

Abstract
To elucidate the roles of the transcription factor NF‐E2‐related factor (Nrf2) in hepatocarcinogenesis induced by 2‐amino‐3‐methylimidazo[4,5‐f]quinoline (IQ), a mutagenic and carcinogenic heterocyclic amine, Nrf2‐deficient mice were treated with 300 p.p.m. IQ in their diet for 1, 4 or 52 weeks. In the long‐term experiment, the multiplicity and incidence of liver tumors in male and female IQ‐treated Nrf2 deficient (–/–) mice were significantly higher than those in their counterpart wild‐type (+/+) mice exposed to IQ. In the short‐term experiment, although IQ exposure to Nrf2(+/+) mice of both sexes did not modify UDP‐glucuronosyltransferase values, glutathione S‐transferase values were significantly increased due to IQ treatment, in contrast to no alteration in male and female Nrf2(–/–) mice. Levels of oxidative stress markers such as 8‐hydroxydeoxyguanosine and thiobarbituric acid reactive substances in the livers of all treated mice were not changed by IQ treatment. IQ‐specific DNA adduct levels were elevated only in female Nrf2(–/–) mice, although the increase was not significant. IQ treatment caused an increase in proliferating cell nuclear antigen labeling indices only in male Nrf2(–/–) mice. The present data clearly show that Nrf2(–/–) mice of both sexes are susceptible to IQ hepatocarcinogenicity, which might result from IQ accumulation due to failure of metabolizing enzyme induction. In addition, inconsistent results concerning IQ‐specific adducts and proliferating cell nuclear antigen labeling indices in male and female Nrf2(–/–) mice suggest the existence of different contributions of Nrf2 to IQ hepatocarcinogenesis between mice of the two sexes. (Cancer Sci 2007; 98: 19–24)
Abbreviations:
- 8‐OHdG
8‐hydroxydeoxyguanosine
- ARE
antioxidant responsive element
- CDNB
l‐chloro‐2,4‐dinitrobenzene
- EDTA
ethylenediaminetetraacetic acid
- GST
glutathione S‐transferase
- HCA
heterocyclic amines
- IQ
2‐amino‐3‐methylimidazo[4,5‐f]quinoline
- NaNO2
sodium nitrite
- Nrf2
NF‐E2‐related factor
- PCNA
proliferating cell nuclear antigen
- PCNA‐LI
proliferating cell nuclear antigen labeling indices
- PCR
polymerase chain reaction
- TBARS
thiobarbituric acid reactive substances
- TLC
thin‐layer chromatography
- UGT
UDP‐glucuronosyltransferase.
2‐Amino‐3‐methylimidazo[4,5‐f]quinoline, a mutagenic and carcinogenic HCA, has been detected in cooked meat and fish,( 1 ) and also in cigarette smoke condensate( 2 ). IQ is carcinogenic to the liver, colon, small intestine, Zymbal's gland, clitoral gland and skin in F344 rats,( 3 ) to the liver, forestomach and lung in CDF1 mice,( 4 ) and to the liver in B6C3F1 mice( 5 ) in long‐term experiments. Recently, we have found that rat lung is also a target of IQ carcinogenicity.( 6 ) Its carcinogenesis is considered to involve mainly IQ bioactivation, that is phase I hepatic CYP1A1/2‐mediated N‐hydroxylation followed by phase II (mainly N‐acetyltransferase and sulfotransferase) esterification of N‐hydroxylamines to reactive ester derivatives that bind to DNA covalently.( 7 , 8 ) In addition, it was suggested recently that oxidative stress may also participate in HCA carcinogenesis, based on the findings that induction of preneoplastic or neoplastic lesions following HCA exposure was significantly inhibited when given simultaneously with antioxidants in rats,( 9 , 10 ) and that superoxide anion radicals were generated during metabolism of HCA by NADPH/cytochrome P‐450 reductase in vitro using a spin‐trapping method.( 11 ) We have demonstrated enhancement of IQ‐induced colon tumor development due to cotreatment with NaNO2 and the participation of oxidative stress such as oxidative DNA damage and lipid peroxidation in this phenomenon. However, IQ‐induced liver tumors were only slightly increased by the addition of NaNO2, the relation to oxidative stress being equivocal.( 12 ) Thus, IQ causes carcinogenesis in a broad spectrum of target organs and involves several contributing factors, making it necessary to investigate the key factors in in vivo defense against IQ carcinogenicity.
NF‐E2‐related factor, a basic leucine zipper transcription factor, has recently been highlighted in terms of important roles for cellular defense against xenobiotics.( 13 ) It is believed that Nrf2 is released from binding to Keap1, a homolog of the Drosophila actin‐binding protein, in response to electrophiles or reactive oxygen species, is translocated to the nucleus and consequently upregulates ARE‐mediated transcription, leading to the induction of antioxidant enzymes such as thioredoxin,( 14 ) heme oxygenase 1( 15 , 16 ) and glutamate cysteine ligase,( 15 , 17 ) and conjugation enzymes such GST( 15 , 18 ) and UGT.( 15 , 19 ) Several reports have shown that acetaminophen hepatotoxicity,( 20 , 21 ) butylated hydroxytoluene pulmonary toxicity,( 22 ) benzo[a]pyrene forestomach carcinogenicity( 23 ) and N‐butyl‐N‐(4‐hydroxybutyl)nitrosamine urinary bladder carcinogenicity( 24 ) are enhanced in Nrf2‐deficient (–/–) mice. We also demonstrated that liver DNA of Nrf2(–/–) mice is subjected to oxidative stress induced by pentachlorophenol, a mouse hepatocarcinogen.( 25 ) A recent report revealed that mutations have been found in the Keap1 gene of human lung cancer cells, which suggests that consequent gain of Nrf2 function induces the expression of cytoprotective enzymes in these cells.( 26 ) Together with the possible participation of oxidative stress in IQ hepatocarcinogenesis, considering that some electrophiles arise from the IQ metabolizing pathway, it is highly probable that Nrf2 plays a crucial role in defense against IQ‐induced carcinogenicity.
In the present experiment, to assess the roles of Nrf2 in IQ liver carcinogenesis, Nrf2(–/–) mice of both sexes were used to examine IQ‐induced liver tumorigenicity. Additionally, the effects of Nrf2 deficiency on IQ exposure‐related parameters such as UGT and GST enzyme activities, oxidative DNA damage, lipid peroxidation, IQ‐specific DNA adduct formation and hepatocyte proliferation were assessed.
Materials and Methods
Chemicals. 2‐Amino‐3‐methylimidazo[4,5‐f]quinoline was purchased from Toronto Research Chemicals (North York, Canada). Monoclonal mouse anti‐PCNA, biotin‐labeled goat antimouse IgG and streptavidin–biotin–peroxidase complex labeled with horseradish peroxidase were purchased from DakoCytomation (Kyoto, Japan). Micrococcal nuclease and spleen phosphodiesterase were purchased from Worthington Biochemicals (Lakewood, NJ, USA). Nuclease P1 for the determination of IQ‐specific DNA adducts and 8‐OHdG levels was purchased from MP Biomedicals (Irvine, CA, USA) and Yamasa Shoyu (Chiba, Japan), respectively. T4 polynucleotide kinase was purchased from Takara Bio (Ootsu, Japan). [γ‐32P]ATP was purchased from Amersham Biosciences (Piscataway, NJ, USA). Potato apyrase, alkaline phosphatase and deferoxamine mesylate were purchased from Sigma Chemical (St Louis, MO, USA). 2‐Thiobarbituric acid, p‐nitrophenol, CDNB and glutathione were purchased from Wako Pure Chemical Industries (Osaka, Japan). UDP‐glucuronic acid was purchased from Nacalai Tesque (Kyoto, Japan).
Animals. Nrf2‐deficient mice were produced as described by Itoh et al.( 27 ) In brief, Nrf2‐deficient mice with an ICR/129SVJ background were crossed with ICR mice (Japan SLC, Shizuoka, Japan). Homozygous (–/–) and wild‐type littermates (+/+) were obtained from the F1 generation and genotypes were confirmed by PCR amplification of genomic DNA isolated from their tails. PCR amplification was carried out using three different primers: 5′‐TGGACGGGACTATTGAAGGCTG‐3′ (sense for both genotypes), 5′‐GCCGCCTTTTCAGTAGATGGAGG‐3′ (antisense for wild type) and 5′‐GCGGATTGACCGTAATGGGATAGG‐3′ (antisense for LacZ). Totals of 60 mice of both sexes for each genotype, at 7 weeks of age, were assigned to two groups consisting of 20 (experiment 1: histopathological examination) and four groups consisting of five (experiment 2: examination of parameters related to carcinogenesis) animals each (six groups in total). They were housed in plastic cages (five mice/cage) with soft chips for bedding in a room with a barrier system, and maintained under the following conditions: temperature, 23 ± 2°C; relative humidity, 60 ± 5%; ventilation frequency, 18 times/h; and illumination, 12:12 h L:D cycle. Normal powdered diet (CRF‐1; Oriental Yeast, Tokyo, Japan) and tap water were available ad libitum. The animal protocols for these studies were approved by the Animal Care and Utilization Committee of the National Institute of Health Sciences, Japan.
Experiment 1
Experimental design From the age of 8 weeks, animals of both sexes for each genotype were fed a powdered diet containing IQ at a concentration of 300 p.p.m. or basal diet alone for 52 weeks. The dose level of IQ was selected according to a report in which liver, forestomach and lung tumors were induced at 300 p.p.m. IQ in a long‐term experiment using CDF1 mice.( 4 ) Bodyweights and food consumption were measured once a week until week 4 and thereafter once every 4 weeks. All surviving animals were killed under ether anesthesia at the end of the experimental period.
Major organs and tissues were examined carefully and the liver was weighed at autopsy. All of the organs and tissues, including the liver, kidneys, lung, stomach, small intestine, colon, Zymbal's glands and any other gross lesions, were fixed and preserved in 10% phosphate‐buffered formalin solution. Organs of animals found dead or killed on becoming moribund were also fixed and preserved.
Histopathology All of the preserved organs and tissues were embedded in paraffin, sectioned and stained with hematoxylin and eosin for routine histopathological examination.
Experiment 2
Experimental design From the age of 8 weeks, animals of both sexes for each genotype were fed a powdered diet containing IQ at a concentration of 300 p.p.m. or basal diet alone for 1 or 4 weeks. All surviving animals were killed under ether anesthesia at the end of the experimental period and the livers were removed. A part of each of the left lateral and medial lobes at week 4 of administration was fixed and preserved in 10% phosphate‐buffered formalin for PCNA staining. The remainder was frozen immediately with liquid nitrogen and stored at −80°C until used for analysis of UGT and GST enzyme activities and 8‐OHdG, TBARS and IQ‐specific DNA adduct levels. For 8‐OHdG levels, liver samples from the animals fed IQ for 1 week were also frozen and preserved.
Liver microsome preparation For preparation of hepatic microsomal suspensions, whole liver samples were homogenized in ice‐cold 0.25 M sucrose solution containing 1 mM EDTA and 10 mM Tris‐HCl (pH 7.4). Hepatic homogenates (approximately 20% w/v) were centrifuged at 9000g for 20 min at 4°C and the supernatants were centrifuged at 105 000g for 60 min at 4°C. The precipitates were homogenized with 1.15% KCl in 25 mM Tris‐HCl (pH 7.4) and centrifuged at 105 000g for 60 min at 4°C. The precipitates were suspended with 20% glycerol containing 0.1 mM EDTA in 10 mM phosphate (pH 7.4). Protein concentrations of the resulting suspensions were determined with a Lowry Protein Assay Kit (Nacalai Tesque, Kyoto, Japan). Aliquots were stored in a deep‐freezer (–80°C) until use.
Measurement of UGT activity p‐Nitrophenol UGT activity of the microsomal preparations was measured using the method of Burchell and Weatherill.( 28 ) In brief, the same volume of microsomal sample solution (2.0 mg/mL), 1.5 mM p‐nitrophenol dissolved in a UGT reaction buffer (75 mM Tris‐HCl [pH 8.0] and 15 mM MgCl2) and 6.0 mM UDP‐glucuronic acid dissolved in UGT reaction buffer were incubated at 37°C for 10 min. The reaction was terminated by incubation in boiled water for 2 min, followed by mixing with 0.1 M KOH and cooling on ice for 30 min. Each mixture was centrifuged at 1660g for 20 min at 4°C and the supernatant obtained was measured with a spectrophotometer (U‐3310; Hitachi High‐Technologies, Tokyo, Japan) at 405 nm with an extinction coefficient of 18.1 mM−1·cm−1.
Liver cytosol preparation For preparation of hepatic cytosol solutions, whole‐liver samples were homogenized in ice‐cold 67 mM phosphate buffer (pH 7.5) containing 1.15% (w/v) KCl. Hepatic homogenates (∼20% w/v) were centrifuged at 9000g for 20 min at 4°C and the supernatants were centrifuged at 105 000g for 60 min at 4°C. The supernatants were dialyzed against 67 mM phosphate buffer (pH 7.5). Protein concentrations of the resulting solutions were determined with a BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA).
Measurement of GST activity Total GST activity of cytosolic preparations was measured using the method of Habig et al.( 29 ) In brief, the substrate CDNB (0.02–l mM) was added to l mM glutathione and cytosolic protein (0.4 mg/mL) in 100 mM potassium phosphate buffer (pH 6.5) to a final volume of 1 mL. The reaction was followed spectrophotometrically at 340 nm with a spectrophotometer for CDNB with an extinction coefficient of 9.6 mM−1·cm−1.
Measurement of 8‐OHdG formation in DNA To prevent 8‐OHdG formation as a byproduct during DNA isolation,( 30 ) the 8‐OHdG level in liver DNA was determined using the method of Nakae et al.,( 31 ) with slight modification using approximately 0.3 g wet weight samples. Nuclear DNA was isolated with a DNA Extracter WB Kit (Wako Pure Chemical Industries, Osaka, Japan) containing an antioxidant NaI solution to dissolve cellular components. For further prevention of autooxidation in the cell lysis step, deferoxamine mesylate was added to the lysis buffer.( 32 ) The DNA was digested into deoxynucleotides by treatment with nuclease P1 and alkaline phosphatase. Levels of 8‐OHdG (8‐OHdG/105 deoxyguanosine) were then assessed by high‐performance liquid chromatography (column: Beckman Ultrasphere‐ODS, 5 µm, 4.6 × 250 mm; Beckman Coulter, Fullerton, CA, USA; elution buffer, 10 mM NaH2PO4 containing 4% methanol) with an electrochemical detection system (Coulochem II; ESA, Bedford, MA, USA; guard cell, model 5020 [0.35 V]; analytical cell, model 5011 [electrode 1, 0.15 V; electrode 2, 0.3 V]).
Measurement of TBARS Malondialdehyde (nmol/g) was assessed as an index of lipid peroxidation using the method of Uchiyama and Mihara.( 33 ) In brief, a 0.15‐g portion of liver was homogenized with 1.35 mL of 1.15% KCl solution. To 0.05 mL of this homogenate, 0.2 mL 8.1% sodium dodecylsulfate and 3.0 mL 0.4% 2‐thiobarbituric acid in 10% acetic acid solution (pH 3.5) were added, followed by heating in a water bath at 95°C for 60 min. After cooling, 5.0 mL of n‐butanol and pyridine (15:1 v/v) and 1.0 mL distilled water were added to the sample, which was centrifuged at 1870 g for 10 min. The TBARS were measured with a Hitachi F‐2500 fluorescence spectrophotometer (Hitachi High‐Technologies, Tokyo, Japan) at 515 nm (excitation) and 553 nm (emission) in the butanol–pyridine phase.
Measurement of IQ‐specific DNA adduct levels IQ‐specific DNA adduct levels were determined using the methods of Ochiai et al. ( 34 ) and Williams et al.,( 35 ) with slight modification. Nuclear DNA (0.5 µg/µL, 20 µL) from each liver was isolated with a Tissue Kit (D‐7000A; Gentra Systems, Minneapolis, MN, USA) and digested into 3′‐mononucleotides by treatment with micrococcal nuclease (4 IU) and spleen phosphodiesterase (0.04 IU) at 37°C for 3.5 h. The DNA digest (2 µg) was incubated with 0.4 µg/µL nuclease P1 and 37 µM ZnCl2 in 50 mM sodium acetate buffer (pH 5.0) at 38°C for 40 min. The reaction was stopped with the addition of 75 mM Tris base and the mixture was dried in a SpeedVac evaporator for 1 h. It was the 32P‐labeled with 3 IU of T4 polynucleotide kinase and [γ‐32P]ATP (111–185 TBq/mmol, 37 MBq/mL) at 38°C for 40 min in a reaction mixture (10 mM MgCl2, 10 mM dithiothreitol, 1 mM spermidine, 30 mM Tris‐HCl [pH 9.5]). The 32P‐labeled samples were treated with 20 µΙU of potato apyrase at 38°C for 20 min and adducted nucleotides were analyzed by TLC thereafter. The adducts were chromatographed on polyethyleneimine‐cellulose TLC plates with the following TLC solvents: D1, 2.3 M sodium phosphate (pH 6.0); D2, 0.45 M lithium formate, 6.4 M urea (pH 3.5); D3, 0.7 M sodium phosphate, 8.5 M urea (pH 8.0); D4, 1.7 M sodium phosphate (pH 6.0). DNA adducts were detected and quantified with a fluoroimage analyzer (FLA‐5000; Fuji Photo Film, Tokyo, Japan). The level of DNA adducts was determined by relating levels of radioactivity in total amounts of 32P‐labeled normal nucleotides and expressed as relative adduct labeling (adducts/107 nucleotides).
Immunohistochemical staining for PCNA Deparaffinized liver sections were heated in 0.01 M citrate buffer (pH 6.0) in a microwave oven for 10 min after boiling and then treated with 3% hydrogen peroxide to block endogenous peroxidase activity. After blocking non‐specific binding sites with normal goat serum, sections were treated with monoclonal mouse anti‐PCNA (1:100), biotin‐labeled goat antimouse IgG (1:200) and streptavidin–biotin–peroxidase complex labeled with horseradish peroxidase. Peroxidase activity was visualized by treatment with a solution of diaminobenzidine tetrahydrochloride containing hydrogen peroxide and the nuclei were counterstained with hematoxylin.
Cell proliferation quantification in liver In the liver, at least 3000 hepatocytes were counted for each animal and PCNA‐LI were calculated as percentages of positive cells.
Statistical analysis. Incidences of lesions were analyzed by the Fisher's exact test. Data for bodyweight, organ weight, lesion multiplicities, UGT and GST activity, 8‐OHdG and TBARS levels and the proportion of PCNA‐positive cells were examined using one‐way ANOVA followed by Tukey's multiple comparison. Data for IQ‐specific DNA adduct levels were analyzed using the Student's t‐test.
Results
Histopathology (experiment 1). The results for final body and relative liver weights are shown in Table 1. No significant differences in final bodyweights regarding IQ administration were observed in either sex. Relative liver weights in the IQ‐treated groups of both genotypes tended to be or were significantly increased as compared with the relevant controls in both sexes. Between the IQ‐treated groups, relative liver weights in males of the Nrf2(–/–) group were significantly increased compared with those of the Nrf2(+/+) group.
Table 1.
Final body and relative liver weights, and incidences and multiplicity of liver tumors (experiment 1)
| Treatment | Mice (n) | BW (g) | Liver (g/100 g BW) | Hepatocellular adenoma | Hepatocellular carcinoma | Total | |||
|---|---|---|---|---|---|---|---|---|---|
| Incidence | Multiplicity (no./mouse) | Incidence | Multiplicity (no./mouse) | Incidence | Multiplicity (no./mouse) | ||||
| Male | |||||||||
| BD, Nrf2(+/+) | 18 | 53.7 ± 7.8 | 3.77 ± 0.55 | 4 (22) | 0.28 ± 0.57 | 1 (6) | 0.11 ± 0.47 | 4 (22) | 0.39 ± 0.98 |
| BD, Nrf2(–/–) | 19 | 51.9 ± 6.4 | 3.96 ± 1.22 | 2 (11) | 0.16 ± 0.50 | 1 (5) | 0.11 ± 0.46 | 3 (16) | 0.26 ± 0.65 |
| IQ, Nrf2(+/+) | 17 | 50.3 ± 7.0 | 4.06 ± 0.61 | 1 (6) | 0.12 ± 0.49 | 7 (41)* | 0.41 ± 0.51 | 7 (41) | 0.53 ± 0.80 |
| IQ, Nrf2(–/–) | 15 | 50.6 ± 7.2 | 7.02 ± 6.44 [Link] , [Link] | 4 (27) | 0.33 ± 0.62 | 10 (67) § | 1.13 ± 0.99 [Link] , [Link] | 11 (73) § | 1.47 ± 1.13 [Link] , [Link] |
| Female | |||||||||
| BD, Nrf2(+/+) | 17 | 44.2 ± 6.3 | 2.98 ± 0.25 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| BD, Nrf2(–/–) | 19 | 37.2 ± 7.2* | 3.21 ± 0.49 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| IQ, Nrf2(+/+) | 18 | 40.7 ± 7.5 | 4.09 ± 1.31 † | 4 (22) | 0.39 ± 0.78 | 2 (11) | 0.28 ± 0.96 | 4 (22) | 0.67 ± 1.57 |
| IQ, Nrf2(–/–) | 19 | 38.4 ± 9.5 | 4.10 ± 2.03 ‡ | 6 (32) § | 0.63 ± 1.12 § | 8 (42) [Link] , [Link] | 0.53 ± 0.70 § | 11 (58) [Link] , [Link] | 1.16 ± 1.42 § |
Data are presented as mean ± SD. IQ (2‐amino‐3‐methylimidazo[4,5‐f]quinoline) was administered at 300 p.p.m. *P < 0.05, † P < 0.01: significantly different from the untreated Nrf2(+/+) group. ‡ P < 0.05, § P < 0.01, significantly different from the untreated Nrf2(–/–) group. ¶ P < 0.05, **P < 0.01; significantly different from the Nrf2(+/+) group treated with 300 p.p.m. IQ. BD, basal diet; BW, bodyweight. Numbers in parentheses indicate the percentage (%) of incidence.
The data for final incidences and multiplicity of liver tumors are summarized in Table 1. In the Nrf2(+/+) group for both sexes, total incidences of liver tumors and multiplicities of adenomas and carcinomas showed no significant difference with or without IQ administration except for the male carcinoma incidence for which a significant increase was observed with IQ treatment. However, treatment with IQ in the Nrf2(–/–) group of both sexes significantly increased the total tumor incidence and multiplicity as compared with the relevant controls, except for hepatocellular adenomas in males. When compared between IQ‐treated groups, multiplicities of carcinomas and total tumors in males and incidences of carcinomas and total tumors in females of the Nrf2(–/–) group were significantly increased compared with the relevant Nrf2(+/+) group.
The data for final incidences of preneoplastic and neoplastic lesions in forestomach and lung are summarized in Table 2. Forestomach squamous cell hyperplasias and papillomas were increased by IQ treatment in both sexes. The incidences of squamous cell hyperplasias and papillomas in males of the Nrf2(–/–) group were significantly increased and tended to increase compared with the respective values for the relevant Nrf2(+/+) group. Lung preneoplastic and neoplastic lesions were found sporadically regardless of IQ treatment.
Table 2.
Incidences of preneoplastic and neoplastic lesions in the forestomach and lung (experiment 1)
| Forestomach | Lung | ||||||
|---|---|---|---|---|---|---|---|
| Treatment | Mice (n) | Squamous cell hyperplasia | Papilloma | Squamous cell carcinoma | Alveolar hyperplasia | Adenoma | Adenocarcinoma |
| Male | |||||||
| BD, Nrf2(+/+) | 18 | 0 (0) | 0 (0) | 0 (0) | 1 (6) | 0 (0) | 2 (11) |
| BD, Nrf2(–/–) | 19 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (21) |
| IQ, Nrf2(+/+) | 17 | 1 (6) | 1 (6) | 1 (6) | 2 (12) | 0 (0) | 1 (6) |
| IQ, Nrf2(–/–) | 15 | 7 (47)[Link], [Link] | 3 (20) | 0 (0) | 0 (0) | 1 (7) | 2 (13) |
| Female | |||||||
| BD, Nrf2(+/+) | 17 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (6) | 1 (6) |
| BD, Nrf2(–/–) | 19 | 0 (0) | 0 (0) | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| IQ, Nrf2(+/+) | 18 | 6 (33)* | 4 (22) | 0 (0) | 1 (6) | 0 (0) | 0 (0) |
| IQ, Nrf2(–/–) | 19 | 8 (42)** | 4 (21) | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
IQ (2‐amino‐3‐methylimidazo[4,5‐f]quinoline) was administered at 300 p.p.m. *P < 0.05, significantly different from the untreated Nrf2(+/+) group. **P < 0.01, significantly different from the untreated Nrf2(–/–) group. ***P < 0.05, significantly different from the Nrf2(+/+) group treated with 300 p.p.m. IQ. BD, basal diet. Numbers in parentheses indicate the percentage (%) of incidence.
Parameters related to carcinogenesis in the liver (experiment 2). The results for UGT and GST activities and 8‐OHdG, TBARS, IQ‐specific DNA adduct and PCNA‐LI levels in the liver are shown in Table 3. For enzyme activities, despite the differences between the genotypes of the males treated with or without IQ being statistically significant, IQ treatment did not modify UGT values in the groups of either sex. In contrast, the treatment significantly increased GST values in Nrf2(+/+) groups in both sexes compared with the relevant controls. However, IQ‐induced GST enzyme activity exhibited no alteration in Nrf2(–/–) groups of either sex as compared with the relevant control, being significantly decreased as compared with the IQ‐treated Nrf2(+/+) groups.
Table 3.
Parameters related to carcinogenesis in the liver (experiment 2)
| Treatment | No. samples | UGT activity (nmol/min/ mg protein) | GST activity (nmol/min/ mg protein) | 8‐OHdG level | TBARS level (nmol MDA/ g liver) | IQ‐adducted DNA (/107 nucleotides) | PCNA‐LI † | |
|---|---|---|---|---|---|---|---|---|
| (8‐OHdG/105 dG) | ||||||||
| Week 1 | Week 4 | |||||||
| Male | ||||||||
| BD, Nrf2(+/+) | 5 | 9.34 ± 0.82 | 415 ± 122 | 0.32 ± 0.07 | 0.33 ± 0.16 | 66.2 ± 13.4 | ND | 0.74 ± 0.71 |
| BD, Nrf2(–/–) | 5 | 6.76 ± 0.97** | 249 ± 146 | 0.37 ± 0.07 | 0.38 ± 0.10 | 91.4 ± 23.4 | ND | 1.07 ± 1.10 |
| IQ, Nrf2(+/+) | 5 (3) | 9.91 ± 0.70 | 630 ± 121* | 0.28 ± 0.10 | 0.31 ± 0.17 | 77.9 ± 3.8 | 1.52 ± 0.49 | 2.86 ± 1.68 |
| IQ, Nrf2(–/–) | 5 (3) | 7.23 ± 0.92***** | 271 ± 57***** | 0.29 ± 0.10 | 0.34 ± 0.13 | 87.2 ± 21.5 | 1.65 ± 0.80 | 6.72 ± 3.98*** |
| Female | ||||||||
| BD, Nrf2(+/+) | 5 | 8.13 ± 1.62 | 163 ± 44 | 0.36 ± 0.15 | 0.35 ± 0.12 | 88.1 ± 14.2 | ND | 1.90 ± 0.78 |
| BD, Nrf2(–/–) | 5 | 6.33 ± 1.22 | 114 ± 55 | 0.29 ± 0.10 | 0.26 ± 0.10 | 112.6 ± 25.9 | ND | 2.53 ± 1.45 |
| IQ, Nrf2(+/+) | 5 (3) | 8.93 ± 0.41 | 365 ± 98** | 0.26 ± 0.07 | 0.27 ± 0.08 | 100.0 ± 14.9 | 2.57 ± 1.07 | 2.10 ± 1.35 |
| IQ, Nrf2(–/–) | 5 (3) | 7.43 ± 0.84 | 137 ± 69***** | 0.24 ± 0.05 | 0.36 ± 0.20 | 155.3 ± 49.3**** | 4.53 ± 1.38 | 4.08 ± 5.05 |
IQ (2‐amino‐3‐methylimidazo[4,5‐f]quinoline) was administered at 300 p.p.m. Numbers in parentheses indicate the number of samples used for the measurement of IQ‐specific DNA adduct formation. Data are presented as mean ± SD. †Average number of proliferating cell nuclear antigen (PCNA)‐positive cells/total cells (%). *P < 0.05, **P < 0.01: significantly different from the untreated Nrf2(+/+) group. ***P < 0.01: significantly different from the untreated Nrf2(–/–) group. ****P < 0.05, *****P < 0.01: significantly different from the Nrf2(+/+) group treated with IQ. 8‐OHdG, 8‐hydroxydeoxyguanosine; BD, basal diet; GST, glutathione S‐transferase; ND, not determined; PCNA‐LI, proliferating cell nuclear antigen labeling indices; TBARS, thiobarbituric acid reactive substances; UGT, UDP‐glucuronosyltransferase.
There were no significant differences in 8‐OHdG levels among any of groups of either sex irrespective of IQ treatment throughout the experimental period. Also, IQ treatment did not increase the TBARS levels independent of Nrf2.
Six and nine different spots of IQ‐specific DNA adducts were observed in the males and females, respectively. The IQ‐specific DNA adduct level in the male Nrf2(–/–) group was almost equivalent to that in the male Nrf2(+/+) group. In contrast, the levels of some spots in the female Nrf2(–/–) group were two or three times higher than those in the female Nrf2(+/+) group (data not shown), the total value in the female Nrf2(–/–) group being approximately 1.8 times higher than in the female Nrf2(+/+) group (Fig. 1).
Figure 1.
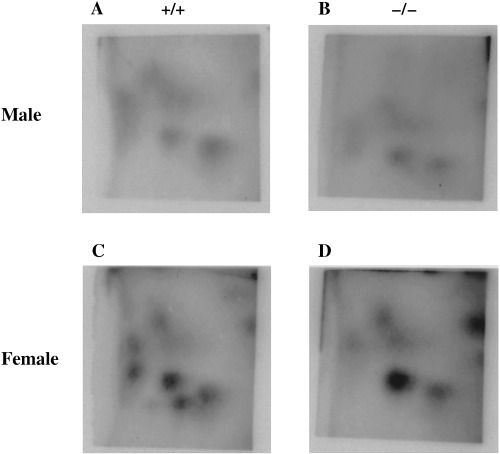
2‐Amino‐3‐methylimidazo[4,5‐f]quinoline (IQ) DNA adduct formation in the livers of NF‐E2‐related factor (Nrf2) (+/+) and Nrf2(–/–) mice. Mice were treated with 300 p.p.m. IQ for 4 weeks. After autopsy, liver DNA was isolated, digested, 32P‐labeled and analyzed by thin‐layer chromatography. Six and nine different spots were found in the males and females, respectively. Male (A) Nrf2(+/+) and (B) Nrf2(–/–) mice treated with IQ. Female (C) Nrf2(+/+) and (D) Nrf2(–/–) mice treated with IQ.
Although treatment with IQ did not affect the PCNA‐LI in males of the Nrf2(+/+) group, the value for the Nrf2(–/–) group males treated with IQ was significantly elevated compared with the relevant control level. There were no statistically significant changes in the females. Those data might account for the fact that the average areas of the tumors on the slide sections in the IQ‐treated male Nrf2(–/–) group were the largest among the groups (data not shown).
Discussion
2‐Amino‐3‐methylimidazo[4,5‐f]quinoline exposure of Nrf2(+/+) mice of both sexes under the present conditions increased the incidence of hepatocellular carcinomas in males with statistical significance, and the other data did not show any significant enhancement by IQ treatment. In contrast, exposure of male and female Nrf2(–/–) mice to IQ did cause liver tumors with significantly higher incidence and multiplicity, except with hepatocellular adenomas in the males. In addition, with regard to hepatocellular carcinomas the multiplicity in the males and the incidence in the female Nrf2(–/–) mice were significantly higher than those in the relevant wild‐type mice treated with IQ. The overall data clearly indicated that Nrf2(–/–) mice of both sexes were more susceptible to IQ hepatocarcinogenicity than the wild‐type mice, which implies a possible participation of Nrf2 in IQ‐induced hepatocarcinogenesis.
Regarding detoxification and excretion, it has been reported that IQ is mainly conjugated by phase II UGT and sulfotransferase in rats and humans.( 7 , 36 ) However, our present data revealed that UGT activity was not induced in IQ‐treated wild‐type mice of either sex, indicating that glucuronidation is not associated with IQ detoxification, at least in this strain of mouse. In contrast, GST activity was clearly induced in IQ‐treated wild‐type mice of both sexes, which supports the fact that IQ (300 p.p.m.) administration for 52 weeks significantly enhanced GST activity as well as UGT conjugating activity in F344 rats.( 37 ) As mentioned above, GST is known as one of the most inducible prototypical phase II enzymes in the presence of Nrf2.( 23 ) In fact, our data demonstrated that induction of GST activities did not occur following IQ exposure to Nrf2(–/–) mice in either sex. These data allow us to hypothesize that accumulation of IQ resulted from a lack of conjugating enzyme induction and this brought about high susceptibility of Nrf2(–/–) mice to IQ hepatocarcinogenicity.
As it has recently been suggested that oxidative stress is involved in IQ carcinogenesis,( 9 , 10 , 11 , 12 ) we measured the levels of 8‐OHdG, a typical marker for oxidative DNA damage, and TBARS, a typical marker for lipid peroxidation, in the livers of Nrf2(–/–) mice exposed to IQ. However, neither 8‐OHdG nor TBARS levels in Nrf2(–/–) or wild‐type mice were altered by IQ administration. Statistical significance in the TBARS levels between the female genotypes treated with IQ seems to arise from the difference in intact levels in the untreated groups. Earlier, we revealed that IQ is unable to increase 8‐OHdG formation in the livers of F344 rats in the absence of sodium nitrite.( 12 ) Thus, it is likely that oxidative cellular damage does not participate in IQ hepatocarcinogenesis, which might imply that the high sensitivity of Nrf2(–/–) mice to IQ hepatocarcinogenicity also did not result from a lack of induction of Nrf2‐regulated antioxidant enzymes.
In the present study, the levels of IQ‐specific DNA adduct, presumed to be a key factor in IQ carcinogenesis,( 7 , 8 ) were approximately 1.7‐fold higher in female wild‐type mice than in the males, consistent with a previous report that IQ‐specific DNA adducts in the liver of CDF1 mice at 4–24 days after a single intragastric dose of IQ were approximately two‐fold higher in females than in males.( 38 ) The fact that induction of CYP1A2 following HCA administration was found in females, but not in males,( 39 , 40 ) has been considered as a major cause of the sex difference in adduct levels.( 38 ) In this respect, our present result showing that Nrf2 deficiency resulted in elevation of adduct levels only in the female, in spite of IQ accumulation occurring in both sexes, might be a consistent outcome. Inconsistency between the susceptibility to IQ carcinogenicity and specific DNA adduct levels in male Nrf2(–/–) mice enables us to speculate that the DNA adducts did not contribute to enhancement of carcinogenicity in males, unlike in the females. In contrast, the effects of Nrf2 deficiency on hepatocyte proliferation activity induced by IQ treatment were more prominent in the males than in the females. Collectively, although the lack of Nrf2 did affect susceptibility to IQ carcinogenicity in both sexes, different modes of action might underlie the enhancement in males as opposed to females.
In other organs, only the forestomach in the male mice was affected by Nrf2 deficiency. Sensitivity to IQ‐pulmonary tumorigenicity was higher in the males, but with no relation to Nrf2. These data suggest that IQ exerts its carcinogenicity through several different mechanisms, and consequently diversity of IQ carcinogenicity among species might be expected.
In conclusion, Nrf2(–/–) mice of both sexes are vulnerable to IQ hepatocarcinogenicity, which might result from a failure of excretion possibly due to the lack of GST induction. Accordingly, the present data made it clear that Nrf2 plays a crucial role in prevention against hepatocarcinogenicity by IQ. Additionally, the results suggest that Nrf2 might exert tumor prevention effects through different pathways between male and female mice.
Acknowledgments
We appreciate the expert technical assistance Ms Machiko Maeda, Ms Ayano Ogose and Ms Ayako Kaneko. This work was supported in part by Health and Labour Science Research Grants from the Ministry of Health, Labour and Welfare of Japan.
References
- 1. Sugimura T. Overview of carcinogenic heterocyclic amines. Mutat Res 1997; 376: 211–19. [DOI] [PubMed] [Google Scholar]
- 2. Yamashita M, Wakabayashi K, Nagao M et al. Detection of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in cigarette smoke condensate. Jpn J Cancer Res 1986; 77: 419–22. [PubMed] [Google Scholar]
- 3. Takayama S, Nakatsuru Y, Masuda M, Ohgaki H, Sato S, Sugimura T. Demonstration of carcinogenicity in F344 rats of 2‐amino‐3‐methyl‐imidazo[4,5‐f]quinoline from broiled sardine, fried beef and beef extract. Gann 1984; 75: 467–70. [PubMed] [Google Scholar]
- 4. Ohgaki H, Kusama K, Matsukura N et al. Carcinogenicity in mice of a mutagenic compound, 2‐amino‐3‐methylimidazo[4,5‐f]quinoline, from broiled sardine, cooked beef and beef extract. Carcinogenesis 1984; 5: 921–4. [DOI] [PubMed] [Google Scholar]
- 5. Dooley KL, Von Tungeln LS, Bucci T, Fu PP, Kadlubar FF. Comparative carcinogenicity of 4‐aminobiphenyl and the food pyrolysates, Glu‐P‐1, IQ, PhIP and MeIQx in the neonatal B6C3F1 male mouse. Cancer Lett 1992; 62: 205–9. [DOI] [PubMed] [Google Scholar]
- 6. Kitamura Y, Umemura T, Kanki K et al. Lung as a new target in rats of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline carcinogenesis − Results of a two stage model initiated with N‐bis (2‐hydroxypropyl) nitrosamine. Cancer Sci 2006; 97: 368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. IARC Monographs Programme. Heterocyclic aromatic amines: IQ (2‐amino‐3‐methylimidazo[4,5‐f]quinoline). IARC Monogr Eval Carcinog Risks Hum 1993; 56: 177–9. [PMC free article] [PubMed] [Google Scholar]
- 8. Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis 1999; 20: 353–68. [DOI] [PubMed] [Google Scholar]
- 9. Hirose M, Futakuchi M, Tanaka H et al. Prevention by antioxidants of heterocyclic amine‐induced carcinogenesis in a rat medium‐term liver bioassay: results of extended and combination treatment experiments. Eur J Cancer Prev 1998; 7: 61–7. [PubMed] [Google Scholar]
- 10. Hirose M, Takahashi S, Ogawa K, Futakuchi M, Shirai T. Phenolics: blocking agents for heterocyclic amine‐induced carcinogenesis. Food Chem Toxicol 1999; 37: 985–92. [DOI] [PubMed] [Google Scholar]
- 11. Sato K, Akaike T, Kojima Y, Ando M, Nagao M, Maeda H. Evidence of direct generation of oxygen free radicals from heterocyclic amines by NADPH/cytochrome P‐450 reductase in vitro . Jpn J Cancer Res 1992; 83: 1204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitamura Y, Umemura T, Okazaki K et al. Enhancing effects of simultaneous treatment with sodium nitrite on 2‐amino‐3‐methylimidazo[4,5‐f]quinoline‐induced rat liver, colon and Zymbal's gland carcinogenesis after initiation with diethylnitrosamine and 1,2‐dimethylhydrazine. Int J Cancer 2006; 118: 2399–404. [DOI] [PubMed] [Google Scholar]
- 13. Ishii T, Itoh K, Takahashi S et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress‐inducible genes in macrophages. J Biol Chem 2000; 275: 16023–9. [DOI] [PubMed] [Google Scholar]
- 14. Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J. Thioredoxin‐dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene 2003; 22: 1860–5. [DOI] [PubMed] [Google Scholar]
- 15. Chen XL, Varner SE, Rao AS et al. Laminar flow induction of antioxidant response element‐mediated genes in endothelial cells: A novel anti‐inflammatory mechanism. J Biol Chem 2003; 278: 703–11. [DOI] [PubMed] [Google Scholar]
- 16. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase‐1 gene. J Biol Chem 1999; 274: 26071–8. [DOI] [PubMed] [Google Scholar]
- 17. Sekhar KR, Crooks PA, Sonar VN et al. NADPH oxidase activity is essential for Keap1/Nrf2‐mediated induction of GCLC in response to 2‐indol‐3‐yl‐methylenequinuclidin‐3‐ols. Cancer Res 2003; 63: 5636–45. [PubMed] [Google Scholar]
- 18. Rushmore TH, King RG, Paulson KE, Pickett CB. Regulation of glutathione S‐transferase Ya subunit gene expression: identification of a unique xenobiotic‐responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci USA 1990; 87: 3826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. King CD, Rios GR, Green MD, Tephly TR. UDP‐glucuronosyltransferases. Curr Drug Metab 2000; 1: 143–61. [DOI] [PubMed] [Google Scholar]
- 20. Enomoto A, Itoh K, Nagayoshi E et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE‐regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 2001; 59: 169–77. [DOI] [PubMed] [Google Scholar]
- 21. Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA 2001; 98: 4611–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci USA 1999; 96: 12731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramos‐Gomez M, Kwak MK, Dolan PM et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor‐deficient mice. Proc Natl Acad Sci USA 2001; 98: 3410–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iida K, Itoh K, Kumagai Y et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 2004; 64: 6424–31. [DOI] [PubMed] [Google Scholar]
- 25. Umemura T, Kuroiwa Y, Kitamura Y et al. A crucial role of Nrf2 in in vivo defense against oxidative damage by an environmental pollutant, pentachlorophenol. Toxicol Sci 2006; 90: 111–19. [DOI] [PubMed] [Google Scholar]
- 26. Padmanabhan B, Tong KI, Ohta T et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 2006; 21: 689–700. [DOI] [PubMed] [Google Scholar]
- 27. Itoh K, Chiba T, Takahashi S et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997; 236: 313–22. [DOI] [PubMed] [Google Scholar]
- 28. Burchell B, Weatherill P. 4‐Nitrophenol UDPglucuronyltransferase (rat liver). Methods Enzymol 1981; 77: 169–77. [DOI] [PubMed] [Google Scholar]
- 29. Habig WH, Pabst MJ, Jakoby WB. Glutathione S‐transferases: The first enzymatic step in mercapturic acid formation. J Biol Chem 1974; 249: 7130–9. [PubMed] [Google Scholar]
- 30. Kasai H. Chemistry‐based studies on oxidative DNA damage: formation, repair, and mutagenesis. Free Radic Biol Med 2002; 33: 450–6. [PubMed] [Google Scholar]
- 31. Nakae D, Mizumoto Y, Kobayashi E, Noguchi O, Konishi Y. Improved genomic/nuclear DNA extraction for 8‐hydroxydeoxyguanosine analysis of small amounts of rat liver tissue. Cancer Lett 1995; 97: 233–9. [DOI] [PubMed] [Google Scholar]
- 32. Helbock HJ, Beckman KB, Shigenaga MK et al. DNA oxidation matters: the HPLC‐electrochemical detection assay of 8‐oxo‐deoxyguanosine and 8‐oxo‐guanine. Proc Natl Acad Sci USA 1998; 95: 288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uchiyama M, Mihara M. Determination of malonaldehyde precursor in tissue by the thiobarbituric acid test. Anal Biochem 1978; 86: 271–8. [DOI] [PubMed] [Google Scholar]
- 34. Ochiai M, Nakagama H, Turesky RJ, Sugimura T, Nagao M. A new modification of the 32P‐post‐labeling method to recover IQ‐DNA adducts as mononucleotides. Mutagenesis 1999; 14: 239–42. [DOI] [PubMed] [Google Scholar]
- 35. Williams JA, Stone EM, Millar BC, Hewer A, Phillips DH. Pathways of heterocyclic amine activation in the breast: DNA adducts of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline (IQ) formed by peroxidases and in human mammary epithelial cells and fibroblasts. Mutagenesis 2000; 15: 149–54. [DOI] [PubMed] [Google Scholar]
- 36. Turesky RJ, Lang NP, Butler MA, Teitel CH, Kadlubar FF. Metabolic activation of carcinogenic heterocyclic aromatic amines by human liver and colon. Carcinogenesis 1991; 12: 1839–45. [DOI] [PubMed] [Google Scholar]
- 37. McPherson RA, Tingle MD, Ferguson LR. Contrasting effects of acute and chronic dietary exposure to 2‐amino‐3‐methyl‐imidazo[4,5‐f]quinoline (IQ) on xenobiotic metabolizing enzymes in the male Fischer 344 rat: implications for chemoprevention studies. Eur J Nutr 2001; 40: 39–47. [DOI] [PubMed] [Google Scholar]
- 38. Zu HX, Schut HA. Sex differences in the formation and persistence of DNA adducts of 2‐amino‐3‐methylimidazo[4,5‐f]quinoline (IQ) in CDF1 mice. Carcinogenesis 1991; 12: 2163–8. [DOI] [PubMed] [Google Scholar]
- 39. Degawa M, Hishinuma T, Yoshida H, Hashimoto Y. Species, sex and organ differences in induction of a cytochrome P‐450 isozyme responsible for carcinogen activation: effects of dietary hepatocarcinogenic tryptophan pyrolysate components in mice and rats. Carcinogenesis 1987; 8: 1913–18. [DOI] [PubMed] [Google Scholar]
- 40. Degawa M, Yamaya C, Hashimoto Y. Hepatic cytochrome P‐450 isozyme(s) induced by dietary carcinogenic aromatic amines preferentially in female mice of DBA/2 and other strains. Carcinogenesis 1988; 9: 567–71. [DOI] [PubMed] [Google Scholar]