

Abstract
(Cancer Sci 2010; 101: 573–578)
Characteristic chromosome translocations are associated with specific disease entities, and are known to play a pivotal role in lymphoma development. Chromosome translocation alone, however, is not sufficient to produce tumors. Factors including the microenvironment and epigenetic and genetic alterations other than chromosome translocations have been shown to play a role in lymphoma development. Follicular lymphoma cells proliferate in close contact with follicular dendritic cells. Mucosa‐associated lymphoid tissue (MALT) lymphoma cells proliferate at the marginal zone area of reactive follicles which are formed by preceding chronic inflammation. The importance of genetic alterations other than chromosome translocation has been recognized since the introduction of array comparative genomic hybridization (array CGH). Variations in the genomic profile among patients with the same disease entity have been found by array CGH analyses. These variations indicate that multiple genetic pathways leading to the development of lymphomas may exist and hence result in the variable clinicopathological features observed.
Chromosome translocation for lymphomagenesis
Chromosomal translocations involving immunoglobulin loci are a hallmark of many types of B‐cell lymphoma.( 1 ) However, transgenic mouse models with various genes associated with chromosome translocation have revealed that chromosome translocation alone is not sufficient for lymphoma development. Transgenic mice with the MYC gene juxtaposed to the Eμ enhancer of the immunoglobulin heavy chain (IgH) gene developed lymphomas after a long period of time.( 2 ) Furthermore, all of the developed lymphomas were monoclonal, indicating that clonal selection occurred in Eμ‐MYC B cells during lymphoma development. If Eμ‐MYC alone is sufficient for lymphoma development, polyclonal tumors should be expected. Eμ‐BCL2 transgenic mice showed polyclonal proliferation of B cells resulting in expansion of lymphoid follicles, but only 20% of the mice developed monoclonal diffuse large‐cell lymphoma after the long latency.( 3 ) The long latency, progression from polyclonal to monoclonal disease, and histological conversion in the BCL2 transgenic mouse model suggested the influence of secondary genetic events for lymphoma development. Importantly, half of the tumors in Eμ‐BCL2 transgenic mice were shown to posses a MYC translocation, indicating that BCL2 and MYC genes collaborated in lymphoma development. Indeed, the collaborative work of BCL2 and MYC has been demonstrated with in vitro experiments using Epstein–Barr virus‐transformed lymphoblastoid cell lines.( 4 )
Another line of evidence showing the importance of secondary genetic events is the presence of B cells with IgH‐BCL2 translocation in normal individuals, as demonstrated by Limpens et al. ( 5 ) That study revealed that IgH‐BCL2 cells were found at a frequency of 1 in 105 cells in lymph nodes and tonsils with follicular hyperplasia in over 50% of cases. Yasukawa et al. reported that IgH‐BCL2 positive cells were also found in 10% of Japanese normal individuals when 1–2 × 106 peripheral blood lymphocytes were used.( 6 ) Given that the annual incidence of follicular lymphoma in the Japanese population is <1 in 100 000, it is obvious that these IgH‐BCL2 cells do not directly develop follicular lymphomas. This correlates well with the transgenic mouse model in that the other events are necessary for cells to become fully malignant.
Microenvironments for lymphoma development
Secondary follicles represent an important apparatus for B‐cell differentiation and maturation. B cells start differentiation by gene rearrangement for D‐J joining of the IgH gene. The introduction of the V region to the rearranged DJ results in VDJ joining, and when the rearrangement is successful it produces the IgH chain protein. The Ig gene has a hierarchical order of rearrangement. After the successful IgH gene rearrangement, Ig light chain (IgL) gene rearrangement follows. The rearrangement for IgL starts from the kappa light chain gene. When both alleles of the kappa chain gene rearrangement fail, the lambda chain gene begins rearrangement.( 7 ) When IgH and IgL genes are productively rearranged, B cells are called naïve B cells because they are not exposed to external antigens, and reside at the mantle zone area of the secondary follicles (Fig. 1).( 8 ) When these cells are stimulated with appropriate antigens, the cells proliferate rapidly at the dark zone area of the germinal center in the secondary follicles. Most of these cells die by the mechanism of apoptosis, and B cells producing high‐affinity antibody are selected and move out of the secondary follicles through the marginal zone area to the peripheral blood circulation. Malignant lymphomas with different entities exist corresponding to these various differentiation stages. These include mantle cell lymphoma, follicular lymphoma corresponding to germinal center cells, and marginal zone lymphoma (MALT lymphoma) (Fig. 1).
Figure 1.
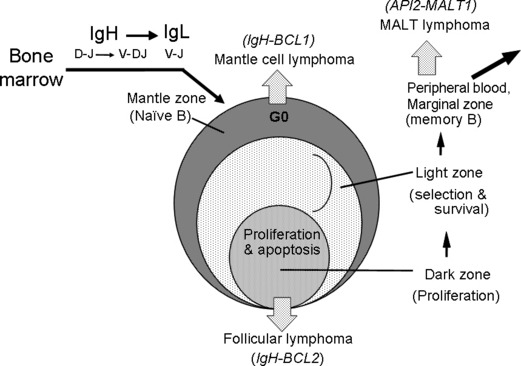
Developmental stages of B‐cell lymphomas and B‐cell differentiation via secondary follicles. The B‐cell stem cell starts differentiation from D‐J joining of the immunoglobulin heavy chain (IgH) gene, then V‐DJ joining take places. If the IgH gene is successful for productive rearrangement, the Ig light chain (IgL) gene undergoes rearrangement in the order of Igk to Igl.( 6 ) B cells with successful rearrangement of IgH and IgL genes come to the mantle zone of the secondary follicles. The B cells at this stage are called naïve B cells because they are not exposed to antigens.( 7 ) When they are stimulated with appropriate antigens, the cells proliferate rapidly at the dark zone, hypermutation takes place, and most cells die through the mechanism of apoptosis. The cells producing high‐affinity antibody are selected and move out of the germinal center. These cells are believed to go to the peripheral circulation through the marginal zone. During these B‐cell maturation steps, malignant lymphomas exist corresponding to the mantle zone, germinal center, and marginal zone.( 8 ) These types of lymphomas are mantle cell lymphoma, follicular lymphoma, and marginal zone (MALT) lymphoma, respectively. Each type has a characteristic chromosome translocation: IgH‐BCL1/CCND1, IgH‐BCL2, and API2‐MALT1, respectively.
These lymphomas have characteristics of their normal counterparts,( 9 ) and they proliferate at their sites of origins. The microenvironments for cell proliferations are important. The dependency of cell growth on the microenvironment was clearly shown in follicular lymphoma by Kagami et al. ( 10 ) They established a follicular dendritic cell (FDC)‐dependent follicular lymphoma cell line, FLK‐1. Although the detailed mechanisms still remain to be elucidated, it was shown that the long‐term growth of the cell line needs direct contact with FDCs. It is particularly important to note that proliferating follicular lymphoma cells in follicular lymphoma patients are found in the FDC area.( 11 ) In this context, it is quite conceivable that mantle cell lymphoma and marginal zone lymphoma may have similar microenvironments as found in follicular lymphoma. It is still unclear, however, what kind of cells make suitable microenvironments for proliferation of mantle cell lymphoma and marginal zone lymphoma. The interaction between lymphoma and the microenvironment should be clarified for a better understanding of lymphoma development.
Genetic alterations other than chromosome translocations
Chromosome translocations can be divided into two types: the fusion gene type and the transcriptional deregulation type.( 1 ) It is well known that the fusion gene type makes chimeric protein whose characteristic is different from donor gene products. The unique characteristic chimeric protein( 12 , 13 ) is believed to play a role in oncogenesis. The transcriptional deregulation type deregulates transcription by the juxtaposed gene. In cases of BCL1/CCND1(cyclin D1) and BCL2 translocations, the juxtaposed IgH gene renders its target gene expressed where these genes are normally down‐regulated, leading to lymphoma development.( 1 ) It is well known that these translocations alone do not produce tumors, indicating that other factors are required.
Genomic copy number aberrations (CNAs) identified by array comparative genomic hybridization (array CGH) analyses have given important insights into understanding the mechanism of lymphoma development. When a variety of disease entities of malignant lymphoma were analyzed, non‐random genomic alterations became evident (Fig. 2), and it was found that each disease entity has characteristic genomic alteration patterns that are different from each other (3, 4).( 14 , 15 , 16 , 17 , 18 ) The fact that each disease entity has a unique genomic profile indicates that these genomic alterations play some role in making each type of lymphoma of the different entities. Thus, explorations of target genes for these genomic regions are one of the most important research areas for an understanding of the molecular mechanisms of lymphoma development. It must be remembered, however, that even if one can identify the target gene from the genetically altered regions, such a target alone is not sufficient to produce tumors. One of the future directions of this research field is to understand the interaction or collaboration of multiple genes identified from genetically altered regions.
Figure 2.
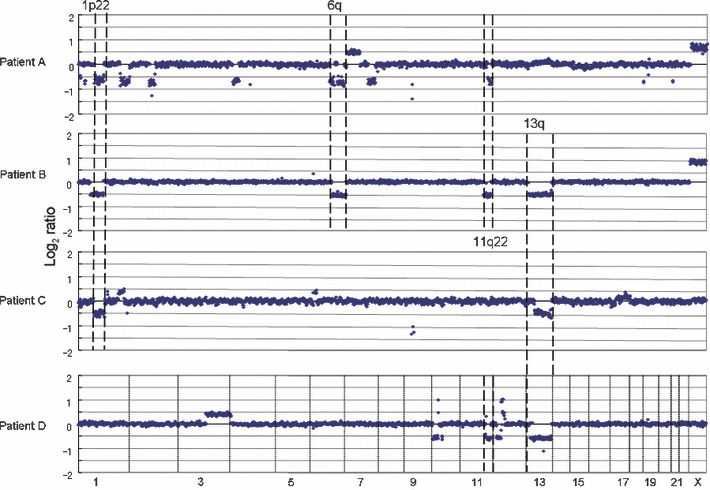
Recurrent genomic aberrations in mantle cell lymphoma patients. Mantle cell lymphoma (MCL) has t(11;14)(q13;q32) resulting in a BCL1/CCND1 translocation with the immunoglobulin heavy chain (IgH) gene (IgH‐BCL1/CCND1).( 11 ) Array comparative genomic hybridization (CGH) analysis revealed that non‐random copy number aberrations also occurred in MCL patients.( 14 )
Figure 3.
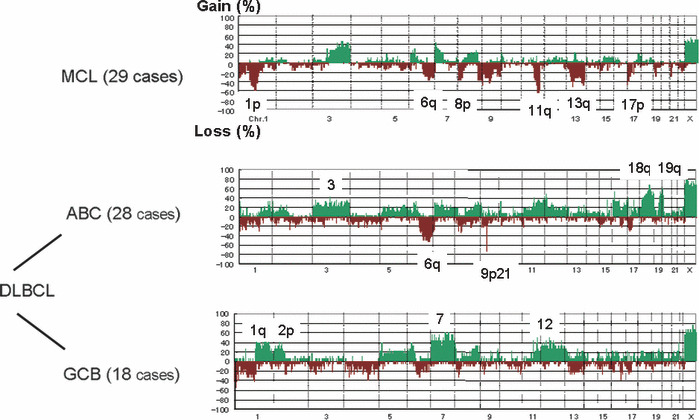
Genome profiles of mantle cell lymphoma and diffuse large B‐cell lymphoma (DLBCL). Frequency of genomic aberrations is summarized for 29 cases of mantle cell lymphoma (MCL) and DLBCL of ABC (28 cases) and GCB (18 cases) subtypes.( 14 , 15 ) The genome profiles of each entity are different, suggesting that these differences play important roles in establishing clinicopathological features of these types of lymphoma.
Figure 4.
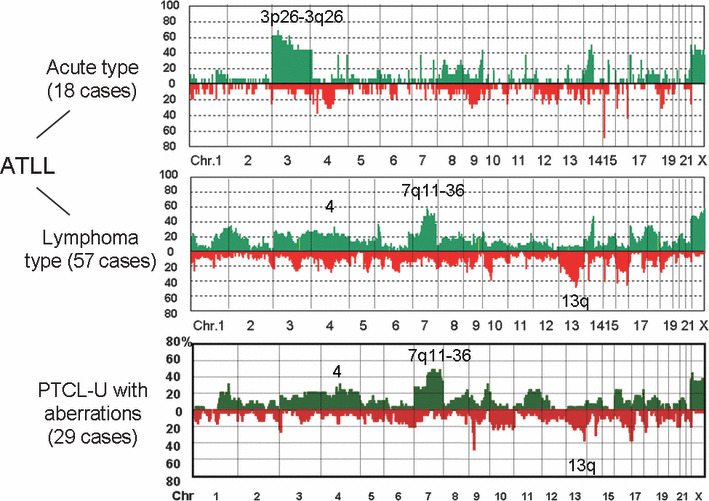
Genome profiles of T‐cell malignancies. Adult T‐cell leukemia/lymphoma (ATLL) is caused by human T‐lymphotrophic virus (HTLV)‐1 integration after a long latency of about 40–50 years.( 19 ) It was shown that acute and lymphoma types have distinct genome profiles.( 17 ) Peripheral T‐cell lymphoma‐unspecified (PTCL‐U) with genomic aberrations has genome profiles similar to those of lymphoma type ATLL.( 18 ) It should be noted that the histology and surface markers are also very similar to each other.
Genome‐wide array CGH analysis provided an important finding regarding T‐cell malignancies. Adult T‐cell leukemia/lymphoma (ATLL) is caused by human T‐lymphotrophic virus (HTLV)‐1 viral integration, but 95% of HTLV‐1 virus carriers do not develop tumors, indicating that HTLV‐1 alone cannot make cells malignant.( 19 ) Oshiro et al. demonstrated that genomic profiles differ between acute and lymphoma types, and they showed the importance of genomic alterations after HTLV‐1 integrations.( 17 ) It was recently revealed by Nakagawa et al. that peripheral T‐cell lymphoma‐unspecified (PTCL‐U) may be divided into two groups, one with genomic alterations and one without.( 18 ) Importantly, PTCL‐U with genomic alterations has clinicopathological features similar to those of the ATLL lymphoma type. Furthermore, it was also shown that both have similar histology and surface markers such as CCR4. In other words, PTCL‐U with a genomic alteration subtype is difficult to differentiate from the ATLL lymphoma type if HTLV‐1 information is not available. These findings suggest the following important facts. First, HTLV‐1 integration is important for lymphoma development, but other genetic events are necessary. Second, PTCL‐U with genomic alteration is not distinguishable from the ATLL lymphoma type without HTLV‐1 information. In this regard, HTLV‐1 negative ATLL( 20 ) may belong to the same disease entity. Our findings might reflect the genetic bases for this entity.
In clinical practice, diagnosis of ATLL has been restricted to cases with HTLV‐1 integration, indicating that HTLV‐1‐negative ATLL cases are omitted from ATLL cases irrespective of the similarity of the clinical manifestation. The clinical subtyping of ATLL by Shimoyama is quite useful and important from a practical aspect,( 21 ) but one needs to keep in mind that the molecular bases for ATLL development also work for the development of HTLV‐1‐negative ATLL or PTCL‐U with genomic alterations. The recognition of such a disease entity might be more important for an understanding of peripheral T‐cell malignancies.
Genetic variations and disease spectrum
Each disease entity has a characteristic genome profile and it is quite conceivable that such characteristic genomic aberrations play important roles in lymphoma development. Mantle cell lymphoma possesses a characteristic chromosome translocation of t(11;14)(q13;q32), resulting in the common presence of IgH‐BCL1/CCND1. ( 22 ) Regarding CNAs, as shown in Figure 3, the genome profile of mantle cell lymphoma in 29 cases revealed that non‐random genomic alterations are involved in mantle cell lymphoma development.( 14 ) However, the genome alteration pattern in each patient is quite variable, although there are some tendencies in genome alteration regions (Fig. 2), indicating that variable combinations of genomic alterations may work for the development of mantle cell lymphoma. Taking this genomic variation into account, it is quite likely that there are multiple genetic pathways from the initial chromosome translocation to the final manifestation of lymphoma (Fig. 5). These genetic variations in a single disease entity are likely to be the cause of clinicopathological heterogeneity; in other words, the so‐called spectrum of the disease. In this regard, it is necessary to identify the number of genes involved in a single disease entity.
Figure 5.
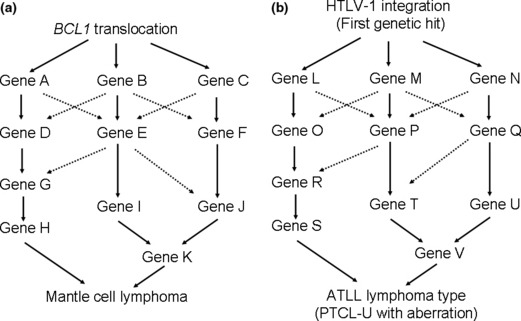
Multiple pathways to establish the same lymphoma type. (a) The initial genetic hit for mantle cell lymphoma is likely to be a BCL1 translocation. The pattern of genomic alteration differs from patient to patient, although there are shared regions as shown in Figure 2, suggesting that there are multiple genetic pathways that form mantle cell lymphoma. (b) In cases of adult T‐cell leukemia/lymphoma (ATLL) lymphoma type and peripheral T‐cell lymphoma‐unspecified (PTCL‐U) with genomic alterations, the initial genetic hit for the former involves human T‐lymphotrophic virus (HTLV)‐1 integration. A genetic hit yet to be found may replace the HTLV‐1 integration for the latter. The genetic pathways other than the first hit may be quite similar to each other.
Recently, Takeuchi et al. reported bioinformatic analyzes to differentially diagnose lymphoma subtypes using array CGH data with 2304 BAC clones.( 23 ) They reported an 88% accuracy of differential diagnosis between mantle cell lymphoma and diffuse large B‐cell lymphoma (DLBCL) when 50 genomic regions were selected by a computer program. The same is true for differential diagnosis between ABC and GCB types of DLBCL, which yielded an accuracy of 83%. These genomic regions are suspected of being regions that are involved in lymphoma development. The next step to further advance these findings would be the identification of target genes by which functional consequences may be clarified.
Indication of genetic variations in a single disease entity
Insights into the meaning of genetic variations in a single disease entity are suggested by the series of mucosa‐associated lymphoid tissue (MALT) lymphoma studies. It is known that MALT lymphoma has variable chromosome translocations. These include t(11;18)(q21;q21), t(14;18)(q32;q21), and t(1;14)(p22q32) (Fig. 6). After t(11;18)(q21;q21) was revealed as the API2‐MALT1 translocation by us and others,( 24 , 25 , 26 ) t(14;18)(q32;q21) and t(1;14)(p22;q32) were identified as IgH‐MALT1 ( 27 ) and IgH‐BCL10,( 28 ) respectively. MALT1 and BCL10 were found to form a complex that activates nuclear factor‐kappa B (NF‐κB).( 29 ) Interestingly, in knockout mouse systems, these molecules were found to be involved in the signal transduction pathway from T‐ and B‐cell receptors to the nucleus resulting in NF‐κB activation.( 30 , 31 , 32 ) Hosokawa et al. reported that API2‐MALT1 chimeric protein bypasses signals from TCR/BCR and activates NF‐κB.( 33 ) The three different chromosome translocations therefore have a common outcome, NF‐κB activation, suggesting that deregulated NF‐κB activation plays a pivotal role in MALT lymphoma development.
Figure 6.
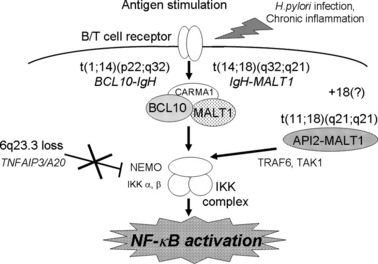
Shared function driving lymphomagenesis of variable genetic alterations. An indication of variable genetic alterations in the same disease entity was revealed by a series of studies on mucosa‐associated lymphoid tissue (MALT) lymphoma. Three kinds of chromosome translocations, t(11;18)(q21;q21), t(14;18)(q32;q21) and t(1;14)(p22;q32) were revealed as ALI2‐MALT1, IgH‐MALT1, and IgH‐BCL10, respectively. All of these translocations were found to deregulate signal transduction pathways to activate nuclear factor‐kappa B (NF‐κB). We have found that 6q23.3 loss is frequent in ocular adnexal MALT lymphoma and identified that the target gene is TNFAIP3/A20, which has a suppressor function on NF‐κB activity.( 34 , 35 ) The loss of TNFAIP3, therefore, would result in NF‐κB activation, indicating that the NF‐κB activation is essential for MALT lymphoma development. In this regard, 18 trisomy and chronic inflammation may work to activate NF‐κB.
Recently, we reported that 6q23.3 loss is characteristic to ocular adnexal MALT lymphoma( 34 ) and that TNFAIP3/A20 is the target gene for this region.( 35 ) Since TNFAIP3 is known to suppress NF‐κB activity, its loss would lead to NF‐κB activation, indicating that the loss of 6q23.3, a distinct genomic alteration from the chromosome translocations found in MALT lymphoma, shares the common function. These findings indicated the importance of NF‐κB activation in MALT lymphoma development. In this context, trisomy 18, which is found in MALT lymphoma, may serve to activate NF‐κB as a result of a gene dosage effect of MALT1.( 36 ) Chronic inflammations associated with MALT lymphoma may also be associated with NF‐κB activation.( 1 ) The indication of genetic variations in MALT lymphoma genetics is a typical example of genetic variations that possess a common function leading to lymphoma development. Therefore, the discovery of functional grouping of genetic variations associated with a specific type of lymphoma is an important step for an understanding of the molecular mechanisms of lymphomagenesis.
The association of TNFAIP3/A20 loss with lymphomas was reported by us and others,( 37 , 38 , 39 , 40 ) especially with ABC‐type DLBCL and mantle cell lymphoma,( 38 ) indicating that TNFAIP3/A20 is not only associated with MALT lymphoma, but also with other types of lymphomas. These findings highlight an important role of NF‐κB activation in the development of several lymphoma types. Genetic variations and their association with specific types of lymphoma development, and elucidation of the variable genetic pathways leading to lymphoma development, are important directions for future cancer research.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Health, Labor and Welfare of Japan; from the Ministry of Education, Culture, Sports, Science and Technology of Japan; from the Japan Society for the Promotion of Science; and from the Foundation for Promotion of Cancer Research.
References
- 1. Seto M. Genetic and epigenetic factors involved in B‐cell lymphomagenesis. Cancer Sci 2004; 95: 704–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams JM, Harris AW, Pinkert CA et al. The c‐myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985; 318: 533–8. [DOI] [PubMed] [Google Scholar]
- 3. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high‐grade malignant lymphoma in mice transgenic for the t(14;18). Nature 1991; 349: 254–6. [DOI] [PubMed] [Google Scholar]
- 4. Nunez G, Seto M, Seremetis S et al. Growth‐ and tumor‐promoting effects of deregulated BCL2 in human B‐lymphoblastoid cells. Proc Natl Acad Sci USA 1989; 86: 4589–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Limpens J, De Jong D, Van Krieken JH et al. Bcl‐2/JH rearrangements in benign lymphoid tissues with follicular hyperplasia. Oncogene 1991; 6: 2271–6. [PubMed] [Google Scholar]
- 6. Yasukawa M, Bando S, Dölken G et al. Low frequency of BCL‐2/J(H) translocation in peripheral blood lymphocytes of healthy Japanese individuals. Blood 2001; 98: 486–8. [DOI] [PubMed] [Google Scholar]
- 7. Korsmeyer SJ, Hieter PA, Ravetch JV, Poplack DG, Waldmann TA, Leder P. Developmental hierarchy of immunoglobulin gene rearrangements in human leukemic pre‐B‐cells. Proc Natl Acad Sci U S A 1981; 78: 7096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu YJ, Johnson GD, Gordon J, MacLennan IC. Germinal centres in T‐cell‐dependent antibody responses. Immunol Today 1992; 13: 17–21. [DOI] [PubMed] [Google Scholar]
- 9. Swerdlow SH, Campo E, Harris NL et al. Pathology and Genetics of Tumours of Hematopoietic and Lymphoid Tissues, Chap. 11.270‐299367. Lyon, France: World Health Organization classification of tumours. International Agency for Research on Cancer Press, 2008. [Google Scholar]
- 10. Kagami Y, Jung J, Choi YS et al. Establishment of a follicular lymphoma cell line (FLK‐1) dependent on follicular dendritic cell‐like cell line HK. Leukemia 2001; 15: 148–56. [DOI] [PubMed] [Google Scholar]
- 11. Dogan A, Du MQ, Aiello A et al. Follicular lymphomas contain a clonally linked but phenotypically distinct neoplastic B‐cell population in the interfollicular zone. Blood 1998; 91: 4708–14. [PubMed] [Google Scholar]
- 12. Izumiyama K, Nakagawa M, Yonezumi M et al. Stability and subcellular localization of API2‐MALT1 chimeric protein involved in t(11;18) (q21;q21) MALT lymphoma. Oncogene 2003; 22: 8085–92. [DOI] [PubMed] [Google Scholar]
- 13. Nakagawa M, Hosokawa Y, Yonezumi M et al. MALT1 contains nuclear export signals and regulates cytoplasmic localization of BCL10. Blood 2005; 106: 4210–6. [DOI] [PubMed] [Google Scholar]
- 14. Tagawa H, Karnan S, Suzuki R et al. Genome‐wide array‐based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005; 24: 1348–58. [DOI] [PubMed] [Google Scholar]
- 15. Tagawa H, Suguro M, Tsuzuki S et al. Comparison of genome profiles for identification of distinct subgroups of diffuse large B‐cell lymphoma. Blood 2005; 106: 1770–7. [DOI] [PubMed] [Google Scholar]
- 16. Nakashima Y, Tagawa H, Suzuki R et al. Genome‐wide array‐based comparative genomic hybridization of natural killer cell lymphoma/leukemia: different genomic alteration patterns of aggressive NK‐cell leukemia and extranodal Nk/T‐cell lymphoma, nasal type. Genes Chromosomes Cancer 2005; 44: 247–55. [DOI] [PubMed] [Google Scholar]
- 17. Oshiro A, Tagawa H, Ohshima K et al. Identification of subtype‐specific genomic alterations in aggressive adult T‐cell leukemia/lymphoma. Blood 2006; 107: 4500–7. [DOI] [PubMed] [Google Scholar]
- 18. Nakagawa M, Nakagawa‐Oshiro A et al. Array comparative genomic hybridization analysis of PTCL‐U reveals a distinct subgroup with genetic alterations similar to lymphoma‐type adult T‐cell leukemia/lymphoma. Clin Cancer Res 2009; 15: 30–8. [DOI] [PubMed] [Google Scholar]
- 19. Taylor GP, Matsuoka M. Natural history of adult T‐cell leukemia/lymphoma and approaches to therapy. Oncogene 2005; 24: 6047–57. [DOI] [PubMed] [Google Scholar]
- 20. Shimoyama M, Kagami Y, Shimotohno K et al. Adult T‐cell leuke‐mia/lymphoma not associated with human T‐cell leukemia virus type I. Proc Natl Acad Sci U S A 1986; 83: 4524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T‐cell leukaemia‐lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haematol 1991; 79: 428–37. [DOI] [PubMed] [Google Scholar]
- 22. Seto M, Yamamoto K, Iida S et al. Gene rearrangement and overexpression of PRAD1 in lymphoid malignancy with t(11;14)(q13;q32) translocation. Oncogene 1992; 7: 1401–6. [PubMed] [Google Scholar]
- 23. Takeuchi I, Tagawa H, Tsujikawa A et al. The potential of copy number gains and losses, detected by array‐based comparative genomic hybridization, for computational differential diagnosis of B‐cell lymphomas and genetic regions involved in lymphomagenesis. Haematologica 2009; 94: 61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akagi T, Motegi M, Tamura A et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low‐grade B‐cell lymphoma of mucosa‐associated lymphoid tissue. Oncogene 1999; 18: 5785–94. [DOI] [PubMed] [Google Scholar]
- 25. Dierlamm J, Baens M, Wlodarska I et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa‐associated lymphoid tissue lymphomas. Blood 1999; 93: 3601–9. [PubMed] [Google Scholar]
- 26. Motegi M, Yonezumi M, Suzuki H et al. API2‐MALT1 chimeric transcripts involved in mucosa‐associated lymphoid tissue type lymphoma predict heterogeneous products. Am J Pathol 2000; 156: 807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Streubel B, Lamprecht A, Dierlamm J et al. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood 2003; 101: 2335–9. [DOI] [PubMed] [Google Scholar]
- 28. Willis TG, Jadayel DM, Du MQ et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999; 96: 35–45. [DOI] [PubMed] [Google Scholar]
- 29. Lucas PC, Yonezumi M, Inohara N et al. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF‐kappa B signaling pathway. J Biol Chem 2001; 276: 19012–9. [DOI] [PubMed] [Google Scholar]
- 30. Ruland J, Duncan GS, Elia A et al. Bcl10 is a positive regulator of antigen receptor‐induced activation of NF‐kappaB and neural tube closure. Cell 2001; 104: 33–42. [DOI] [PubMed] [Google Scholar]
- 31. Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity 2003; 19: 749–58. [DOI] [PubMed] [Google Scholar]
- 32. Xue L, Morris SW, Orihuela C et al. Defective development and function of Bcl10‐deficient follicular, marginal zone and B1 B cells. Nat Immunol 2003; 4: 857–65. [DOI] [PubMed] [Google Scholar]
- 33. Hosokawa Y, Suzuki H, Suzuki Y, Takahashi R, Seto M. Antiapoptotic function of apoptosis inhibitor 2‐MALT1 fusion protein involved in t(11;18)(q21;q21) mucosa‐associated lymphoid tissue lymphoma. Cancer Res 2004; 64: 3452–7. [DOI] [PubMed] [Google Scholar]
- 34. Kim WS, Honma K, Karnan S et al. Genome‐wide array‐based comparative genomic hybridization of ocular marginal zone B cell lymphoma: comparison with pulmonary and nodal marginal zone B cell lymphoma. Genes Chromosomes Cancer 2007; 46: 776–83. [DOI] [PubMed] [Google Scholar]
- 35. Honma K, Tsuzuki S, Nakagawa M et al. TNFAIP3 is the target gene of chromosome band 6q23.3‐q24.1 loss in ocular adnexal marginal zone B cell lymphoma. Genes Chromosomes Cancer 2008; 47: 1–7. [DOI] [PubMed] [Google Scholar]
- 36. Sanchez‐Izquierdo D, Buchonnet G, Siebert R et al. MALT1 is deregulated by both chromosomal translocation and amplification in B‐cell non‐Hodgkin lymphoma. Blood 2003; 101: 4539–46. [DOI] [PubMed] [Google Scholar]
- 37. Honma K, Tsuzuki S, Nakagawa M et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non‐Hodgkin lymphomas. Blood 2009; 114: 2467–75. [DOI] [PubMed] [Google Scholar]
- 38. Compagno M, Lim WK, Grunn A et al. Mutations of multiple genes cause deregulation of NF‐kappaB in diffuse large B‐cell lymphoma. Nature 2009; 459: 717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kato M, Sanada M, Kato I et al. Frequent inactivation of A20 in B‐cell lymphomas. Nature 2009; 459: 712–6. [DOI] [PubMed] [Google Scholar]
- 40. Schmitz R, Hansmann ML, Bohle V et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 2009; 206: 981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]