

Abstract
Metronomic chemotherapy has been advocated recently as a novel chemotherapeutic regimen. Polyethylene glycol (PEG)‐coated liposomes are well known to accumulate in solid tumors by virtue of the highly permeable angiogenic blood vessels characteristic for growing tumor tissue, the so‐called “enhanced permeability and retention (EPR) effect”. To expand the range of applications and investigate the clinical value of the combination strategy, the therapeutic benefit of metronomic S‐1 dosing in combination with oxaliplatin (l‐OHP)‐containing PEG‐coated liposomes was evaluated in a murine colon carcinoma‐bearing mice model. S‐1 is an oral fluoropyrimidine formulation and metronomic S‐1 dosing is a promising alternative to infused 5‐FU in colorectal cancer therapy. Therefore, the combination of S‐1 with l‐OHP may be an alternative to FOLFOX (infusional 5‐FU/leucovorin (LV) in combination with l‐OHP), which is a first‐line therapeutic regimen of a colorectal carcinoma. The combination of oral metronomic S‐1 dosing with intravenous administration of liposomal l‐OHP formulation exerted excellent antitumor activity without severe overlapping side‐effects, compared with either metronomic S‐1 dosing, free l‐OHP or liposomal l‐OHP formulation alone or metronomic S‐1 dosing plus free l‐OHP. We confirmed that the synergistic antitumor effect is due to prolonged retention of l‐OHP in the tumor on account of the PEG‐coated liposomes, presumably via alteration of the tumor microenvironment caused by the metronomic S‐1 treatment. The combination regimen proposed here may be a breakthrough in treatment of intractable solid tumors and an alternative to FOLFOX in advanced colorectal cancer therapy with acceptable tolerance and preservation of quality of life (QOL). (Cancer Sci 2010; 101: 2470–2475)
Oxaliplatin (l‐OHP), an innovative third generation platinum compound, has powerful anti‐neoplastic competence with no cross drug resistance with cisplatin and carboplatin( 1 , 2 ) However, l‐OHP shows relatively low anticancer effectivity when it is administered alone, because it shows poor accumulation in tumor tissues due to a high plasma protein binding ratio and high partitioning to erythrocytes, while in addition displaying peripheral neurotoxicity due to high protein binding in the tissue.( 3 ) These features stand in the way of an effective continuous treatment with l‐OHP. Oxaliplatin is frequently used for treatment of advanced colorectal cancer when combined with fluorouracil (5‐FU) and leucovorin (LV) (FOLFOX).( 4 , 5 )
Chemotherapy using nanocarriers as a delivery system has been developed to improve the success of clinical treatment of solid tumors by achieving high accumulation of the chemotherapeutic agent in tumor tissues but with limited accumulation in healthy tissues. Polyethylene glycol (PEG)‐modified (PEG‐coated) liposomes show prolonged circulating times and thereby enhanced accumulation in solid tumors by virtue of the increased vascular permeability observed in tumor angiogenic blood vessels (the so‐called “enhanced permeability and retention (EPR) effect”).( 6 ) Therefore, it is reasonable to assume that PEG‐coated liposomes may improve the pharmacokinetic features of l‐OHP and enhance its anticancer efficiency. We and other groups have shown therapeutic improvement of l‐OHP by encapsulation in PEG‐coated targeted liposomes.( 7 , 8 , 9 )
However, nanocarriers of various designs have repeatedly shown insufficient delivery of their payloads to solid tumors. One of the major limitations is insufficiency of the EPR effect due to a disordered intratumoral microenvironment represented for instance by hypovascularity. Recently, Kano et al. ( 10 ) reported that treatment with transforming growth factor‐β type 1 receptor (TβR‐1) inhibitor resulted in increased accumulation of nanocarriers accompanied by a pronounced antitumor response in a murine solid tumor model. ten Hagen and co‐workers( 11 , 12 ) have reported a similar observation with tumor necrosis factor α (TNF‐α) in both rat and murine models. These approaches are regarded as a breakthrough that can overcome the insufficient EPR effect for nanocarriers in solid tumors. An obvious drawback of this approach is that TβR‐1 inhibitor and TNF‐α cannot be readily applied because these compounds have not yet been approved for clinical use worldwide.
Metronomic chemotherapy, which refers to the frequent administration of chemotherapeutics at doses significantly below the maximum tolerated dose (MTD) without prolonged drug‐free breaks, is a novel approach to the control of advanced cancer.( 13 , 14 ) The therapy shows a potent anti‐angiogenetic effect by targeting genetically stable endothelial cells within the tumor vascular bed, rather than tumor cells with a high mutation rate. Drugs that can be administered orally, such as cyclophosphamide (CPA), capecitabine, UFT and S‐1, would meet the requirements of prolonged daily administration schedules. Recently, we showed that metronomic CPA dosing augments intratumoral accumulation of co‐administered doxorubicin (DXR)‐containing PEG‐coated liposomes and this combination exerted an excellent antitumor activity in a murine tumor model without overlapping severe side‐effects.( 15 , 16 ) This favorable therapeutic effect might be attributed to the following mechanism: metronomic dosing with CPA makes the newly forming tumor vessels leaky, and thereby enhances intratumoral accumulation of PEG‐coated liposomes.
Oxaliplatin administered together with infusions of 5‐FU and LV (FOLFOX) has become a standard treatment regimen for advanced colorectal cancer.( 4 , 5 ) To extend our approach described above, in the present study we evaluated l‐OHP/5‐FU synergy by combination of metronomic S‐1 dosing (orally, daily) with l‐OHP‐containing PEG‐coated liposomes (intravenously, once a week) in a murine colorectal cancer model. S‐1 consists of tegaful (a prodrug of 5‐FU), 5‐chloro‐2, 4‐dihydroxypyridine (CDHP: an inhibitor of 5‐FU degradation) and potassium oxonate (Oxo: a reducer of gastrointestinal toxicity) at a molar ratio of 1:0.4:1. S‐1 is one of the most frequently used drugs for oral administration in Japan and shows less toxic side‐effects than 5‐FU.( 17 , 18 , 19 ) The biochemical modulation of S‐1 leads to prolonged retention of 5‐FU in the blood, which mimics the pharmacokinetic profile of infusional 5‐FU. Daily oral administration with S‐1 meets the concept of metronomic dosing and is assumed to enhance intratumoral accumulation of PEG‐coated liposomes and l‐OHP associated with the liposome.
Materials and Methods
Materials. Hydrogenated soy phosphatidylcholine (HSPC) and 1, 2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐n‐(methoxy[polyethylene glycol]‐2000) (mPEG2000‐DSPE) were generously donated by NOF (Tokyo, Japan). Cholesterol (CHOL) was purchased from Wako Pure Chemical (Osaka, Japan). S‐1 and l‐OHP were generously donated by Taiho Pharmaceutical (Tokyo, Japan). DiI (1, 1′‐dioctadecyl‐3, 3, 3′,3′‐tetramethyl‐indocarbocyanine perchlorate) and DiD (1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl‐indodicarbocyanine perchlorate) were purchased from Invitrogen (Paisley, UK). 3H‐Cholesterylhexadecyl ether (3H‐CHE) was purchased from Perkin Elmer Japan (Yokohama, Japan). All other reagents were of analytical grade.
Preparation of l‐OHP‐containing PEG‐coated liposomes. l‐OHP‐containing PEG‐coated liposomes, composed of HSPC/CHOL/mPEG2000‐DSPE (2/1/0.2, molar ratio), were prepared using a reverse‐phase evaporation method as described earlier.( 9 ) Unencapsulated, free l‐OHP was removed by dialysis by means of a dialysis cassette (Slyde‐A‐Lyzer, 10000MWCO; Pierce, Rockford, IL, USA) against 5% dextrose. Encapsulated l‐OHP was quantified using an atomic absorption photometer (Z‐5700; Hitachi, Tokyo, Japan). The phospholipid concentration was determined by colorimetric assay.( 20 ) The particle size of the liposomes was 180 ± 52 nm, as determined with a NICOMP 370 HPL submicron particle analyzer (Particle Sizing System, Mountain View, CA, USA). The encapsulation efficiency of l‐OHP was calculated by dividing the drug to lipid ratio after the dialysis by the initial drug to lipid ratio and was approximately 20%. These values are three times higher than that reported recently by another group.
Animal and tumor cell. Male BALB/c mice, 5 weeks old, were purchased from Japan SLC (Shizuoka, Japan). All animal experiments were evaluated and approved by the Animal and Ethics Review Committee of the University of Tokushima.
The Colon 26 (C26) murine colorectal carcinoma cell line was purchased from Cell Resource Center for Biomedical Research (Institute of Development, Aging and Cancer, Tohoku University, Sendai, Japan). To develop tumor‐bearing mice, C26 cells (2 × 106) were inoculated subcutaneously into the back of BALB/c mice.
Combination therapy with S‐1 and l‐OHP formulation. Treatments began when the tumor volumes reached a volume of 40–60 mm3. The day treatment began was defined as day 0. The dosing schedule of each chemotherapeutic treatment was as follows:
1 Metronomic S‐1 dosing: S‐1 (6.9 mg tegafur/kg/dose) was administered orally every day from day 0 to day 21.
2 Free or liposomal l‐OHP dosing: Free or liposomal l‐OHP (4.2 mg/kg/dose) was intravenously administered at day 0, 7 and 14.
3 Combination dosing (S‐1 plus free or liposomal l‐OHP): S‐1 (6.9 mg tegafur/kg per dose) was orally administered daily from day 0 to day 21. Either free or liposomal l‐OHP (4.2 mg/kg per dose) was intravenously administered at day 0, 7 and 14.
Tumors were measured externally every third day. Tumor volume was approximated by using formula A below. The antitumor activity was determined by evaluating the change of the relative tumor volume (formula B) and the tumor growth inhibition rate was calculated through formula C.
 |
Effect of S‐1 dosing on blood clearance and tumor accumulation of l‐OHP encapsulated in PEG‐coated lipo‐somes. Treatment with S‐1 (6.9 mg tegafur/kg, daily, 7 days) was started when the tumor volumes reached a volume of 40–60 mm3. To evaluate blood clearance and tumor accumulation of l‐OHP encapsulated in PEG‐coated liposomes, the liposomal l‐OHP formulation (4.2 mg l‐OHP/kg) was intravenously injected right after the final S‐1 administration. At 6, 12, 24, 36, 48, 72 and 120 h post‐injection of l‐OHP formulation, plasma was collected, and then the mice were killed to remove the tumor. Tumors were harvested and weighed. Thereafter, concentrated nitric acid was added to the tumor or plasma (100 μL), which was then digested in a microwave oven (600 W for 25 min at 50°C ETHOS TC; Milestone general, Kanagawa, Japan). The content of platinum (Pt) in the plasma and tumor was measured using an ICP‐MS (Agilent 7500 series; YOKOKAWA analytical systems, Tokyo, Japan). Europium was added to the assay mixture and calibration standards, respectively. The l‐OHP concentrations were calculated from ion counts Pt using the calibration method with internal standard correction. The l‐OHP concentration in the tumor was expressed as μg l‐OHP per g tissue. Pharmacokinetic parameters were calculated on the basis of l‐OHP concentration using poly‐exponential curve fitting and the least‐squares parameter estimation program SAAM II (SAAM Institute, Seattle, WA, USA).
Effect of S‐1 dosing on biodistribution of PEG‐coated lipo‐somes. Treatment with S‐1 (6.9 mg tegafur/kg, daily, 7 days) was started when the tumor volumes had reached a volume of 40–60 mm3. To assess the biodistribution of PEG‐coated liposomes, 3H‐CHE‐labeled liposomes (25 mg total lipid/kg) were intravenously injected right after the final S‐1 administration. At 24 h after liposome injection, samples (tumor, blood [100 μL], heart, lung, liver, spleen, kidney) were collected. Tissue samples were washed and weighed after removing excess fluid. Radioactivity in the samples was assayed as described previously.( 22 )
Effect of S‐1 dosing on tumor accumulation and distribution of PEG‐coated liposomes. Treatment with S‐1 (6.9 mg tegafur/kg, daily, 7 days) was started when the tumor volumes had reached a volume of 40–60 mm3. In order to assess the effect of S‐1 dosing on intratumoral accumulation of PEG‐coated liposomes, DiI‐ or DiD‐labeled PEG‐coated liposomes (25 mg phospholipids/kg) were intravenously injected right after the final S‐1 administration. At defined time points (6, 12 and 24 h) after injection, fluorescence imaging was performed with Fluorescence Image Analyzer LAS‐4000 IR (Fujifilm, Tokyo, Japan). The fluorescence images were acquired with a 1/100 s exposure time. For the intratumoral liposome distribution study, at 24 h post‐injection, the tumors were harvested and snap‐frozen in Optical Cutting Compound (OCT) compound (Sakura Fintechnical, Tokyo, Japan) with dry‐iced acetone. Sections of frozen samples (5 μm thick) were directly observed using a fluorescence microscope (Axiovert 200 M; Zeiss, Oberkohen, Germany). Three tumors per group were studied. Thirty images from 10 randomly selected sections per tumor (three images from one section) were analyzed using AxioVision software (Zeiss).
Statistics. All values are expressed as the mean ± SD. Statistical analysis was performed with a two‐tailed unpaired t‐test using GraphPad InStat software (GraphPad Software, La Jola, CA, USA). The level of significance was set at P < 0.05.
Results
Tumor growth suppressive effect of metronomic S‐1 dosing plus l‐OHP‐containing PEG‐coated liposomes. As shown in Figure 1, free l‐OHP showed relatively low antitumor activity compared with other treatments and the TGI was only 19.0%. Metronomic S‐1 dosing showed a higher tumor growth suppressive effect than free l‐OHP (36.5%, TGI). Liposomal l‐OHP showed even a much higher tumor suppressive effect (52.9%, TGI) compared with free l‐OHP, and the therapeutic efficiency was very similar to that of conventional combination therapy (metronomic S‐1 dosing plus free l‐OHP) (57.7%, TGI). Metronomic S‐1 dosing plus liposomal l‐OHP showed by far the strongest tumor growth suppressive effect (87.0%, TGI) of all treatments. This result indicates that metronomic S‐1 dosing combined with l‐OHP‐containing PEG‐coated liposomes produces a superior tumor growth suppressive effect in a murine colorectal tumor model.
Figure 1.
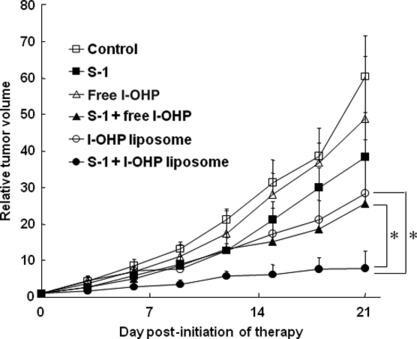
Antitumor effect of mono‐ or combination chemotherapy in colorectal tumor‐bearing mice. Control (non‐treated, □); S‐1 dosing (daily, ); free l‐OHP (weekly, △); S‐1 dosing (daily) plus free oxaliplatin (l‐OHP) (weekly) (); l‐OHP‐containing polyethylene glycol (PEG)‐coated liposomes (weekly, ○); S‐1 dosing (daily) plus l‐OHP‐containing PEG‐coated liposomes (weekly) (•). Data represent mean ± SD (n = 5). *P < 0.05.
To assess toxicity of each mono‐ or combination therapy, change of bodyweight and blood cells (white cells, red cells and platelets) were determined. Only combination chemotherapy of metronomic S‐1 dosing plus l‐OHP formulations showed a slight suppression of bodyweight increase. However, there was no significant difference between S‐1 plus free l‐OHP and S‐1 plus liposomal l‐OHP (data not shown). In addition there were no significant changes in blood cell counts between each mono‐ and combination therapy (data not shown).
Effect of metronomic S‐1 dosing on clearance and tumor accumulation of l‐OHP associated with PEG‐coated lipo‐somes. The plasma clearance of l‐OHP in S‐1 treated mice was very similar to that in control mice; the t1/2 in S‐1 treated mice amounting to 18.5 h and the t1/2 in control mice to 18.1 h. In the tumor without S‐1 treatment, l‐OHP concentration reached the maximum level (approximately 1500 ng/g tissue) at 24 h after injection, and then precipitously decreased (Fig. 2). In the S‐1‐treated tumor, l‐OHP concentration reached the maximum level, similar to the control, at 24 h after injection, being retained at this level until 48 h and then gradually decreasing (Fig. 2). The area under the concentration–time curve (AUC) of l‐OHP in the S‐1‐treated tumor was approximately 1.4‐fold higher than that in the control tumor; 131.6 (μg/g tissue h) in S‐1‐treated tumor vs 96.2 (μg/g tissue h) in the control. These results indicate that the S‐1 treatment prolonged the retention of l‐OHP within the tumor tissue.
Figure 2.
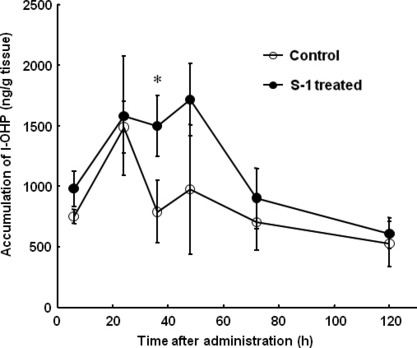
Effect of S‐1 dosing on tumor accumulation of oxaliplatin (l‐OHP) delivered by polyethylene glycol (PEG)‐coated liposomes. Oxaliplatin‐containing PEG‐coated liposomes were intravenously administered into tumor‐bearing mice that were pre‐treated with or without S‐1 dosing for 7 days. At various time points, tumor tissue was collected and then l‐OHP in the tissue was determined. Data represent mean ± SD (n = 3). *P < 0.05 vs control.
Effect of metronomic S‐1 dosing on biodistribution and tumor accumulation of PEG‐coated liposomes. To gain more insight into the underlying mechanism of the improved tumor suppressive effect (Fig. 1) and in the prolonged l‐OHP retention within the tumor (Fig. 2), we investigated the effect of metronomic S‐1 dosing on the biodistribution and tumor accumulation of co‐administered PEG‐coated liposomes. The effect of daily metronomic S‐1 dosing (for 7 days) on the biodistribution of PEG‐coated liposomes was investigated with a radio‐labeled liposome. The S‐1 treatment yielded significant enhancement of accumulation of PEG‐coated liposomes (1.3‐fold) in tumor at 24 h following administration (Fig. 3). Interestingly, in the S‐1‐treated mice, there appeared to be a discrepancy between the tumor accumulation of l‐OHP (Fig. 2) and liposomes at 24 h following administration. Although the mechanism is uncertain, l‐OHP that leaked from the liposome and then bound to plasma proteins and partitioned to erythrocytes might affect the platinum concentration in the tumor. In addition, the treatment did not affect accumulation of PEG‐coated liposomes in the major organs (Fig. 3). This observation indicates that the S‐1 treatment does not affect the biodistribution of PEG‐coated liposomes and the permeability of blood vessels towards the liposomes already existing in normal tissues.
Figure 3.
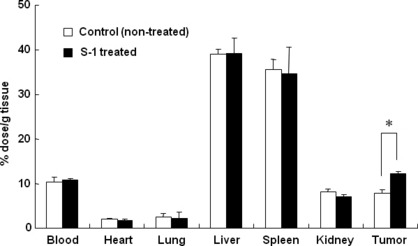
Effect of S‐1 dosing on biodistribution of polyethylene glycol (PEG)‐coated liposomes. Biodistribution of PEG‐coated liposomes was determined at 24 h following intravenous injection in tumor‐bearing mice pretreated with or without S‐1 dosing for 7 days. Data represent mean ± SD (n = 3). *P < 0.05.
In addition, in vivo imaging studies indicated a similar tendency of intratumor accumulation of PEG‐coated liposomes as a function of time following injection (Fig. 4). Both the control and S‐1‐treated mice showed time‐dependent augmentation of PEG‐coated liposome accumulation. These findings indicate that PEG‐coated liposomes accumulated in tumor tissue due to the EPR effect, and S‐1 treatment facilitated the EPR effect towards PEG‐coated liposomes, resulting in further accumulation of PEG‐coated liposomes in solid tumor.
Figure 4.
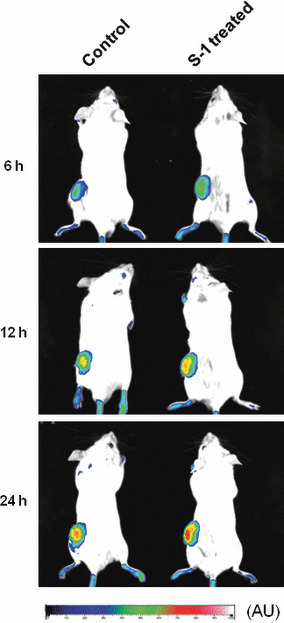
In vivo optical imaging of tumor accumulation of polyethylene glycol (PEG)‐coated liposomes. Tumor‐bearing mice, pretreated with S‐1 dosing for 7 days, received an intravenous injection of DiD (1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl‐indodicarbocyanine perchlorate)‐labeled PEG‐coated liposomes. At 6, 12 and 24 h post‐injection, in vivo optical images were recorded. AU, arbitrary unit.
To investigate the intratumoral distribution of PEG‐coated liposomes, a histological analysis was carried out. Fluorescence associated with PEG‐coated liposomes was observed in the section of both control and S‐1‐treated tumor (Fig. 5A). The number and size of fluorescence spots in the section of S‐1‐treated tumor were substantially larger than those in the section of the control tumor, indicating that the S‐1 treatment enhanced liposome distribution in tumor tissue. The area density of fluorescence in the tumor section indicated that the sections of S‐1 treated tumor contain a much larger amount of PEG‐coated liposomes than the section of control tumor (Fig. 5B).
Figure 5.
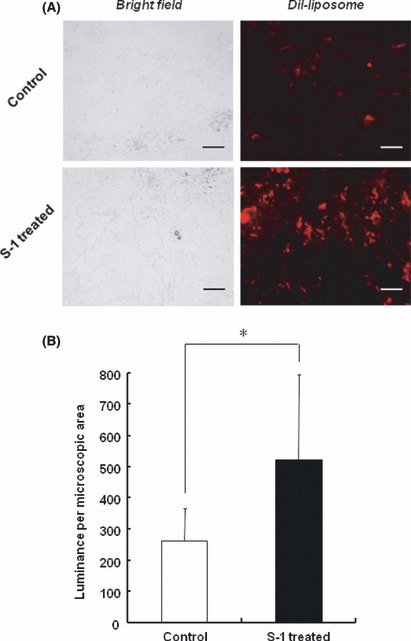
Effect of S‐1 dosing on intratumoral distribution of polyethylene glycol (PEG)‐coated liposomes. Tumor‐bearing mice, pretreated with S‐1 dosing for 7 days, received DiI (1, 1′‐dioctadecyl‐3, 3, 3′,3′‐tetramethyl‐indocarbocyanine perchlorate)‐labeled PEG‐coated liposomes. At 24 h post‐injection, the section of tumor was examined with fluorescence microscopy. (A) Intratumoral distribution of PEG‐coated liposomes. Red spots represent liposomal distribution. Bar, 100 μm. Original magnification, ×200. (B) Mean fluorescence intensity per microscopic area. Data represent mean ± SD. *P < 0.05.
Discussion
In the foregoing section we showed that the combination of oral metronomic S‐1 dosing with oxaliplatin (l‐OHP)‐containing PEG‐coated liposomes exerts improved antitumor activity in a murine colorectal tumor model without causing severe side‐effects, as compared with conventional combination therapy (metronomic S‐1 dosing plus free l‐OHP) (Fig. 1). This improvement resulted from enhanced accumulation of l‐OHP‐containing PEG‐coated liposomes and prolonged retention of l‐OHP in the tumor induced by metronomic S‐1 treatment (2, 3, 4, 5). The FOLFOX regimen (5‐FU/LV plus l‐OHP) has frequently been used for treatment of advanced colorectal cancer in the clinic.( 4 , 5 ) However, extended periods of infusional 5‐FU (≈48 h) have the disadvantage of increased inconvenience and morbidity of patients related to the use of a portable infusion pump and a central venous catheter. Daily oral administration of S‐1 can mimic the pharmacokinetic profile of infusional 5‐FU and overcome the problems related to infusional 5‐FU treatment. In fact, Yamada et al. ( 23 ) recently reported in a Phase I/II trial that the combination of S‐1 with free l‐OHP (SOX) is a preferable alternative to the FOLFOX regimen in metastatic colorectal cancer. In contrast to cisplatin, l‐OHP has no renal toxicity, only mild hematological and gastrointestinal toxicity, while neurotoxicity is the dose‐limiting toxicity.( 24 , 25 ) The selective delivery of l‐OHP to tumors by PEG‐coated liposomes raises the possibility of reducing the side‐effects of l‐OHP in the FOLFOX and SOX regimens. Accordingly, the proposed combination regimen (i.e. addition of S‐1 dosing to l‐OHP‐containing PEG‐coated liposomes) may be an alternative to FOLFOX and SOX in advanced colorectal cancer therapy.
Accumulation of nanocarriers into solid tumor after systemic administration is thought to involve the following three processes: (i) distribution through the vascular compartment; (ii) transport across the angiogenic vascular wall (extravasation from neo‐vasculature); and (iii) diffusion within the tumor interstitium.( 26 ) It is generally believed that the major target of metronomic chemotherapy is endothelial cells of the growing vasculature in the solid tumor. Ooyama et al. ( 27 ) recently demonstrated that metronomic S‐1 dosing damages endothelial cells of tumor vasculature. Loss of the endothelial lining of vessels may make tumor vasculature much leakier. On the basis of our current and previous results,( 16 ) we conclude that the therapy enhances the EPR effect towards PEG‐coated liposomes in the following manner: the therapy causes blood vessels in the tumor to become more leaky, resulting in enhanced extravasation of PEG‐coated liposomes from the vasculature into the interstitial space of the tumor (3, 4). Moreover, metronomic therapy with S‐1 may also exert a cytotoxic effect on viable tumor cells and stromal cells and thus bring about a decrease in the number of both cell types and, consequently, a decrease in the tumor interstitial pressure and enlargement of tumor interstitial space, which, in turn, will allow deeper penetration of the extravasated PEG‐coated liposomes (Fig. 5). A similar observation was recently reported by Nagano et al.;( 28 ) paclitaxel‐induced tumor cell death enhanced the penetration and distribution of virus vector and microspheres in tumor tissue.
In addition to intratumoral accumulation of PEG‐coated liposomes, the liposome distribution to major organs was investigated. Metronomic S‐1 dosing did not affect accumulation of the liposomes in major organs and blood clearance of the liposomes (Fig. 3). This finding suggests that S‐1 treatment does not affect normal vasculature pre‐existing in normal tissues, but only the vasculature in tumors, although the mechanism by which S‐1 changes only tumor vascular permeability remains unclear. This clearly relates to a safety issue in the proposed combination therapy. In addition, it appears that S‐1 treatment does not affect the essential phagocytic uptake activity of hepatic and splenic macrophages, because the treatment did not affect blood clearance of PEG‐coated liposomes. Daemen et al. ( 29 ) have previously reported that injection of DXR‐loaded PEG‐coated liposomes has a toxic effect on liver macrophages, both in terms of specific phagocytic activity and cell numbers. It is known that defects in the phagocytic uptake mechanism of macrophages can enhance metastatic growth, as reported in numerous animal studies.( 30 ) The cytotoxic effect of l‐OHP‐containing PEG‐coated liposomes on macrophages has not been elucidated yet. Hence, further experiments are in progress to ascertain the alteration of the tumor microenvironment (such as vascular permeability and compressive mechanical force of growing tumor cells) induced by metronomic S‐1 dosing and cumulative toxicity of combination therapy of S‐1 with l‐OHP‐containing PEG‐coated liposomes.
Anticancer chemotherapy using nanocarriers has shown marked therapeutic effects in many tumor models; however, nanocarriers do not always accumulate effectively in solid tumors, probably due to barriers generated by the tumor microenvironment. Recently, a number of approaches have been introduced that render chemotherapeutics associated with a nanocarrier more efficient. Iyer et al. ( 31 ) demonstrated that hypertension induced by infusion of angiotensin‐II (AT‐II) increased the blood flow volume and generated a pressure gradient between the intra‐ and extravascular space in tumor tissue, resulting in increased extravasation from the tumor vessel of an anticancer agent associated with a nanocarrier. They assumed that the selective accumulation in a solid tumor can be attributed to the absence of a vascular smooth‐muscle layer in tumor vasculature. This approach also did not increase the amount of nanocarrier accumulating in healthy organs because of vasoconstriction and tighter endothelial gap junctions of the vasculature. Kano et al. ( 10 ) demonstrated that low‐dose treatment with transforming growth factor‐β type 1 receptor inhibitor resulted in further enhanced accumulation of PEG‐coated liposomes and micelles to a solid tumor. Seynhaeve et al. ( 12 ) showed similar results with low‐dose TNF‐α administration. Our approach also has a potential to achieve enhanced accumulation of PEG‐coated liposomes in solid tumors. Hence, the approach that actively causes alteration of the tumor microenvironment by treatment with vaso‐active agents or anticancer agents may become a breakthrough in improved delivery of anticancer agents associated with nanocarriers.
Disclosure Statement
No potential conflicts of interest are disclosed.
Acknowledgments
We thank Dr G.L. Scherphof for his helpful advice on writing the English manuscript. This study was supported in part by a Grant‐in‐Aid for Young Scientists (A) (21689002), the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Kidani Y, Noji M, Tashiro T. Antitumor activity of platinum(II) complexes of 1,2‐diamino‐cyclohexane isomers. Gann 1980; 71: 637–43. [PubMed] [Google Scholar]
- 2. Mathe G, Kidani Y, Noji M, Maral R, Bourut C, Chenu E. Antitumor activity of l‐OHP in mice. Cancer Lett 1985; 27: 135–43. [DOI] [PubMed] [Google Scholar]
- 3. Pendyala L, Creaven PJ. In vitro cytotoxicity, protein binding, red blood cell partitioning, and biotransformation of oxaliplatin. Cancer Res 1993; 53: 5970–6. [PubMed] [Google Scholar]
- 4. Saltz LB, Cox JV, Blanke C et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med 2000; 343: 905–14. [DOI] [PubMed] [Google Scholar]
- 5. Goldberg RM, Sargent DJ, Morton RF et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004; 22: 23–30. [DOI] [PubMed] [Google Scholar]
- 6. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 7. Suzuki R, Takizawa T, Kuwata Y et al. Effective anti‐tumor activity of oxaliplatin encapsulated in transferrin‐PEG‐liposome. Int J Pharm 2008; 346: 143–50. [DOI] [PubMed] [Google Scholar]
- 8. Abu Lila AS, Kizuki S, Doi Y, Suzuki T, Ishida T, Kiwada H. Oxaliplatin encapsulated in PEG‐coated cationic liposomes induces significant tumor growth suppression via a dual‐targeting approach in a murine solid tumor model. J Control Release 2009; 137: 8–14. [DOI] [PubMed] [Google Scholar]
- 9. Abu‐Lila AS, Suzuki T, Doi Y, Ishida T, Kiwada H. Oxaliplatin targeting to angiogenic vessels by PEGylated cationic liposomes suppresses the angiogenesis in a dorsal air sac mouse model. J Control Release 2009; 134: 18–25. [DOI] [PubMed] [Google Scholar]
- 10. Kano MR, Bae Y, Iwata C et al. Improvement of cancer‐targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF‐beta signaling. Proc Natl Acad Sci USA 2007; 104: 3460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ten Hagen TL, Van Der Veen AH, Nooijen PT, Van Tiel ST, Seynhaeve AL, Eggermont AM. Low‐dose tumor necrosis factor‐alpha augments antitumor activity of stealth liposomal doxorubicin (DOXIL) in soft tissue sarcoma‐bearing rats. Int J Cancer 2000; 87: 829–37. [DOI] [PubMed] [Google Scholar]
- 12. Seynhaeve AL, Hoving S, Schipper D et al. Tumor necrosis factor alpha mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res 2007; 67: 9455–62. [DOI] [PubMed] [Google Scholar]
- 13. Kerbel RS, Kamen BA. The anti‐angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 2004; 4: 423–36. [DOI] [PubMed] [Google Scholar]
- 14. Laquente B, Vinals F, Germa JR. Metronomic chemotherapy: an antiangiogenic scheduling. Clin Transl Oncol 2007; 9: 93–8. [DOI] [PubMed] [Google Scholar]
- 15. Shiraga E, Barichello JM, Ishida T, Kiwada H. A metronomic schedule of cyclophosphamide combined with PEGylated liposomal doxorubicin has a highly antitumor effect in an experimental pulmonary metastatic mouse model. Int J Pharm 2008; 353: 65–73. [DOI] [PubMed] [Google Scholar]
- 16. Ishida T, Shiraga E, Kiwada H. Synergistic antitumor activity of metronomic dosing of cyclophosphamide in combination with doxorubicin‐containing PEGylated liposomes in a murine solid tumor model. J Control Release 2009; 134: 194–200. [DOI] [PubMed] [Google Scholar]
- 17. Shirasaka T, Nakano K, Takechi T et al. Antitumor activity of 1 M tegafur‐0.4 M 5‐chloro‐2,4‐dihydroxypyridine‐1 M potassium oxonate (S‐1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res 1996; 56: 2602–6. [PubMed] [Google Scholar]
- 18. Sakata Y, Ohtsu A, Horikoshi N, Sugimachi K, Mitachi Y, Taguchi T. Late phase II study of novel oral fluoropyrimidine anticancer drug S‐1 (1 M tegafur‐0.4 M gimestat‐1 M otastat potassium) in advanced gastric cancer patients. Eur J Cancer 1998; 34: 1715–20. [DOI] [PubMed] [Google Scholar]
- 19. Sakuramoto S, Sasako M, Yamaguchi T et al. Adjuvant chemotherapy for gastric cancer with S‐1, an oral fluoropyrimidine. N Engl J Med 2007; 357: 1810–20. [DOI] [PubMed] [Google Scholar]
- 20. Bartlett GR. Colorimetric assay methods for free and phosphorylated glyceric acids. J Biol Chem 1959; 234: 469–71. [PubMed] [Google Scholar]
- 21. Cabral H, Nishiyama N, Kataoka K. Optimization of (1,2‐diamino‐cyclohexane)platinum (II)‐ loaded polymeric micelles directed to improved tumor targeting and enhanced antitumor activity. J Control Release 2007; 121: 146–55. [DOI] [PubMed] [Google Scholar]
- 22. Harashima H, Yamane C, Kume Y, Kiwada H. Kinetic analysis of AUC‐dependent saturable clearance of liposomes: mathematical description of AUC dependency. J Pharmacokinet Biopharm 1993; 21: 299–08. [DOI] [PubMed] [Google Scholar]
- 23. Yamada Y, Tahara M, Miya T et al. Phase I/II study of oxaliplatin with oral S‐1 as first‐line therapy for patients with metastatic colorectal cancer. Br J Cancer 2008; 98: 1034–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grothey A. Oxaliplatin‐safety profile: neurotoxicity. Semin Oncol 2003; 30: 5–13. [DOI] [PubMed] [Google Scholar]
- 25. Pietrangeli A, Leandri M, Terzoli E, Jandolo B, Garufi C. Persistence of high‐dose oxaliplatin‐induced neuropathy at long‐term follow‐up. Eur Neurol 2006; 56: 13–6. [DOI] [PubMed] [Google Scholar]
- 26. Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther 2007; 322: 80–8. [DOI] [PubMed] [Google Scholar]
- 27. Ooyama A, Oka T, Zhao HY, Yamamoto M, Akiyama S, Fukushima M. Anti‐angiogenic effect of 5‐Fluorouracil‐based drugs against human colon cancer xenografts. Cancer Lett 2008; 267: 26–36. [DOI] [PubMed] [Google Scholar]
- 28. Nagano S, Perentes JY, Jain RK, Boucher Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res 2008; 68: 3795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daemen T, Regts J, Meesters M, Ten Kate MT, Bakker‐Woudenberg IA, Scherphof GL. Toxicity of doxorubicin entrapped within long‐circulating liposomes. J Control Release 1997; 44: 1–9. [Google Scholar]
- 30. Phillips NC. Kupffer cells and liver metastasis. Optimization and limitation of activation of tumoricidal activity. Cancer Metastasis Rev 1989; 8: 231–52. [DOI] [PubMed] [Google Scholar]
- 31. Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today 2006; 11: 812–8. [DOI] [PubMed] [Google Scholar]