

Abstract
Hepatocellular carcinoma (HCC), one of the most common cancers worldwide, is resistant to anticancer drugs. Hypoxia is a major cause of tumor resistance to chemotherapy, and hypoxia‐inducible factor (HIF)‐1 is a key transcription factor in hypoxic responses. We have previously demonstrated that gene transfer of an antisense HIF‐1α expression vector downregulates expression of HIF‐1α and vascular endothelial growth factor (VEGF), and synergizes with immunotherapy to eradicate lymphomas. The aim of the present study was to determine whether gene transfer of antisense HIF‐1α could enhance the therapeutic efficacy of doxorubicin to combat HCC. Both antisense HIF‐1α therapy and doxorubicin suppressed the growth of subcutaneous human HepG2 tumors established in BALB/c nude mice, tumor angiogenesis, and cell proliferation, and induced tumor cell apoptosis. The combination therapy with antisense HIF‐1α and doxorubicin was more effective in suppressing tumor growth, angiogenesis, and cell proliferation, and inducing cell apoptosis than the respective monotherapies. Gene transfer of antisense HIF‐1α downregulated the expression of both HIF‐1α and VEGF, whereas doxorubicin only downregulated VEGF expression. Antisense HIF‐1α and doxorubicin synergized to downregulate VEGF expression. Both antisense HIF‐1α and doxorubicin inhibited expression of proliferating cell nuclear antigen, and combined to exert even stronger inhibition of proliferating cell nuclear antigen expression. Antisense HIF‐1α therapy warrants investigation as a therapeutic strategy to enhance the efficacy of doxorubicin for treating HCC. (Cancer Sci 2008; 99: 2055–2061)
Abbreviations:
- Ab
antibody
- HCC
hepatocellular carcinoma
- HIF
hypoxia‐inducible factor
- HRE
hypoxia‐response element
- OD
optical density
- PBS
phosphate‐buffered saline
- PCNA
proliferating cell nuclear antigen
- siRNA
small interference RNA
- TUNEL
terminal deoxynucleotidyl transferase biotin‐dUTP nick end labeling
- VEGF
vascular endothelial growth factor
Hepatocellular carcinoma, accounting for up to 90% of primary liver cancers, is characterized by a very poor prognosis and is associated with high mortality.( 1 , 2 ) Currently available therapeutic modalities for HCC are largely inadequate. Surgical approaches, including resection and transplantation, are the treatment of choice for HCC; however, few patients are suitable for resection, and the utility of transplantation is limited by organ availability. Chemotherapy offers unsatisfactory response rates. In a recent review by Zhu,( 3 ) no chemotherapeutic agent was identified as being particularly effective against HCC. Among the approved anticancer drugs, doxorubicin is perhaps the most widely used agent to treat HCC. An initial report by Uganda( 3 ) on the use of doxorubicin to combat HCC was encouraging, but subsequent studies have been disappointing. Doxorubicin failed to induce responses in 109 patients with advanced HCC,( 4 ) and there was only a 16% response rate in 475 patients.( 5 ) In a recent study involving a large number of patients, doxorubicin only showed response rates ranging from 4 to 10.5% in HCC patients, and had significant side effects.( 6 ) Therefore, new strategies to enhance the therapeutic efficacy of doxorubicin to treat HCC are needed.
Hypoxic microenvironments are frequent in solid tumors. Hypoxia orchestrates a wide spectrum of molecular pathways that frustrate therapy, and is a major cause of tumor resistance to radiotherapy and chemotherapy.( 7 , 8 ) HIF‐1, a transcription factor,( 9 ) mediates the adaptation of cancer cells to the hypoxic environment by controlling the expression of hundreds of genes,( 10 ) including VEGF,( 11 ) glycolytic enzymes, and glucose transporters.( 12 ) HIF‐1, formed by the assembly of HIF‐1α and HIF‐1β, binds HRE in the promoters of the above genes.( 12 , 13 ) Although HIF‐1β is expressed constitutively, HIF‐1α is degraded rapidly during normoxia and dramatically stabilized and activated during hypoxia by oxygen‐sensing and signaling processes.( 14 , 15 ) HIF‐1‐deficient hepatoma cells( 16 ) and lung cancer cells treated with siRNA targeting HIF‐1α( 17 ) are more susceptible to radiotherapy and chemotherapy, respectively, than parental and untreated cells. Hypoxia stimulates the migration of human HCC Hep3B, an effect that is abolished by HIF‐1α siRNA and the HIF inhibitor YC‐1, which also effectively inhibits cell invasion and metastases in vivo.( 18 )
We recently reported that gene therapy using the antiangiogenic agent endostatin enhances the efficacy of doxorubicin to treat HCC.( 19 ) Gene transfer of endostatin downregulates the expression of HIF‐1α and synergizes with doxorubicin to downregulate the expression of VEGF, thereby inhibiting tumor growth and angiogenesis.( 19 ) Therefore, we reasoned that the simplest means of inhibiting tumor angiogenesis would be to target HIF‐1α. We have already demonstrated that intratumoral injection of antisense HIF‐1α expression vectors suppresses the growth of lymphomas by downregulating expression of HIF‐1α and VEGF, and synergizes with immunotherapy to eradicate tumors.( 20 ) We subsequently revealed that antisense HIF‐1α synergizes with von Hippel‐Lindau tumor‐suppressor protein to eradicate solid tumors.( 21 ) Here we investigated whether gene transfer of antisense HIF‐1α could enhance the therapeutic efficacy of doxorubicin to combat HCC.
Materials and Methods
Mice and cell line. Male nude BALB/c mice (H‐2b), 6–8 weeks old, were obtained from the Animal Research Center, Shandong University, China. The human HCC cell line HepG2 was obtained from the American Type Culture Collection (Rockville, MD, USA). The cells were cultured routinely at 37°C in RPMI‐1640 medium supplemented with 10% fetal calf serum.
Gene expression vector and antibodies. The antisense HIF‐1α expression vector (aHIF‐pcDNA3.1) containing a 320‐bp antisense cDNA fragment of HIF‐1α (nucleotides 152–454; GenBank no. AF003695) has been described previously.( 20 , 21 ) The Ab used in the present study include an anti‐HIF‐1α Ab (Boster Biological Technology, Wuhan, China), anti‐VEGF Ab (Laboratory Vision Corporation, Fremont, CA, USA), anti‐CD31 (MEC13.3) Ab (Pharmingen, San Diego, CA, USA), anti‐Ki‐67 (clone B56) Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti‐PCNA Ab (Zhongshan Golden Bridge Biotechnology, Beijing, China).
Animal experimental protocols. All surgical procedures and care administered to the animals were in accordance with institutional guidelines. Tumors were established by subcutaneous injection of 1 × 106 HepG2 tumor cells into the flanks of mice. Tumor volumes were estimated according to the formula:
| p/6 × a 2 × b, |
where a is the short axis and b the long axis. When tumors reached approximately 100 mm3 (at approximately 3 weeks), the mice were assigned randomly to four treatment groups to receive pcDNA3.1, aHIF‐pcDNA3.1, doxorubicin, or aHIF‐pcDNA3.1 + doxorubicin. To standardize the experiments, mice in each group received both intratumoral and intraperitoneal injections. In the pcDNA3.1 and aHIF‐pcDNA3.1 groups, mice received intraperitoneal injection of 200 µL PBS, and intratumoral injection of 200 µg pcDNA3.1 or aHIF‐pcDNA3.1 diluted in 100 µL FuGENE 6 transfection reagent (Roche, Shanghai, China), respectively. In the doxorubicin group, mice received intraperitoneal injection of 200 µL doxorubicin (diluted in PBS) at a dose of 12.5 mg/kg and intratumoral injection of 200 µg pcDNA3.1 diluted in 100 µL FuGENE 6. In the aHIF‐pcDNA3.1 + doxorubicin group, mice received intraperitoneal injection of 200 µL doxorubicin (diluted in PBS) at a dose of 12.5 mg/kg and intratumoral injection of 200 µg aHIF‐pcDNA3.1 diluted in 100 µL FuGENE 6. All experiments included 12 mice per treatment group. FuGENE 6 was shown to be an efficient in vivo transfection reagent in our previous study.( 19 , 22 )
Immunohistochemistry. The procedure of immunohistochemistry has been described in our previous report.( 19 ) Briefly, tumor sections were fixed, blocked, and incubated overnight with primary antibodies. They were subsequently incubated with appropriate secondary antibodies, and immunoreactivity developed with 3,3‐diaminobenzidine (DAB), followed by counterstaining.
Assessment of tumor vascularity. The methodology used to determine tumor vascularity has been described previously.( 19 , 23 ) Briefly, tumor sections were immunostained with an anti‐CD31 Ab, as described above. Stained vessels were counted in 10 blindly chosen random fields at ×400 magnification, and the mean microvessel density was recorded.
Quantitation of the Ki‐67 proliferation index. Tumor sections were immunostained with an anti‐Ki‐67 Ab as above, and the Ki‐67‐positive cells were counted in 10 randomly selected ×400 high‐power fields by microscopy. The Ki‐67 proliferation index was calculated according to the following formula:
| no. Ki‐67‐positive cells/total cell count × 100%. |
Western blot analysis. The methodology to detect protein expression in tumor homogenates has been described previously.( 19 ) Briefly, tumor tissues were minced and homogenized in protein lysate buffer, and debris was removed by centrifugation. The lysates were resolved on 12% polyacrylamide sodium dodecylsulfate gels, and transferred electrophoretically to polyvinylidene difluoride membranes. The membranes were blocked with 3% bovine serum albumin (BSA), incubated with primary antibodies, and subsequently with alkaline phosphatase‐conjugated secondary antibody. They were developed with 5‐bromo‐4‐chloro‐3‐indolyl phosphate/nitro blue tetrazolium (Tiangen Biotech, Beijing, China). Blots were stained with an antitubulin antibody to confirm that each lane contained similar amounts of tumor homogenate.
In situ detection of apoptotic cells. The method of in situ detection of apoptotic cells has been described previously.( 24 ) Briefly, serial 6‐µm tumor sections were stained with the TUNEL agent (Roche) and examined by fluorescence microscopy. Adjacent sections were counterstained with hematoxylin. The total number of apoptotic cells in 10 randomly selected fields was counted. The apoptosis index was calculated as the percentage of positive‐staining cells, namely:
| apoptosis index = no. apoptotic cells × 100/total no. nucleated cells. |
In vitro assay. HepG2 cells were grown to 60–70% confluence in 10‐cm plastic dishes, and transfected with 4 µg (each dish) aHIF‐pcDNA3.1 or empty pcDNA3.1 plasmid using Lipofectamine Plus (Life Technologies, Shanghai, China). CoCl2 was added to one dish at the concentration of 200 µmol/L 48 h after transfection. Three hours later, the cells were harvested and cell lysates were subjected to western blot analysis as described previously( 25 ) to confirm the downregulation of HIF‐1α by aHIF‐pcDNA3.1. The cells in other dishes were harvested, counted, reseeded in 200 µL RPMI‐1640 in the presence or absence of CoCl2 into 96‐well plates (5 × 103 cells per well), and cultured for a further 6 h. 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (20 µL) was added to each well followed by a 4‐h incubation, and cells were processed to record the OD value at 570 nm. The untreated cells were used as controls. The proliferation index was calculated according to the formula:
| proliferation index = experimental OD value/control OD value × 100%. |
The experiments were repeated three times.
Statistical analysis. The growth patterns of tumors were compared using the analysis of variance (ANOVA) test. Other results were expressed as mean values ± SD, and Student's t‐test was used to evaluate statistical significance. A P‐value of less than 0.05 was used for statistical significance.
Results
Antisense HIF‐1α gene transfer downregulates expression of HIF‐1α and VEGF. The initial goal was to investigate whether gene transfer of antisense HIF‐1α could downregulate expression of HIF‐1α and its downstream effector VEGF in HepG2 tumors. HepG2 tumors of 100 mm3 in volume were injected with a DNA–FuGENE 6 transfection vehicle containing 200 µg of either empty pcDNA3.1 vector or aHIF‐pcDNA3.1, and 4 days later the tumors were excised. Gene transfer of aHIF‐pcDNA3.1 led to downregulation of HIF‐1α (Fig. 1a,b) and VEGF (Fig. 1c,d) expression in tumor cells compared with pcDNA3.1‐injected tumors, as revealed by immunohistochemistry and supported by western blot analysis (Fig. 1e).
Figure 1.
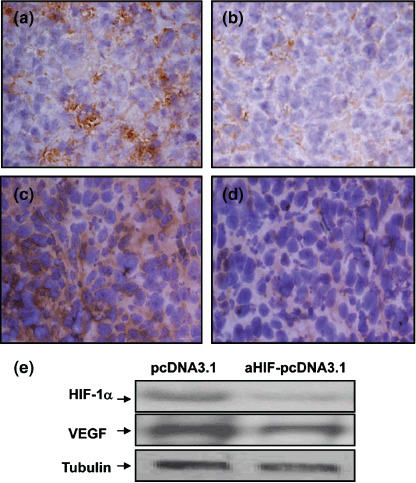
Antisense hypoxia inducible factor (HIF)‐1α gene transfer downregulates expression of HIF‐1α and vascular endothelial growth factor (VEGF). Tumors of approximately 100 mm3 in volume were injected with either (a,c) pcDNA3.1, or (b,d) aHIF‐pcDNA3.1 plasmids. Representative tumor sections prepared 4 days after gene transfer, stained brown with antibodies against (a,b) HIF‐1α and (c,d) VEGF (×400 magnification). (e) Homogenates of tumors prepared 4 days after intratumoral injection of the pcDNA3.1 or aHIF‐pcDNA3.1 plasmids were western blotted with antibodies against HIF‐1α (upper panel), VEGF (middle panel), and tubulin (lower panel), as indicated.
Antisense HIF‐1α gene therapy synergizes with doxorubicin to suppress hepatomas. HepG2 tumors were established in nude mice. Three weeks later, when the tumors reached approximately 100 mm3, the mice were assigned randomly to four treatment groups to receive pcDNA3.1, aHIF‐pcDNA3.1, doxorubicin, or aHIF‐pcDNA3.1 + doxorubicin. Untreated mice served as an additional control. As shown in Figure 2, tumors treated with empty vector pcDNA3.1 grew remarkably fast, reaching 2032 ± 312 mm3 in volume 6 weeks after implantation, which was not significantly different from the growth of the untreated tumors (2191 ± 245 mm3, P > 0.05). In contrast, the tumors of mice treated with doxorubicin were significantly (P < 0.01) smaller than control tumors, reaching only 897 ± 178 mm3 in volume 6 weeks after implantation. Antisense HIF‐1 therapy also resulted in a significant (P < 0.05) reduction in tumor volume (1107 ± 211 mm3) compared with control tumors. The combination of aHIF‐pcDNA3.1 and doxorubicin further suppressed tumor growth such that tumors reached only 359 ± 134 mm3 in volume, which is significantly (P < 0.001) smaller than control tumors, and significantly smaller than the tumors treated with the aHIF‐pcDNA3.1 and doxorubicin monotherapies (both P < 0.05) (Fig. 2).
Figure 2.
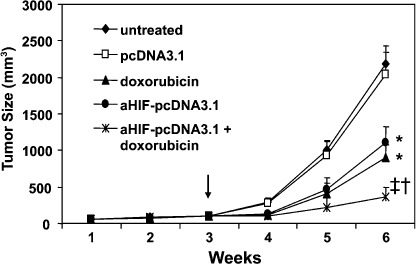
Antisense hypoxia inducible factor (HIF)‐1α gene therapy synergizes with doxorubicin to suppress tumor growth. HepG2 hepatomas were established subcutaneously in the flanks of mice. When the tumors reached approximately 100 mm3 in volume (arrow), they were injected with the pcDNA3.1, and aHIF‐pcDNA3.1 plasmids, either alone or in combination with intraperitoneal injection of mice with doxorubicin, as indicated. Untreated tumors served as controls. The sizes (mm3) of tumors were monitored and recorded. *Significant and †highly significant difference in tumor volumes compared with control. ‡Significant difference compared with aHIF‐pcDNA3.1 or doxorubicin treatments.
Antisense HIF‐1α synergizes with doxorubicin to inhibit tumor angiogenesis. Five mice were killed 2 weeks after the above treatments, and tumors were sectioned and stained with an anti‐CD31 Ab to visualize microvessels. There were fewer microvessels in tumors treated with aHIF‐pcDNA3.1 (Fig. 3b) or doxorubicin (Fig. 3c), compared with pcDNA3.1‐treated tumors (Fig. 3a), and there were even fewer microvessels in tumors treated with the combination therapy with aHIF‐pcDNA3.1 and doxorubicin (Fig. 3d) compared with tumors treated with the monotherapies. In addition, blood vessels present in tumors treated with aHIF‐pcDNA3.1 monotherapy or the combination therapy were small and poorly formed. Tumor microvessels in sections were counted in blindly chosen random fields to record microvessel density. Gene transfer of antisense HIF‐1α resulted in a significant 36% (P < 0.01) reduction in tumor microvessel density compared with the density of microvessels in control tumors. Doxorubicin also reduced tumor microvessel density by 29% compared with control tumors (P < 0.05). Furthermore, the combination therapy with aHIF‐pcDNA3.1 and doxorubicin very significantly (P < 0.001) reduced microvessel density by 68%, compared with treatment with the pcDNA3.1 control plasmid, and significantly reduced microvessel density compared to the aHIF‐pcDNA3.1 or doxorubicin monotherapy groups (both P < 0.05). Thus, antisense HIF‐1α and doxorubicin demonstrate synergism in inhibiting tumor angiogenesis (Fig. 3e).
Figure 3.
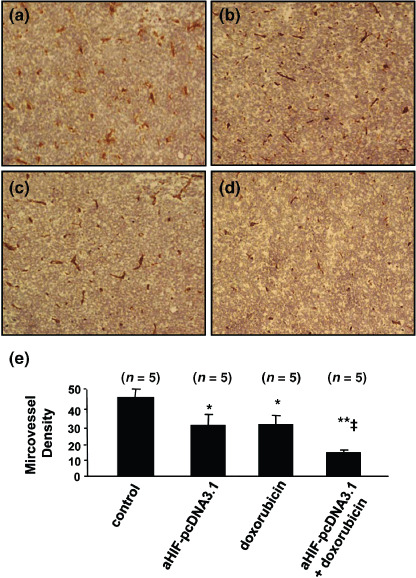
Antisense hypoxia inducible factor (HIF)‐1α synergizes with doxorubicin to inhibit tumor angiogenesis. Representative tumor sections prepared 2 weeks after treatment from mice receiving (a) pcDNA3.1 (control), (b) aHIF‐pcDNA3.1, (c) doxorubicin, or (d) aHIF‐pcDNA3.1 + doxorubicin treatment. Tumor sections were stained with an anti‐CD31 antibody to view microvessels. (e) Tumor microvessels in sections were counted in blindly chosen random fields to record microvessel density. n, number of tumors assessed. *Significant difference in microvessel density for tumors treated with doxorubicin, or aHIF‐pcDNA3.1 versus control; **highly significant difference for tumors treated with doxorubicin + aHIF‐pcDNA3.1 versus control; ‡significant difference for the combination therapy versus aHIF‐pcDNA3.1 or doxorubicin monotherapy.
Antisense HIF‐1α synergizes with doxorubicin to inhibit cell proliferation in situ. We have previously reported that doxorubicin inhibits the proliferation of HepG2 cells in vitro.( 19 ) Here we have further demonstrated that doxorubicin inhibits the proliferation of HepG2 tumor cells in situ, and synergizes with antisense HIF‐1α in inhibiting cell proliferation. Tumor sections from the above experiments were stained with an Ab that detects the cell proliferation marker Ki‐67. There were fewer Ki‐67‐positive cells in tumors treated with aHIF‐pcDNA3.1 (Fig. 4b) and doxorubicin (Fig. 4c) compared with pcDNA3.1‐treated tumors (Fig. 4a) and there were even fewer Ki‐67‐positive cells in tumors treated with combination therapy with aHIF‐pcDNA3.1 and doxorubicin (Fig. 4d) compared with tumors treated with the monotherapies. Ki‐67‐positive cells in sections were counted to record the proliferation index. Gene transfer of antisense HIF‐1α resulted in a significant 31% (P < 0.01) reduction in the proliferation index compared with transfer of the pcDNA3.1 control plasmid. Doxorubicin reduced the proliferation index by 42% compared with the control (P < 0.05). Combination therapy with aHIF‐pcDNA3.1 and doxorubicin very significantly (P < 0.001) reduced the proliferation index by 71%, compared with treatment with the pcDNA3.1 plasmid (Fig. 4e).
Figure 4.
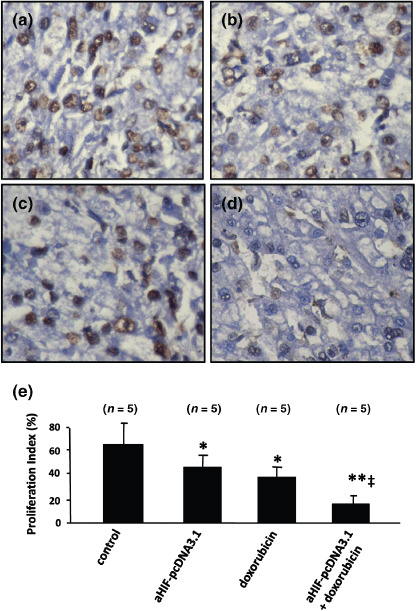
Antisense hypoxia inducible factor (HIF)‐1α synergizes with doxorubicin to inhibit tumor cell proliferation. Representative tumor sections prepared 2 weeks after treatment from mice receiving (a) pcDNA3.1 (control), (b) aHIF‐pcDNA3.1, (c) doxorubicin, or (d) aHIF‐pcDNA3.1 + doxorubicin treatment. Tumor sections were stained with an anti‐Ki‐67 antibody to detect proliferating cells. (e) Cells expressing Ki‐67 were counted to calculate the proliferation index. n, number of tumors assessed. *Significant difference in the proliferation index for tumors treated with doxorubicin, or aHIF‐pcDNA3.1 versus control; **highly significant difference for tumors treated with doxorubicin + aHIF‐pcDNA3.1 versus control; ‡significant difference for the combination therapy versus aHIF‐pcDNA3.1 or doxorubicin monotherapy.
Antisense HIF‐1α synergizes with doxorubicin to induce cell apoptosis in situ. We have previously demonstrated that intratumoral injection of antisense HIF‐1α induces the apoptosis of lymphoma cells in situ.( 21 ) Doxorubicin has been shown to induce tumor apoptosis.( 26 , 27 ) Here we have demonstrated that antisense HIF‐1α synergizes with doxorubicin to induce the apoptosis of HepG2 cells in situ. Tumor sections from the above experiments were stained with the TUNEL agent and examined by fluorescence microscopy. A small number of apoptotic cells were detected in tumors injected with pcDNA3.1 (Fig. 4a), whereas a greater number of apoptotic cells were detected in tumors treated with either aHIF‐pcDNA3.1 (Fig. 4b) or doxorubicin (Fig. 4c). Tumors treated with the combination of aHIF‐pcDNA3.1 and doxorubicin contained the greatest number of apoptotic cells (Fig. 4d). The apoptotic cells in sections were counted to record the apoptosis index. The apoptosis index for tumors treated with either aHIF‐pcDNA3.1 or doxorubicin was significantly (both P < 0.05) higher than that of tumors treated with pcDNA3.1 by 44 and 92%, respectively (Fig. 5e). Thus, despite the finding that antisense HIF‐1α was superior at inhibiting tumor angiogenesis, doxorubicin was more effective at inducing tumor apoptosis, but there was no significant difference in the apoptosis index of tumors treated with the two therapies. The apoptosis index for tumors treated with the combination of aHIF‐pcDNA3.1 and doxorubicin was very significantly higher (by 143%, P < 0.001) than for tumors treated with the pcDNA3.1 control vector, and significantly higher (both P < 0.05) than the apoptosis index of tumors treated with the aHIF‐pcDNA3.1 and doxorubicin monotherapies (Fig. 5e).
Figure 5.
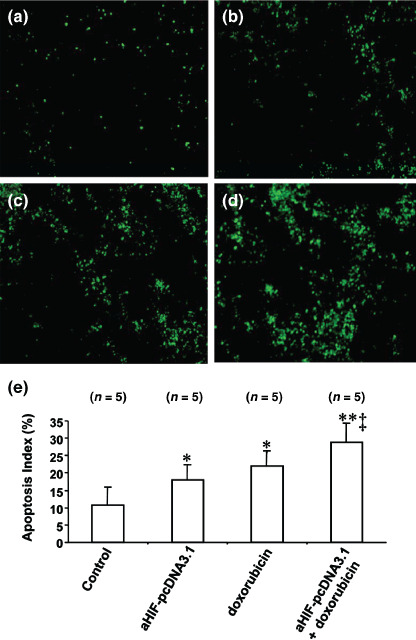
Antisense hypoxia inducible factor (HIF)‐1α synergizes with doxorubicin to induce cell apoptosis. Representative tumor sections prepared 2 weeks after treatment from mice receiving (a) pcDNA3.1 (control), (b) aHIF‐pcDNA3.1, (c) doxorubicin, or (d) aHIF‐pcDNA3.1 + doxorubicin treatment. Tumor sections were stained with the terminal deoxynucleotidyl transferase biotin‐dUTP nick end labeling agent to view apoptotic cells. (e) Cells stained by the TUNEL agent were counted to calculate the apoptosis index. n, number of tumors assessed. *Significant difference in the apoptosis index for tumors treated with doxorubicin, or aHIF‐pcDNA3.1 versus control; **highly significant difference for tumors treated with doxorubicin + aHIF‐pcDNA3.1 versus control; and ‡significant difference for the combination therapy versus aHIF‐pcDNA3.1 or doxorubicin monotherapy.
Antisense HIF‐1α and doxorubicin downregulates gene expression. The tumor tissues from the above experiments were collected and subjected to western blot analysis to detect the expression of HIF‐1α, VEGF, and PCNA. As shown in Figure 6a, doxorubicin treatment reduced the expression of VEGF, but not HIF‐1α, in accordance with our previous report.( 19 ) Rather, the expression of HIF‐1α was slightly upregulated in tumors treated with doxorubicin compared with controls, but the difference did not reach significance after quantitative analysis (Fig. 6b). Gene transfer of antisense HIF‐1α downregulated the expression of both VEGF and HIF‐1α, as confirmed by quantitative analysis of band densities in Figure 6b. The combination therapy with doxorubicin and antisense HIF‐1α further downregulated the expression of VEGF but not HIF‐1α, compared to antisense HIF‐1α monotherapy. Both doxorubicin and antisense HIF‐1α downregulated the tumoral expression of the cell proliferation marker PCNA, and they synergized to further reduce the expression of PCNA in HepG2 tumors (Fig. 6a,b).
Figure 6.
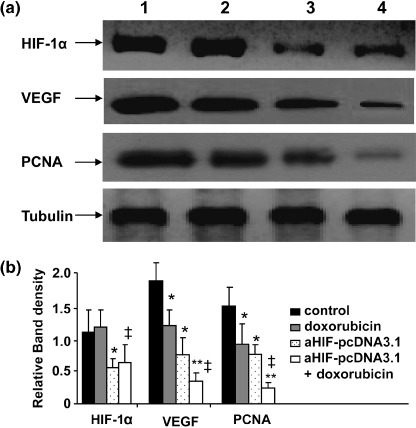
Antisense hypoxia inducible factor (HIF)‐1α and doxorubicin downregulate gene expression. (a) Homogenates of tumors from mice treated with pcDNA3.1 (lane 1), doxorubicin (lane 2), aHIF‐pcDNA3.1 (lane 3), or doxorubicin + aHIF‐pcDNA3.1 (lane 4) were western blotted with antibodies against HIF‐1α, vascular endothelial growth factor (VEGF), proliferating cell nuclear antigen (PCNA), and tubulin. The density of each band was measured and compared to that of the internal control tubulin. Significant difference in the band intensities of each protein for *pcDNA3.1 versus doxorubicin or aHIF‐pcDNA3.1; ‡doxorubicin versus doxorubicin + aHIF‐pcDNA3.1; and **aHIF‐pcDNA3.1 versus doxorubicin + aHIF‐pcDNA3.1.
Antisense HIF‐1α downregulates HIF‐1a expression and inhibits the proliferation of HepG2 cells subjected to hypoxia. Transfection of HepG2 cells with aHIF‐pcDNA3.1 led to the downregulation of HIF‐1α expression in HepG2 cells cultured in the presence of CoCl2 to induce hypoxia, as revealed by western blot analysis (Fig. 7a). As shown in Figure 7b, there was no significant difference in the rate of proliferation of HepG2 cells transfected with aHIF‐pcDNA3.1 and pcDNA3.1, when the cells were cultured under normoxic conditions. However, when the latter cells were exposed to hypoxia induced by CoCl2, the cells transfected with aHIF‐pcDNA3.1 grew significantly more slowly than those transfected with pcDNA3.1 (Fig. 7b).
Figure 7.
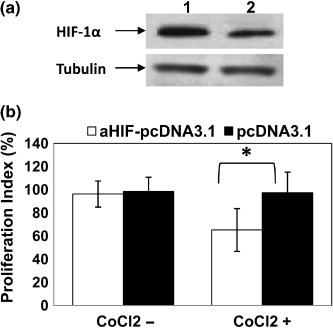
Antisense hypoxia inducible factor (HIF)‐1α gene transfection downregulates HIF‐1α expression and inhibits cell proliferation in vitro. (a) Lysates of HepG2 cells transfected with aHIF‐pcDNA3.1 (lane 2) and pcDNA3.1 (lane 1) were western blotted with antibodies against HIF‐1α and tubulin. (b) HepG2 cells transfected with aHIF‐pcDNA3.1 or pcDNA3.1 were cultured in the absence or presence of CoCl2 to mimic hypoxia. Untreated cells served as controls. Cell proliferation was assessed by the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide method to calculate the proliferation index (% inhibition of cell proliferation). *Significant difference in the proliferation index.
Discussion
The present study has demonstrated that antisense HIF‐1α therapy synergizes with doxorubicin to suppress the growth of subcutaneous human HCC tumors in mice. HCC is extremely insensitive to chemotherapy. Even doxorubicin, the anticancer drug most widely used in the clinic, displays only a 4–10.5% patient response rate, and is associated with significant side effects.( 6 ) The hypoxic microenvironment inside solid tumors is a major cause of tumor resistance to chemotherapy. HIF‐1 is central to the hypoxia response of tumors as it regulates a wide range of hypoxia‐related molecules.( 10 ) In seeking potential strategies to combat the resistance of HCC to doxorubicin, the present study has shown that antisense HIF‐1α exerts its antitumor effect by downregulating the expression of HIF‐1α, VEGF, and PCNA, resulting in inhibition of tumor angiogenesis and cell proliferation, and induction of cell apoptosis. Doxorubicin also downregulated expression of VEGF, but had a negligible effect on the expression of HIF‐1α. In accordance with this, a previous study reported that doxorubicin downregulates VEGF expression in human ovarian cancer cells, and although it inhibits hypoxic activation of HIF‐1, it has no significant effect on the expression levels of HIF‐1α.( 28 ) The slight increase in HIF‐1α expression in tumors treated with doxorubicin may be caused by increased tumor hypoxia.( 14 ) In the present study, antisense HIF‐1α and doxorubicin synergistically suppressed tumor angiogenesis by downregulating VEGF. Thus, combination therapy with doxorubicin and antisense HIF‐1α might be advantageous, as antisense HIF‐1α could be used to reduce the dose of chemotherapeutic agents, thereby sparing the patient from the side effects of cytotoxic drugs without necessarily impairing antitumor efficacy.
It has been well documented that doxorubicin exerts its antitumor activity by inhibiting the proliferation of HCC cells.( 29 , 30 ) Here we have shown that doxorubicin and antisense HIF‐1α synergize to inhibit the proliferation of HCC cells and downregulate the expression of PCNA, a key marker of cell proliferation. Antisense HIF‐1 gene transfection also inhibited the proliferation of HepG2 cells in vitro under hypoxic conditions. In accordance with this, the expression of HIF‐1α mRNA was found to closely correlate with the expression of PCNA in pancreatic cancers,( 31 ) and HIF‐1α protein levels correlated with the PCNA index in transitional‐cell carcinomas of the upper urinary tract.( 32 ) Antisense HIF‐1α may inhibit the proliferation of cells in part by disturbing their energy metabolism, as HIF‐1 controls the expression of the two key glycolysis factors: glucose transporter 1 and lactate dehydrogenase A.( 33 )
The present study has demonstrated that doxorubicin enhances the apoptosis of HepG2 cells in vivo, in accordance with several previous studies.( 26 , 27 ) Antisense HIF‐1α was shown to induce the apoptosis of HepG2 cells in situ, and synergized with doxorubicin to exert increased cell apoptosis. By downregulating VEGF and inhibiting tumor angiogenesis, antisense HIF‐1α may restrict the supply of tumor cell survival factors provided either by endothelial cells or by the circulation, such as platelet‐derived growth factor, interleukin‐6, and heparin‐binding epithelial growth factor.( 34 ) The tumor cells deprived of nutrients and survival factors due to the loss of an adequate vasculature may then subsequently undergo apoptosis. In contrast to most normal cells where the mitochondria are the major ATP producers, cancer cells rely on both mitochondria and glycolysis, and the latter contributes nearly half of the ATP even in the presence of oxygen,( 35 ) and is the main energy‐providing form under hypoxia. HIF‐1‐regulated glucose metabolism is regarded as a key factor in apoptosis resistance.( 11 ) Disruption of HIF‐1α has been shown to impair DNA synthesis and aerobic glycolysis, and inhibit the proliferation of human colon carcinoma HCT116 cells.( 36 )
In summary, antisense HIF‐1α therapy has promising utility in the treatment of HCC as it inhibits tumor angiogenesis, disturbs the energy metabolism of cancer cells, induces tumor cell apoptosis, and enhances the pro‐apoptotic effects of doxorubicin.
Acknowledgments
This work was supported in part by grants from the National Natural Scientific Foundation of China (30571808), the Scientific and Technological Bureau of Heilongjiang Province, China (QC06C075), and the Natural Scientific Foundation of Shandong Province, China (Y2007C107). F. Liu, P. Wang and X. Jiang contributed equally to this work.
References
- 1. Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology 2002; 122: 1609–19. [DOI] [PubMed] [Google Scholar]
- 2. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 3. Zhu AX. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist 2006; 11: 790–800. [DOI] [PubMed] [Google Scholar]
- 4. Sciarrino E, Simonetti RG, Le Moli S et al . Adriamycin treatment for hepatocellular carcinoma. Experience with 109 patients. Cancer 1985; 56: 2751–5. [DOI] [PubMed] [Google Scholar]
- 5. Nerenstone SR, Ihde DC, Friedman MA. Clinical trials in primary hepatocellular carcinoma: current status and future directions. Cancer Treat Rev 1988; 15: 1–31. [DOI] [PubMed] [Google Scholar]
- 6. Yeo W, Mok TS, Zee B et al . A randomized phase III study of doxorubicin versus cisplatin/interferon α‐2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst 2005; 97: 1532–8. [DOI] [PubMed] [Google Scholar]
- 7. Bussink J, Kaanders JH, Van Der Kogel AJ. Tumor hypoxia at the micro‐regional level: clinical relevance and predictive value of exogenous and endogenous hypoxic cell markers. Radiother Oncol 2003; 67: 3–15. [DOI] [PubMed] [Google Scholar]
- 8. Brizel DM, Scully SP, Harrelson JM et al . Tumor oxygenation predicts the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res 1996; 56: 941–3. [PubMed] [Google Scholar]
- 9. Fedele AO, Whitelaw ML, Peet DJ. Regulation of gene expression by the hypoxia‐inducible factors. Mol Interv 2002; 2: 229–43. [DOI] [PubMed] [Google Scholar]
- 10. Semenza GL. Life with oxygen. Science 2007; 318: 62–4. [DOI] [PubMed] [Google Scholar]
- 11. Fulda S, Debatin KM. HIF‐1‐regulated glucose metabolism: a key to apoptosis resistance? Cell Cycle 2007; 6: 790–2. [DOI] [PubMed] [Google Scholar]
- 12. Semenza GL, Jiang BH, Leung SW et al . Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia‐inducible factor 1. J Biol Chem 1996; 271: 32 529–37. [DOI] [PubMed] [Google Scholar]
- 13. Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia‐inducible factor 1. J Biol Chem 1996; 271: 17 771–8. [DOI] [PubMed] [Google Scholar]
- 14. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 2001; 93: 266–76. [DOI] [PubMed] [Google Scholar]
- 15. Unruh A, Ressel A, Mohamed HG et al . The hypoxia‐inducible factor‐1 α is a negative factor for tumor therapy. Oncogene 2003; 22: 3213–20. [DOI] [PubMed] [Google Scholar]
- 16. Williams KJ, Telfer BA, Xenaki D et al . Enhanced response to radiotherapy in tumours deficient in the function of hypoxia‐inducible factor‐1. Radiother Oncol 2005; 75: 89–98. [DOI] [PubMed] [Google Scholar]
- 17. Song X, Liu X, Chi W et al . Hypoxia‐induced resistance to cisplatin and doxorubicin in non‐small cell lung cancer is inhibited by silencing of HIF‐1 α gene. Cancer Chemother Pharmacol 2006; 58: 776–84. [DOI] [PubMed] [Google Scholar]
- 18. Shin DH, Kim JH, Jung YJ et al . Preclinical evaluation of YC‐1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett 2007; 255: 107–16. [DOI] [PubMed] [Google Scholar]
- 19. Liu F, Tan G, Li J, Dong X, Krissansen GW, Sun X. Gene transfer of endostatin enhances the efficacy of doxorubicin to suppress human hepatocellular carcinomas in mice. Cancer Sci 2007; 98: 1381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun X, Kanwar JR, Leung E, Lehnert K, Wang D, Krissansen GW. Gene transfer of antisense hypoxia inducible factor‐1α enhances the therapeutic efficacy of cancer immunotherapy. Gene Ther 2001; 8: 638–45. [DOI] [PubMed] [Google Scholar]
- 21. Sun X, Kanwar JR, Leung E, Vale M, Krissansen GW. Regression of solid tumors by engineered overexpression of von Hippel‐Lindau tumor suppressor protein and antisense hypoxia‐inducible factor‐1α. Gene Ther 2003; 10: 2081–9. [DOI] [PubMed] [Google Scholar]
- 22. Li J, Tan H, Dong X et al . Antisense integrin αV and β3 gene therapy suppresses subcutaneously implanted hepatocellular carcinomas. Dig Liver Dis 2007; 39: 557–65. [DOI] [PubMed] [Google Scholar]
- 23. Ma L, Luo L, Qiao H et al . Vasostatin synergizes with B7H3‐mediated immunotherapy to eradicate hepatocellular carcinomas. J Hepatol 2006; 46: 98–106. [DOI] [PubMed] [Google Scholar]
- 24. Xu R, Sun X, Chan D et al . Long‐term expression of angiostatin suppresses liver metastatic cancer in mice. Hepatology 2003; 37: 1451–60. [DOI] [PubMed] [Google Scholar]
- 25. Sun X, Liu M, Wei Y et al . Overexpression of von Hippel–Lindau tumor suppressor protein and antisense HIF‐1α eradicates gliomas. Cancer Gene Ther 2006; 13: 428–35. [DOI] [PubMed] [Google Scholar]
- 26. Manov I, Bashenko Y, Eliaz‐Wolkowicz A, Mizrahi M, Liran O, Iancu TC. High‐dose acetaminophen inhibits the lethal effect of doxorubicin in HepG2 cells: the role of P‐glycoprotein and mitogen‐activated protein kinase p44/42 pathway. Pharmacol Exp Ther 2007; 322: 1013–22. [DOI] [PubMed] [Google Scholar]
- 27. Pan YY, Xu SP, Jia XY et al . Combination of cyclooxygenase‐2 inhibitor and doxorubicin increases the growth inhibition and apoptosis in human hepatocellular carcinoma cells. Exp Oncol 2007; 29: 23–9. [PubMed] [Google Scholar]
- 28. Duyndam MC, Van Berkel MP, Dorsman JC, Rockx DA, Pinedo HM, Boven E. Cisplatin and doxorubicin repress vascular endothelial growth factor expression and differentially down‐regulate hypoxia‐inducible factor I activity in human ovarian cancer cells. Biochem Pharmacol 2007; 74: 191–201. [DOI] [PubMed] [Google Scholar]
- 29. Chuu JJ, Liu JM, Tsou MH et al . Effects of paclitaxel and doxorubicin in histocultures of hepatocelular carcinomas. J Biomed Sci 2007; 14: 233–44. [DOI] [PubMed] [Google Scholar]
- 30. Sutter AP, Maaser K, Grabowski P et al . Peripheral benzodiazepine receptor ligands induce apoptosis and cell cycle arrest in human hepatocellular carcinoma cells and enhance chemosensitivity to paclitaxel, docetaxel, doxorubicin and the Bcl‐2 inhibitor HA14‐1. J Hepatol 2004; 41: 799–807. [DOI] [PubMed] [Google Scholar]
- 31. Wei H, Wang C, Chen L. Proliferating cell nuclear antigen, survivin, and CD34 expressions in pancreatic cancer and their correlation with hypoxia‐inducible factor 1α. Pancreas 2006; 32: 159–63. [DOI] [PubMed] [Google Scholar]
- 32. Nakanishi K, Hiroi S, Tominaga S et al . Expression of hypoxia‐inducible factor‐1α protein predicts survival in patients with transitional cell carcinoma of the upper urinary tract. Clin Cancer Res 2005; 11: 2583–90. [DOI] [PubMed] [Google Scholar]
- 33. Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor‐associated stroma. Cancer Res 2006; 66: 632–7. [DOI] [PubMed] [Google Scholar]
- 34. Rak JW, St Croix BD, Kerbel RS. Consequences of angiogenesis for tumour progression, metastasis and cancer therapy. Anticancer Drugs 1995; 6: 3–18. [DOI] [PubMed] [Google Scholar]
- 35. Pedersen PL. The cancer cell's ‘power plants’ as promising therapeutic targets: An overview. J Bioenerg Biomembr 2007; 39: 1–12. [DOI] [PubMed] [Google Scholar]
- 36. Dang DT, Fang C, Gardner LB et al . Hypoxia‐inducible factor‐1α promotes nonhypoxia‐mediated proliferation in colon cancer cells and xenografts. Cancer Res 2006; 66: 1684–93. [DOI] [PubMed] [Google Scholar]