemo‐attractant factors (ligands and receptors) and actin, a positive control, was performed for 27 cycles. The sense/antisense primers and the predicted sizes of the PCR products are presented in Table 1 and have been described previously.( 21 )
Table 1.
The oligonucleotide sequences of the primers used in this study and the predicted sizes of the PCR products
| mRNA | Oligo‐sequences (5′–3′) | Expected size (bp) |
|---|---|---|
| CD | ||
| Sense | GCGCGAGTCACCGCCAGCCACACCACGGC | 559 |
| Antisense | GTTTGTAATCGATGGCTTCTGGCTGC | |
| CE (rabbit) | ||
| Sense | TGCTGGGCTATCCACTCTCT | 237 |
| Antisense | CTCCAGCATCTCTGTGGTGA | |
| CE (human) | ||
| Sense | CACTCCTGCTGACTTGACCA | 182 |
| Antisense | CATCC CCTGT GCTGA AGAAT | |
| SCF | ||
| Sense | ACTTGGATTCTCACTTGCATTT | 505 |
| Antisense | CTTTCTCAGGACTTAATGTTGAAG | |
| c‐Kit | ||
| Sense | GCCCACAATAGATTGGTATTT | 570 |
| Antisense | AGCATCTTTACAGCGACAGTC | |
| CXCR4 | ||
| Sense | CTCTCCAAAGGAAAGCGAGGTGGACAT | 558 |
| Antisense | AGACTGTACACTGTAGGTGCTGAAATCA | |
| VEGF | ||
| Sense | AAGCCATCCTGTGTGCCCCTGATG | 377 |
| Antisense | GCTCCTTCCTCCTGCCCGGCTCAC | |
| VEGFR2 | ||
| Sense | ACGCTGACATGTACGGTCTAT | 438 |
| Antisense | GCCAAGCTTGTACCATGTGAG | |
| GAPDH | ||
| Sense | ATGTTCGTCATGGGTGTGAACCA | 351 |
| Antisense | TGGCA GGTTT TTCTA GACGG CAG | |
| Actin | ||
| Sense | GCCCAGAGCAAGAGAGGCAT | 509 |
| Antisense | GGCCATCTCTTGCTCGAAGT | |
Cell growth assay. Cell proliferation was monitored by [3H]thymidine incorporation, as previously described.( 25 ) To investigate the effect of 5‐FC and 5‐FU in ovarian cancer cells, cells were seeded in 24‐well plates and cultured in 0.5 mL medium with 10% FBS. After a 24 h preincubation, HB1.F3.CD cells were added to the cultures in medium containing 10% FBS and incubated for 24 h before treatment with 5‐FC or 5‐FU. On the day of treatment, 5‐FC and 5‐FU were diluted appropriately with medium and the cells were treated for 4 days, as described previously.( 21 ) To investigate the effect of CPT‐11 and SN‐38 on ovarian cancer cells, HB1.F3.CE cells in medium containing 10% FBS were incubated with SKOV‐3 cells for 24 h. The cells were then treated CPT‐11 or SN‐38 for 24 h. Following the drug treatment, cells were then incubated with medium containing 1 μCi [3H]thymidine (0.5 Ci/mmol; Amersham Pharmacia Biotech Inc.) for 16 h. The cells were washed three times with PBS and precipitated with 0.5 mL 10% trichloroacetic acid for 20 min at 4°C. The precipitate was washed in methanol twice and solubilized in 0.5 mL 0.1 N NaOH. The radioactivity was measured using a Tri‐Carb Liquid Scintillation Analyzer (Model 2100TR; Packard Instrument Com., Meriden, CT, USA).
In vitro migration assay. To investigate whether GESTECs are capable of migrating to ovarian cancer cells, SKOV‐3 cells (1 × 106) were plated in 100‐mm dishes and incubated for 24 h. The medium was changed to serum‐free medium and incubated for 24 h to prepare conditioned medium, which was added to 24‐well plates coated with fibronectin (250 μg/mL; Sigma‐Aldrich Corp., St. Louis, MO, USA) and incubated overnight. Transwell plates (8 μm; Falcon; Becton Dickinson, Franklin Lakes, NJ, USA) were placed in the 24‐well plates and HB1.F3.CD or CE cells were plated in the upper chambers of the transwell plates and cultured for 24 h. The upper side of the transwell membrane was then scraped off to remove cells that had not migrated into the membrane, and immersed in pre‐cooled (−20°C) methanol for 20 min. To visualize cells in the membrane, the inserts were placed in 0.2% crystal‐violet (Sigma‐Aldrich Corp.) dissolved in 2% methanol for 10 min at 37°C. The membranes were then rapidly washed three times with distilled water and examined microscopically.
HB1.F3.CE cells were labeled with CM‐DiI (chloromethylbenzamido‐1,1′‐dioctadecyl‐3,3,3′‐tetramethylindocarbocyanine perchlorate, Invitrogen, Burlington, ON) according to the manufacturer’s suggested procedure. Sterilized cover glasses were placed in 24‐well plates (Falcon; Becton Dickinson) and seeded with SKOV‐3 cells at a density of 1 × 104 cells and cultured for 24 h. The cells were treated with CPT‐11 for 24 h and stained by adding DAPI (4′,6‐diamidino‐2‐phenylindole, Invitrogen, Burlington, ON, USA). The cells stained with CM‐DiI and DAPI were examined by fluorescence microscopy.
Statistical analysis. The results of three independent experiments are presented as the mean ± SD. Statistical analysis was performed by one‐way anova followed by Dunnett’s or Tukey’s multiple comparison test. P < 0.05 was considered statistically significant.
Results
Identification of CD/CE in HB1.F3.CD/CE cells. The expression of CD was confirmed by RT‐PCR. The predicted PCR product of the CD mRNA was detected at 559 bp. As demonstrated in Figure 1a, the CD mRNA was detected in HB1.F3.CD, but not in HB1.F3 cells, indicating that HB1.F3.CD cells express the CD gene, enabling these cells to activate a prodrug (5‐FC) to the active form (5‐FU). In addition, the expression of rabbit CE mRNA in HB1.F3.CE cells was confirmed by touchdown RT‐PCR. HB1.F3 and SKOV‐3 cells were used as negative controls. The predicted PCR product of the CE mRNA was detected at 237 bp in HB1.F3.CE cells, but not in the parental HB1.F3 cells or in SKOV‐3 cells (Fig. 1b).
Figure 1.
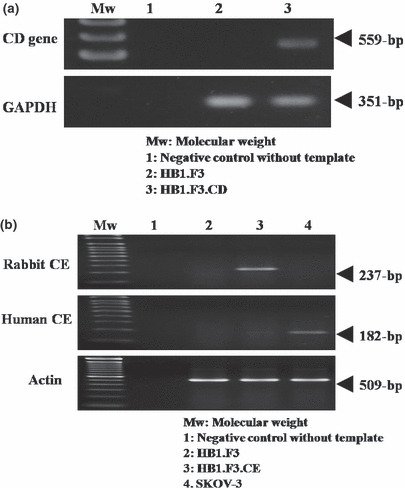
Transcriptional expression of a bacterial CD gene in HB1.F3.CD cells and a rabbit CE gene in HB1.F3.CE cells. The predicted 559 bp (CD) and 237 bp (CE) PCR products of the CD and CE genes were obtained by touchdown PCR. HB1.F3 CE cells. HB1. F3 and SKOV‐3 cells were prepared as described in Materials and Methods. (a) The expression of bacterial CD mRNA was confirmed in HB1.F3.CD cells. The HB1.F3 cells were used as negative control. (b) The expression of transfected rabbit CE mRNA was confirmed in HB1.F3 CE cells, but not in HB1. F3 and SKOV‐3 cells. GAPDH and actin were used as controls. Mw, Molecular weight standards.
In vitro migration assay. A modified transwell migration assay was performed to measure the capacity of GESTECs to migrate toward ovarian cancer cells. Conditioned medium from SKOV‐3 cells significantly increased cell migration of HB1.F3.CD and HB1.F3.CE, compared to that of NIH 3T3 cells (Fig. 2a). The number of cells which migrated toward ovarian cancer cell conditioned medium was quantified and analyzed in Figure 2b. Furthermore, the migrated cell number of GESTECs was compared to that of IOSE to determine that GESTECs have no migratory ability towards non‐tumorigenic ovarian cells. HB1.F3.CD cells significantly migrated to conditioned medium derived from SKOV‐3 cells, compared to the number of cell migrated to IOSE (Fig. 2c). We counted the number of migrated NIH3T3 cells in order to determine that only stem cells migrate toward ovarian cancer cells. The migrated cell number were counted and described in Figure 2d.
Figure 2.
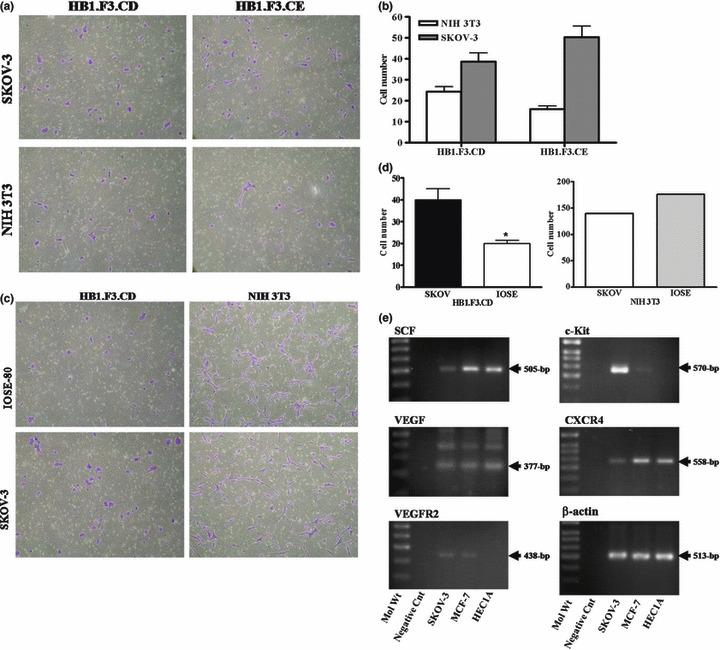
In vitro migration of HB1.F3.CD toward ovarian cancer cells. The migratory capacity was assessed by a modified transwell migration assay. The 24 well plates were pre‐coated with fibronectin and HB1.F3.CD or CE cells were added to the upper chamber of the insert after placing the transwell chamber in conditioned medium. (a and c) The conditioned medium from SKOV‐3, NIH 3T3, and IOSE‐80 cells was placed in the lower well of 24 well plates and HB1.F3.CD and CE cells (50 000/0.5 mL) were seeded in the upper well of the insert. The inserts were collected and stained as previously described. (b and d) The numbers of cells migrating into the membrane were counted using a light microscope (100×). Values are presented as the mean ± SD for three independent experiments. *P < 0.05 vs control. (e) Expression of factors potentially involved in chemo‐attractant or cell growth in cancers of the female reproductive tract. The PCR products of SCF, VEGF, VEGFR2, CXCR4, and c‐kit were obtained by RT‐PCR. Values are the mean ± SD for three independent experiments. *P < 0.05 vs NIH 3T3 cells.
This result indicates that the conditioned medium from the ovarian cancer cells may contain chemo‐attractant factors which accelerate the migration of the GESTECs HB1.F3.CD and HB1.F3.CE, thus enhancing the delivery of a therapeutic enzyme to human tumors in situ. To examine whether these ovarian cancer cells express chemo‐attractant factors, the expression of several chemo‐attractant ligands and their associated receptors was examined in SKOV‐3 cells by RT‐PCR. As seen in Figure 2e, chemo‐attractant ligands and receptors, i.e., SDF‐1/CXCR4, SCF/c‐Kit, and VEGF/VEGFR2, were strongly expressed in human ovarian cancer cells (SKOV‐3). Thus, these chemo‐attractant molecules and their respective receptors may play a role in the tumor‐tropic effects of GESTECs.
Effect of 5‐FC/5‐FU on ovarian cancer cells and GESTECs. To determine the effect of 5‐FC and 5‐FU on ovarian cancer cells, SKOV‐3 cells (2 × 104) were seeded and treated with 5‐FC at increasing concentrations (1, 10, and 100 μg/mL), and the proliferative index was measured using a thymidine incorporation assay. Treatment with 5‐FC for 4 days did not alter cell growth of SKOV‐3 cells at the doses used, compared to untreated controls (Fig. 3a). We also examined the cytotoxic effect of 5‐FC (a prodrug) on HB1.F3 and HB1.F3.CD cells. Treatment with 5‐FC induced a significant reduction in cell growth in HB1.F3.CD cells, but not in HB1.F3 cells, compared to untreated controls (Fig. 3a). To confirm the cytotoxic effects of the active drug in SKOV‐3 cells, cells were treated with 5‐FU (the active form), which resulted in a significant decrease of cell proliferation of SKOV‐3 cells (Fig. 3b).
Figure 3.
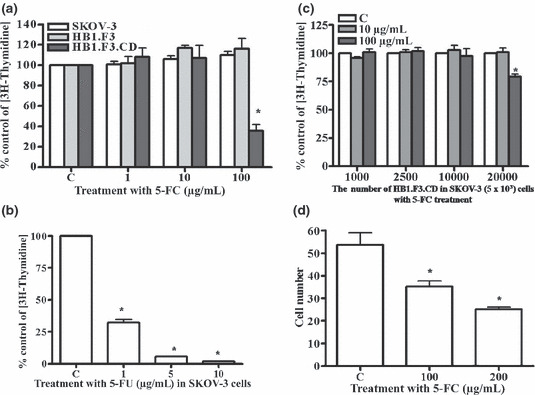
Effect of 5‐FC/5‐FU on cell proliferation. Proliferation levels at each concentration of 5‐FC or 5‐FU are expressed as relative fold change compared to controls. (a) SKOV‐3, HB1.F3, and HB1.F3.CD cells (2 × 104 cells per well) were placed in plates and treated with 5‐FC at the concentrations of 1, 10, or 100 μg/mL for 4 days. (b) SKOV‐3 cells were treated with increasing concentrations of 5‐FU (1, 5, or 10 μg/mL for 4 days). (c) SKOV‐3 cells (5 × 103) were seeded in 24 well plates. Following incubation for 24 h, increasing numbers of HB1.F3.CD cells were placed on top of inserts. After 24 h, the cells were treated with 5‐FC at a concentration of 10 μg/mL or 100 μg/mL for 4 days. (d) HB1.F3.CD cells (2 × 104 cells) cultured with SKOV‐3 cells (5 × 103 cells) were treated with different concentrations of 5‐FC (100 μg/mL or 200 μg/mL) for 4 days. Values are the mean ± SD for three independent experiments. *P < 0.05 compared with a control.
To investigate whether treatment with 5‐FC induces anti‐proliferative effects in ovarian cancer cells cultured in the presence of HB1.F3.CD cells, SKOV‐3 cells (5 × 103) were co‐cultured with different numbers of HB1.F3.CD cells and treated with 5‐FC (10 and 100 μg/mL). Treatment with 5‐FC (100 μg/mL) appeared to decrease cell growth of SKOV‐3 cells in the presence of a high density of HB1.F3.CD cells (Fig. 3c). To confirm the cell growth inhibition, cell numbers were counted following 5‐FC treatment. Treatment with 5‐FC resulted in a dose‐dependent inhibition of cell growth (Fig. 3d). These results demonstrate that HB1.F3.CD cells induce cell growth inhibition of ovarian cancer cells in the presence of a non‐toxic prodrug, 5‐FC, following conversion of the prodrug to the 5‐FU active form.
Effect of CPT‐11/SN‐38 on ovarian cancer cells and GESTECs. To determine the effect of CPT‐11 and SN‐38 on ovarian cancer cells and GESTECs, cells were treated with CPT‐11 or SN‐38 at low or high concentrations (1 and 100 μg/mL). The levels of proliferation of the CPT‐11/SN‐38 treated groups are expressed as percent change relative to the controls. Treatment with a low concentration of CPT‐11 (1 μg/mL) for 24 h does not alter the proliferation of SKOV‐3 cells compared to untreated controls (Fig. 4a), but treatment with a high concentration of CPT‐11 (100 μg/mL) decreased cell proliferation. Treatment of HB1.F3.CE cells with a low concentration of CPT‐11 (1 μg/mL) decreased proliferation compared to untreated controls (Fig. 4b). However, treatment with SN‐38 suppressed cell proliferation at all concentrations in SKOV‐3, HB1.F3, and HB1.F3.CE cells. To investigate whether co‐culture of CE‐producing GESTECs has an anti‐proliferative effect on ovarian cancer cells, SKOV‐3 cells (1 × 104 cells per well) were cultured with different numbers of HB1.F3.CE cells, or HB1.F3 cells. Treatment of SKOV‐3 cells with CPT‐11 in the presence of increasing numbers of HB1.F3.CE cells resulted in a significant decrease of proliferation in an HB1.F3.CE cell dependent manner (Fig. 4c).
Figure 4.
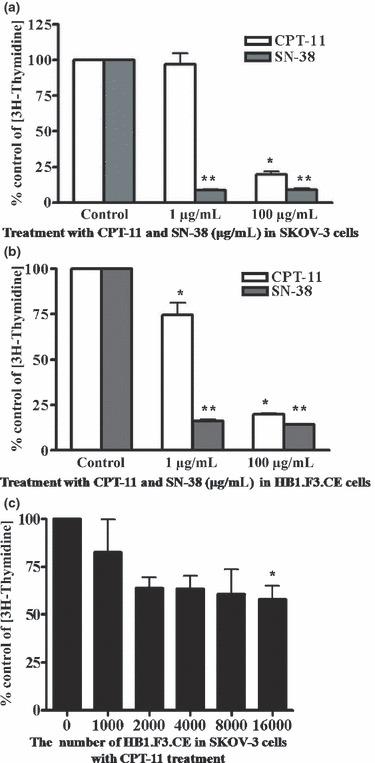
Effect of CPT‐11/SN‐38 on cell proliferation. (a) SKOV‐3 cells were treated with CPT‐11 or SN‐38 at a concentration of 1 μg/mL or 100 μg/mL for 24 h. (b) HB1.F3.CE cells were treated with CPT‐11. Proliferation levels at each concentration of CPT‐11 and SN‐38 are expressed as relative fold change. Values are the mean ± SD of two independent experiments. *P < 0.05 compared CPT‐11 controls; **P < 0.05 compared SN‐38 controls. (c) SKOV‐3 cells were seeded in 24 well plates. Following incubation for 24 h, increasing numbers of HB1.F3.CE cells were placed on the membrane of inserts. After 24 h, the cells were treated with CPT‐11 at a concentration of 1 μg/mL for 24 h. Values of (c) are presented as the mean ± SD of three independent experiments. *P < 0.05 compared to controls.
To confirm the effect of CPT‐11 on cell growth inhibition, the percentages of HB1.F3.CE cell and SKOV‐3 cell were examined by CM‐DiI staining following treatment with CPT‐11. HB1.F3.CE cells were labeled with CM‐DiI, and then co‐cultured with SKOV‐3 cells. Cells were treated with increasing doses of CPT‐11 and cell nuclei were stained with DAPI after 24 h. As seen in Figure 5a, the HB1.F3.CE cells stained with CM‐DiI were detected by fluorescence microscopy and the numbers of HB1.F3.CE and SKOV‐3 cells were quantified. Treatment with CPT‐11 at concentrations of 1 μg/mL or 10 μg/mL decreased the proliferation of both cell types (Fig. 5b). The number of HB1.F3.CE cells was compared to that of SKOV‐3 cells in order to determine the percentage of HB1.F3.CE and SKOV‐3 cells. No significant difference (P > 0.05) was seen in cell number for samples treated with CPT‐11 versus samples treated with vehicle (Fig. 5c).
Figure 5.
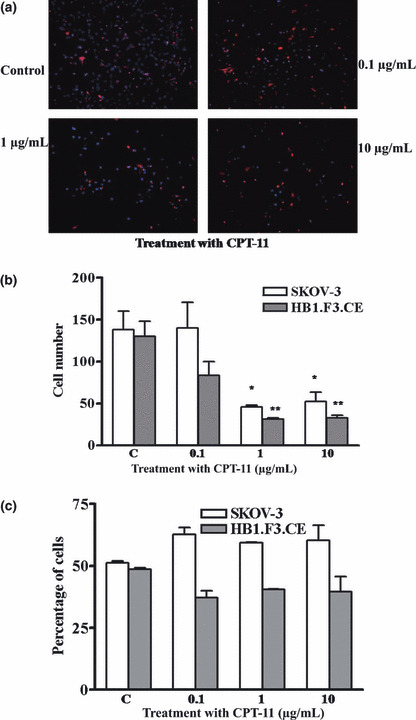
Effect of CPT‐11 on the percentage of HB1.F3.CE and SKOV‐3 cells. HB1.F3.CE cells were labeled with CM‐DiI and co‐cultured with SKOV‐3 cells at a density of 1 × 104 cells, respectively. (a) Cells were treated with CPT‐11 for 24 h in a dose‐dependent manner and stained with DAPI. CM‐DiI‐labeled HB1.F3.CE cells (red cells) are shown. (b) The numbers of HB1.F3.CE and SKOV‐3 cells were quantified and presented. (c) The percentage of HB1.F3.CE and SKOV‐3 in each treatment group of cells is shown. Values are the mean ± SD for three independent experiments. *P < 0.05 vs SKOV‐3 cell controls; **P < 0.05 vs HB1.F3.CE cell controls.
Discussion
Recent studies have found that immortalized GESTECs have advantages that may be useful for gene therapy and cell replacement therapy approaches to the treatment of neurological diseases and injuries.( 23 , 26 , 27 , 28 , 29 , 30 , 31 ) These GESTECs selectively migrate to brain tumors and reduced tumor growth both in vitro and in vivo.( 20 , 21 ) A previous study demonstrated that HB1.F3.CD cells expressing the mRNA of E. coli CD respond to the administration of 5‐FC and did not show any toxicity in an animal model.( 21 ) A recent study also reported that CE‐producing NSCs (HB1.F3.CE cells), which convert the prodrug CPT‐11 to SN‐38 selectively migrate to tumor cells and have a therapeutic effect on brain tumors.( 20 )
The CD/5‐FC derived GEPT system has been tested for therapy of some types of cancers, including breast, prostate, and colon,( 9 , 16 , 32 ) but the effect of GESTECs on many other types of cancer cells remains largely unknown. Thus, in this study, we examined whether GESTECs expressing CD/CE can be a used to selectively treat ovarian cancer. First, we confirmed the expression of the bacterial CD/rabbit CE genes in HB1.F3.CD/CE cells, which can convert 5‐FC to 5‐FU or CPT‐11 to SN‐38, respectively. We further performed a modified transwell migration assay to examine whether these GESTECs expressing CD/CE are capable of migrating to ovarian cancer cells. The migration of these cells increased in SKOV‐3 conditioned medium compared to NIH 3T3 conditioned medium, suggesting that these GESTECs likely respond to factors secreted by ovarian cancer cells. These results are in agreement with a previous report demonstrating that GESTECs migrate to brain tumors in an animal model.( 33 ) Additionally, HB1.F3 cells, the parent cell line of the HB1.F3.CD/CE cell lines, migrate to subcutaneous xenografts of diverse solid tumors, including prostate, breast, melanoma, glioma, and neuroblastoma, indicating that these cell lines do not possess a tissue‐specific homing tendency, but might be exploited for therapeutic use in treatment of many types of cancer.( 20 ) Although several factors, such as scatter factor (SCF; hepatocyte growth factor), stromal cell‐derived factor‐1 (SDF‐1), and vascular endothelial growth factor (VEGF), play a chemo‐attractive role( 34 , 35 , 36 , 37 , 38 ) in tumor cells, the molecular mechanism of the tumor‐tropism of GESTECs is not clearly understood. Thus, we further examined the expression of these genes in ovarian and breast cancer cells and found that chemo‐attractant ligands and receptors, i.e., SCF/c‐Kit, SDF‐1/CXCR4, and VEGF/VEGFR2, were strongly expressed in human ovarian cancer cells (SKOV‐3), as well as breast (MCF‐7) and endometrial (HEC1A) cancer cells. Thus, these chemo‐attractant molecules and their respective receptors may play a role in the tumor‐tropic effects that enable GESTECs to selectively deliver a suicide enzyme to the tumor. These factors also play important roles in the biology of ovarian cancer.( 39 , 40 , 41 ) A further study is required to confirm the role of these genes in the mechanism of tumor cell recognition and/or tumor tropism by GESTECs.
Although 5‐FU, a potent inhibitor of thymidylate synthetase,( 42 ) has been used in treatment of cancer patients over the past few decades, the systemic application of 5‐FU in therapy has been limited by its toxic effects, including myelosuppression and stomatitis, that can occur prior to achieving a therapeutic response. Therefore, the non‐toxic metabolite 5‐FC which is converted to 5‐FU by E. coli CD has received much attention in the last few years.( 21 , 43 ) In the present study, treatment with 5‐FC resulted in a decrease in cell growth with increasing numbers of HB1.F3.CD cells in co‐culture with SKOV‐3, indicating that the application of the CD/5FC GEPT system may have potential as a therapeutic application for the treatment of ovarian cancer. Since an earlier report showed that a small number of CD‐transfected cells could induce anti‐tumor effects through a bystander effect,( 44 ) we examined whether the number of GESTECs is important in modulating proliferation of ovarian cancer cells. Different numbers of GESTECs were co‐cultured with SKOV‐3 cells and treated with 5‐FC at various doses. HB1.F3.CD cells expressing CD gene decreased cell growth of the SKOV‐3 cells in the 1:4 ratios of SKOV‐3 cells/HB1.F3.CD cells, suggesting that the number of GESTECs in the enzyme/prodrug system needs to taken into consideration, in order to maximize the therapeutic benefit in a cancer therapy. To our knowledge, this is the first report of GESTEC‐induced anti‐proliferation using a GEPT system with ovarian cancer cells. On the other hand, treatment with 5‐FC at a higher dose resulted in a significant decrease in SKOV‐3 cell growth, suggesting a high dose of 5‐FC treatment can be associated with ovarian cancer cell‐toxicity. Interestingly, the same level of cell inhibition was not observed in the co‐culture of SKOV‐3 cells and HB1.F3.CD cells. We have no good explanation for this observation but this result shows that the ovarian cancer cell and neural stem cell may have a reciprocal action and further research is required.
Although SN‐38 and CPT‐11 constitute the first‐ and second‐line therapies for metastatic colorectal cancer,( 17 ) the direct application of SN‐38 can cause severe diarrhea and neutropenia causing extreme suppression of the immune system.( 18 , 45 ) Treatment with CPT‐11 at a lower concentration decreased cell proliferation in HB1.F3.CE cells, but not in SKOV‐3 cells. However, treatment of SKOV‐3 cells with CPT‐11 at high concentrations decreased cell proliferation, suggesting that even the prodrug CPT‐11 may have toxic effects at high doses. Moreover, treatment with CPT‐11 in SKOV‐3 cells culture in the presence of increasing numbers of HB1.F3.CE cells decreased the proliferation and the number of HB1.F3.CE and SKOV‐3 cells, indicating that NSCs transduced with the CE gene may enable the ablation of ovarian cancer cells following CPT‐11 administration. The CE approach for therapeutic effect seems to be more promising than the CD approach because the CE approach decreased proliferation with a lower HB1.F3.CE cell number at a lower concentration of CPT‐11 compared to that of the CD approach. This result may help to explain why CPT‐11 induced the cell inhibition of ovarian carcinoma cell lines ( 46 , 47 ) and has been used as the second line therapy for ovarian cancers.( 48 , 49 )
To confirm the “bystander effect”, in which not only the vehicle cells transduced with the suicide gene but also the untransfected neighboring target cells are eradicated,( 50 , 51 ) we counted HB1.F3.CE and SKOV‐3 cells, respectively, following treatment with CPT‐11. In this study, treatment with CPT‐11 not only decreased the number of HB1.F3.CE cells but also inhibited the growth of the untransfected neighboring ovarian cancer cells. Previous studies have reported that immune responses or apoptotic factors may be involved in GEPT induced bystander effects,( 52 , 53 ) and further approaches are needed to clarify the mechanism of the GEPT‐induced bystander effect in ovarian cancer cells. In our previous studies using neuroblastoma or intracranial medulloblastoma, CD/5FC or CE/CPT‐11 GEPT system induced anti‐tumor effect under in vivo conditions.( 20 , 21 ) A further in vivo study warranties to evaluate the clinical potential of GEPT system for ovarian cancers.
In summary, the results of this present study have shown that GESTECs expressing CD/CE have a potent advantage of selective migration toward ovarian cancer cells. Moreover, this GEPT system resulted in an anti‐proliferative effect on ovarian cancer cells, suggesting that these GESTECs expressing suicide genes combined with application of prodrugs may have therapeutic potential to selectively treat ovarian cancers.
Acknowledgments
This work was supported by the research grant of the Chungbuk National University in 2009. In addition, the authors appreciate National Cancer Institute of Canada (NCIC) and Canadian Breast Cancer Research Alliance (CBCRA) for research funding.
References
- 1. Egami T, Ohuchida K, Miyoshi K et al. Chemotherapeutic agents potentiate adenoviral gene therapy for pancreatic cancer. Cancer Sci 2009; 100: 722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fu W, Lan H, Liang S, Gao T, Ren D. Suicide gene/prodrug therapy using salmonella‐mediated delivery of Escherichia coli purine nucleoside phosphorylase gene and 6‐methoxypurine 2′‐deoxyriboside in murine mammary carcinoma 4T1 model. Cancer Sci 2008; 99: 1172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tubiana M. Tumor cell proliferation kinetics and tumor growth rate. Acta Oncol 1989; 28: 113–21. [DOI] [PubMed] [Google Scholar]
- 4. Saukkonen K, Hemminki A. Tissue‐specific promoters for cancer gene therapy. Expert Opin Biol Ther 2004; 4: 683–96. [DOI] [PubMed] [Google Scholar]
- 5. Evoy D, Hirschowitz EA, Naama HA et al. In vivo adenoviral‐mediated gene transfer in the treatment of pancreatic cancer. J Surg Res 1997; 69: 226–31. [DOI] [PubMed] [Google Scholar]
- 6. Hirschowitz EA, Ohwada A, Pascal WR, Russi TJ, Crystal RG. In vivo adenovirus‐mediated gene transfer of the Escherichia coli cytosine deaminase gene to human colon carcinoma‐derived tumors induces chemosensitivity to 5‐fluorocytosine. Hum Gene Ther 1995; 6: 1055–63. [DOI] [PubMed] [Google Scholar]
- 7. Lan KH, Kanai F, Shiratori Y et al. Tumor‐specific gene expression in carcinoembryonic antigen‐‐producing gastric cancer cells using adenovirus vectors. Gastroenterology 1996; 111: 1241–51. [DOI] [PubMed] [Google Scholar]
- 8. Kanai F, Lan KH, Shiratori Y et al. In vivo gene therapy for alpha‐fetoprotein‐producing hepatocellular carcinoma by adenovirus‐mediated transfer of cytosine deaminase gene. Cancer Res 1997; 57: 461–5. [PubMed] [Google Scholar]
- 9. Li Z, Shanmugam N, Katayose D et al. Enzyme/prodrug gene therapy approach for breast cancer using a recombinant adenovirus expressing Escherichia coli cytosine deaminase. Cancer Gene Ther 1997; 4: 113–7. [PubMed] [Google Scholar]
- 10. Austin EA, Huber BE. A first step in the development of gene therapy for colorectal carcinoma: cloning, sequencing, and expression of Escherichia coli cytosine deaminase. Mol Pharmacol 1993; 43: 380–7. [PubMed] [Google Scholar]
- 11. Mullen CA, Kilstrup M, Blaese RM. Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5‐fluorocytosine: a negative selection system. Proc Natl Acad Sci U S A 1992; 89: 33–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Etienne MC, Cheradame S, Fischel JL et al. Response to fluorouracil therapy in cancer patients: the role of tumoral dihydropyrimidine dehydrogenase activity. J Clin Oncol 1995; 13: 1663–70. [DOI] [PubMed] [Google Scholar]
- 13. Pinedo HM, Peters GF. Fluorouracil: biochemistry and pharmacology. J Clin Oncol 1988; 6: 1653–64. [DOI] [PubMed] [Google Scholar]
- 14. Crystal RG, Hirschowitz E, Lieberman M et al. Phase I study of direct administration of a replication deficient adenovirus vector containing the E. coli cytosine deaminase gene to metastatic colon carcinoma of the liver in association with the oral administration of the pro‐drug 5‐fluorocytosine. Hum Gene Ther 1997; 8: 985–1001. [DOI] [PubMed] [Google Scholar]
- 15. Freytag SO, Khil M, Stricker H et al. Phase I study of replication‐competent adenovirus‐mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res 2002; 62: 4968–76. [PubMed] [Google Scholar]
- 16. Chung‐Faye GA, Chen MJ, Green NK et al. In vivo gene therapy for colon cancer using adenovirus‐mediated, transfer of the fusion gene cytosine deaminase and uracil phosphoribosyltransferase. Gene Ther 2001; 8: 1547–54. [DOI] [PubMed] [Google Scholar]
- 17. Fuchs C, Mitchell EP, Hoff PM. Irinotecan in the treatment of colorectal cancer. Cancer Treat Rev 2006; 32: 491–503. [DOI] [PubMed] [Google Scholar]
- 18. Conti JA, Kemeny NE, Saltz LB et al. Irinotecan is an active agent in untreated patients with metastatic colorectal cancer. J Clin Oncol 1996; 14: 709–15. [DOI] [PubMed] [Google Scholar]
- 19. Aboody KS, Brown A, Rainov NG et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A 2000; 97: 12846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aboody KS, Bush RA, Garcia E et al. Development of a tumor‐selective approach to treat metastatic cancer. PLoS ONE 2006; 1: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim SK, Kim SU, Park IH et al. Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression. Clin Cancer Res 2006; 12: 5550–6. [DOI] [PubMed] [Google Scholar]
- 22. Kim SU, Nakagawa E, Hatori K, Nagai A, Lee MA, Bang JH. Production of immortalized human neural crest stem cells. Methods Mol Biol 2002; 198: 55–65. [DOI] [PubMed] [Google Scholar]
- 23. Kim SU. Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology 2004; 24: 159–71. [DOI] [PubMed] [Google Scholar]
- 24. Lee HJ, Kim KS, Kim EJ et al. Brain transplantation of immortalized human neural stem cells promotes functional recovery in mouse intracerebral hemorrhage stroke model. Stem Cells 2007; 25: 1204–12. [DOI] [PubMed] [Google Scholar]
- 25. Choi K‐C, Kang SK, Tai C‐J, Auersperg N, Leung PCK. Estradiol Up‐Regulates Antiapoptotic Bcl‐2 Messenger Ribonucleic Acid and Protein in Tumorigenic Ovarian Surface Epithelium Cells. Endocrinology 2001; 142: 2351–60. [DOI] [PubMed] [Google Scholar]
- 26. Rosser AE, Zietlow R, Dunnett SB. Stem cell transplantation for neurodegenerative diseases. Curr Opin Neurol 2007; 20: 688–92. [DOI] [PubMed] [Google Scholar]
- 27. Meng XL, Shen JS, Ohashi T, Maeda H, Kim SU, Eto Y. Brain transplantation of genetically engineered human neural stem cells globally corrects brain lesions in the mucopolysaccharidosis type VII mouse. J Neurosci Res 2003; 74: 266–77. [DOI] [PubMed] [Google Scholar]
- 28. Jeong SW, Chu K, Jung KH, Kim SU, Kim M, Roh JK. Human neural stem cell transplantation promotes functional recovery in rats with experimental intracerebral hemorrhage. Stroke 2003; 34: 2258–63. [DOI] [PubMed] [Google Scholar]
- 29. Kim SU, Park IH, Kim TH et al. Brain transplantation of human neural stem cells transduced with tyrosine hydroxylase and GTP cyclohydrolase 1 provides functional improvement in animal models of Parkinson disease. Neuropathology 2006; 26: 129–40. [DOI] [PubMed] [Google Scholar]
- 30. Ryu JK, Kim J, Cho SJ et al. Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiol Dis 2004; 16: 68–77. [DOI] [PubMed] [Google Scholar]
- 31. Lee ST, Chu K, Park JE et al. Intravenous administration of human neural stem cells induces functional recovery in Huntington’s disease rat model. Neurosci Res 2005; 52: 243–9. [DOI] [PubMed] [Google Scholar]
- 32. Boucher PD, Im MM, Freytag SO, Shewach DS. A novel mechanism of synergistic cytotoxicity with 5‐fluorocytosine and ganciclovir in double suicide gene therapy. Cancer Res 2006; 66: 3230–7. [DOI] [PubMed] [Google Scholar]
- 33. Barresi V, Belluardo N, Sipione S, Mudo G, Cattaneo E, Condorelli DF. Transplantation of prodrug‐converting neural progenitor cells for brain tumor therapy. Cancer Gene Ther 2003; 10: 396–402. [DOI] [PubMed] [Google Scholar]
- 34. Ehtesham M, Yuan X, Kabos P et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia 2004; 6: 287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun L, Hui AM, Su Q et al. Neuronal and glioma‐derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006; 9: 287–300. [DOI] [PubMed] [Google Scholar]
- 36. Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest 2004; 113: 1364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmidt NO, Przylecki W, Yang W et al. Brain tumor tropism of transplanted human neural stem cells is induced by vascular endothelial growth factor. Neoplasia 2005; 7: 623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beppu K, Jaboine J, Merchant MS, Mackall CL, Thiele CJ. Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J Natl Cancer Inst 2004; 96: 46–55. [DOI] [PubMed] [Google Scholar]
- 39. Martin L, Schilder R. Novel approaches in advancing the treatment of epithelial ovarian cancer: the role of angiogenesis inhibition. J Clin Oncol 2007; 25: 2894–901. [DOI] [PubMed] [Google Scholar]
- 40. Ueoka Y, Kato K, Wake N. Hepatocyte growth factor modulates motility and invasiveness of ovarian carcinomas via ras mediated pathway. Mol Cell Endocrinol 2003; 202: 81–8. [DOI] [PubMed] [Google Scholar]
- 41. Scotton CJ, Wilson JL, Scott K et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res 2002; 62: 5930–8. [PubMed] [Google Scholar]
- 42. Hartmann KU, Heidelberger C. Studies on fluorinated pyrimidines. XIII. Inhibition of thymidylate synthetase. J Biol Chem 1961; 236: 3006–13. [PubMed] [Google Scholar]
- 43. Miller CR, Williams CR, Buchsbaum DJ, Gillespie GY. Intratumoral 5‐fluorouracil produced by cytosine deaminase/5‐fluorocytosine gene therapy is effective for experimental human glioblastomas. Cancer Res 2002; 62: 773–80. [PubMed] [Google Scholar]
- 44. Huber BE, Austin EA, Richards CA, Davis ST, Good SS. Metabolism of 5‐fluorocytosine to 5‐fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc Natl Acad Sci U S A 1994; 91: 8302–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ando Y, Saka H, Ando M et al. Polymorphisms of UDP‐glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 2000; 60: 6921–6. [PubMed] [Google Scholar]
- 46. Miettinen S, Ylikomi T. Concomitant exposure of ovarian cancer cells to docetaxel, CPT‐11 or SN‐38 and adenovirus‐mediated p53 gene therapy. Anticancer Drugs 2009; 20: 589–600. [DOI] [PubMed] [Google Scholar]
- 47. Itamochi H, Kigawa J, Sultana H et al. Sensitivity to anticancer agents and resistance mechanisms in clear cell carcinoma of the ovary. Jpn J Cancer Res 2002; 93: 723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sugiyama T. [Second‐line chemotherapy for recurrent ovarian cancer]. Gan To Kagaku Ryoho 2005; 32: 28–32. [PubMed] [Google Scholar]
- 49. Kita T, Kikuchi Y, Kudoh K et al. Exploratory study of effective chemotherapy to clear cell carcinoma of the ovary. Oncol Rep 2000; 7: 327–31. [DOI] [PubMed] [Google Scholar]
- 50. Greco O, Dachs GU. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol 2001; 187: 22–36. [DOI] [PubMed] [Google Scholar]
- 51. Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986; 46: 5276–81. [PubMed] [Google Scholar]
- 52. Consalvo M, Mullen CA, Modesti A et al. 5‐Fluorocytosine‐induced eradication of murine adenocarcinomas engineered to express the cytosine deaminase suicide gene requires host immune competence and leaves an efficient memory. J Immunol 1995; 154: 5302–12. [PubMed] [Google Scholar]
- 53. Freeman SM, Abboud CN, Whartenby KA et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res 1993; 53: 5274–83. [PubMed] [Google Scholar]