

Abstract
The polyphenol epigallocatechin‐3 gallate (EGCG) in green tea suppresses tumor growth by direct action on tumor cells and by inhibition of angiogenesis, but it is not known whether it specifically inhibits tumor angiogenesis. We examined the anti‐angiogenic effect of EGCG on tumor‐associated endothelial cells (TEC), endothelial progenitor cells (EPC), and normal endothelial cells (NEC). EGCG suppressed the migration of TEC and EPC but not NEC. EGCG also inhibited the phosphorylation of Akt in TEC but not in NEC. Furthermore, vascular endothelial growth factor‐induced mobilization of EPC into circulation was inhibited by EGCG. MMP‐9 in the bone marrow plasma plays key roles in EPC mobilization into circulation. We observed that expression of MMP‐9 mRNA was downregulated by EGCG in mouse bone marrow stromal cells. In an in vivo model, EGCG suppressed growth of melanoma and reduced microvessel density. Our study showed that EGCG has selective anti‐angiogenic effects on TEC and EPC. It is suggested that EGCG could be a promising angiogenesis inhibitor for cancer therapy. (Cancer Sci 2009; 100: 1963–1970)
Tea is one of the most popular beverages consumed worldwide. Drinking tea, especially green tea, inhibits the growth of several tumors in animal models, including cancers of the skin, lung, esophagus, breast, stomach, small intestine, colon, liver, pancreas, and mammary glands,( 1 , 2 ) and is associated with a lower incidence of cancer in humans.( 1 , 3 , 4 ) The components of green tea responsible for these effects are catechins, which are polyphenols with potent antioxidant capacity that have been shown to inhibit mutation, tumor cell growth, tumor initiation, tumor progression, and the activity of urokinase and MMP, which are crucial for cancer growth.( 5 , 6 , 7 ) Among these catechins, epigallocatechin‐3 gallate (EGCG) has the highest antioxidant capacity and is an effective inhibitor of corneal vascularization in vivo.( 8 )
The tumor microenvironment has recently been regarded as a target of cancer chemoprevention because it plays an important role in tumorigenesis and tumor progression.( 9 ) Tumor angiogenesis is one of the key processes within the tumor microenvironment, and controlling this process is a very important strategy for preventing invasive cancers.( 10 ) Many molecules regulating tumor angiogenesis have been identified and characterized recently, including vascular endothelial growth factor (VEGF).( 11 , 12 )
EGCG has also been shown to inhibit cell proliferation,( 13 ) binding of VEGF to its receptors,( 14 ) phosphorylation of VEGF receptor (VEGFR) 2,( 15 ) MMP activity,( 13 ) and interleukin‐8 production( 16 ) in normal endothelial cells such as human umbilical vein endothelial cells.
An important concept in tumor angiogenesis is that tumor blood vessels contain endothelial cells that are genetically normal and stable, whereas tumor cells are typically genetically unstable.( 17 ) However, tumor vessels and tumor‐associated endothelial cells (TEC) differ from their normal counterparts in many respects.( 18 , 19 , 20 ) Tumor vessels show structural changes such as fewer pericytes and leakiness.( 19 , 21 ) Moreover, some studies have reported that TEC possess molecular characteristics distinct from those of normal endothelial cells (NEC). Endothelial cells derived from human renal cell carcinoma express biological features different from those of NEC.( 22 ) We have reported that endothelial cells in malignant tumors (melanomas and liposarcomas) are cytogenetically abnormal.( 23 , 24 ) In addition, TEC showed enhanced responsiveness to Epidermal Growth Factor (EGF), with alteration of EGF receptor expression.( 25 ) These results suggest that TEC are different from NEC, contrary to the conventional assumption. We therefore decided to investigate the differential anti‐angiogenic effects of EGCG on TEC and NEC.
In the tumor microenvironment, endothelial progenitor cells (EPC) also play a critical role in tumor angiogenesis. The mobilization of EPC from the bone marrow into peripheral blood is regulated by tumor‐derived cytokines.( 26 , 27 ) In response to these cytokines, EPC can contribute to tumor angiogenesis and the growth of certain tumors.( 26 , 28 , 29 ) EPC derived from the bone marrow are thus considered to be an important target for anti‐angiogenic therapy. Strategies that block the mobilization of EPC into circulation might provide a new approach to inhibiting tumor angiogenesis.( 26 ) However, the effects of EGCG on EPC mobilization have not been investigated. It is of great interest to investigate whether EGCG specifically acts on TEC and EPC rather than NEC.
To investigate the effects of EGCG on TEC and EPC, TEC were isolated and cultured from human tumor xenografts in nude mice and EPC were isolated from nude mouse peripheral blood. NEC were cultured from the skin of nude mice as a control. We also investigated the effect of EGCG on EPC mobilization in VEGF‐treated nude mice. In addition, we isolated bone marrow stromal cells (BMSC) from oral carcinoma‐bearing mice and investigated the expression of MMP‐9 mRNA in the presence and absence of EGCG in BMSC to determine whether EGCG affects the expression of MMP‐9 in the bone marrow space, which plays an important role in the mobilization of EPC into peripheral circulation.( 26 , 30 ) Finally, we analyzed the effects of EGCG on in vivo tumor growth.
Materials and Methods
Chemicals. EGCG was purchased from Sigma Chemical Co. (St Louis, MO, USA) and dissolved in sterile DMSO. The Phosphoinositide 3‐kinase (PI3K) inhibitor LY294002 was purchased from Calbiochem (San Diego, CA, USA)
Cell lines and culture conditions. Cells of the super‐metastatic human malignant melanoma cell line A375SM, kindly gifted by Dr I. J. Fidler (MD Anderson Cancer Center, Houston, TX, USA), were cultured in a humidified atmosphere of 5% CO2 and 95% air at 37°C in Minimum Essential Medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat‐inactivated FBS. The medium was changed every 3 days. The human oral carcinoma cell line HSC‐3 was supplied by the Japanese Cancer Research Bank (Tokyo, Japan). The cells were cultured in DMEM (Sigma Chemical Co.) supplemented with 10% FBS.
Antibodies. The antibodies purchased were: rat antimouse CD31 and FITC–antimouse CD31 (eBioscience, San Diego, CA, USA); FITC–antimouse CD133 (eBioscience); FITC–Bandeirea simplicifolia lectin 1‐B4 (BS1‐B4; Vector Laboratories, Burlingame, CA, USA); FITC–goat antirat IgG; phycoerythin (PE)–goat antirat IgG (Molecular Probes, Invitrogen, OR, USA); normal rat IgG (BD Biosciences, San Jose, CA); PE–antimouse VEGFR2 (BD Biosciences, San Jose, CA); and FITC–stem cell antigen (Sca)‐1 (BD Biosciences, San Jose, CA) for immunohistochemistry and Flow Cytometry.
Isolation of TEC, peripheral blood‐derived ECs (EPC) and NEC. All animal procedures were carried out in compliance with Hokkaido University guidelines, and the protocols were approved by the Institutional Animal Care and Use Committee. Endothelial cells (EC) were isolated as previously described.( 23 ) In brief, TEC were isolated from melanoma (A375SM) and oral carcinoma (HSC‐3) xenografts in nude mice aged 8–12 weeks (Sankyo Labo, Tokyo, Japan). NEC were isolated from the dermal tissue as a control. EC were isolated using a magnetic cell sorting system (Miltenyi Biotec, Tokyo, Japan) according to the manufacturer's instructions using FITC–anti‐CD31 antibody. CD31‐positive cells were sorted and plated onto 1.5% gelatin‐coated culture plates and grown in EGM‐2 MV (Clonetics, Walkersville, MD, USA) and 15% FBS. Diphtheria toxin (500 ng/mL; Calbiochem) was added to TEC subcultures to kill any remaining human tumor cells( 31 ) and to NEC to ensure technical consistency. The isolated EC were purified by a second round of purification, using FITC–BS1‐B4, and purity was determined by flow cytometry.( 23 ) Peripheral blood‐derived ECs (EPC) were isolated and grown as previously described with modifications.( 32 ) Briefly, mouse peripheral blood was obtained by cardiocentesis. Peripheral blood mononuclear cells (PBMC) were isolated using Histopaque 1077 (Sigma Chemical Co.) and centrifuged as per the manufacturer's instruction. The intermediate phase was collected and washed with HBSS before resuspension in EGM‐2 MV supplemented with 15% FBS. PBMC (1 × 106 per well) were seeded onto 12‐well tissue culture plates coated with 0.5% gelatin (Sigma Chemical Co.) and 2 µg/mL of human fibronectin (Chemicon International, Temecula, CA, USA). Cells were passaged at a 1:3 ratio until they reached 90% confluence. Peripheral blood‐derived ECs were regarded as EPC as they were Sca‐1‐positive in culture.
Cell migration assay. Migration of TEC, NEC, and EPC was measured by a migration assay using a Boyden chamber as previously described with modifications.( 33 , 34 ) TEC, EPC, and NEC were treated with and without EGCG (0, 25, 50 µM) in Endothelial Basal Medium‐2 (EBM‐2; Clonetics) + 0.5% FBS for 24 h. EC and EPC (1.5 × 104) were seeded into the upper chambers in EBM‐2 + 0.5% FBS and EGCG was then added. VEGF (10 ng/mL) was added in the lower chambers as a chemoattractant. Similarly, after overnight starvation in EBM‐2 + 0.5% FBS, EC and EPC were pretreated with LY294002 (20 µM) for 2 h and seeded in EBM‐2 + 0.5% FBS, including LY294002. After 4 h at 37°C, the cells that migrated through a fibronectin‐coated polycarbonate membrane having 8‐µm pores (Corning Costar, Nagog Park, MA, USA) were fixed in 2% paraformaldehyde and stained with DAPI. The numbers of cells that migrated to the lower side of the membrane were counted in seven high‐power fields. The experiment was carried out three times with similar results.
Western blotting. EC were harvested in serum‐free medium after 16 h and were then treated with EGCG (100 µM) in EBM‐2 + 0.5% FBS for 24 h. Similarly, after overnight starvation in EBM‐2 + 0.5% FBS, EC were pretreated with LY294002 (20 µM) for 2 h in EBM‐2 + 0.5%. EGCG was not toxic at these concentrations in these cells. After EGCG treatment, VEGF (10 ng/mL) was added to the cell cultures for 30 min at room temperature. EC were lysed as described previously with some modification.( 25 ) Total protein was measured using a BCA protein assay kit (Pierce, Rockford, IL, USA). Western blotting was also carried out for the detection of Akt and phosphorylated Akt, as previously described with modifications.( 35 ) After electrotransfer, the membranes were probed with anti‐phospho‐Akt (Cell Signaling Technology, Beverly, MA, USA) in Solution 1 (Can Get Signal Immunoreaction Enhancer Solution; Toyobo, Osaka, Japan) overnight at 4°C. After washing, the membrane was incubated with antirabbit IgG secondary antibody (Cell Signaling Technology) in Solution 2 (Toyobo) and chemiluminescence reagents (PerkinElmer, Boston, MA, USA). The membranes were reblotted with anti‐total Akt antibody (Cell Signaling Technology) and β‐actin (Sigma Chemical Co.). The level of phosphorylated Akt was normalized to total Akt by scanning densitometry using Image J software from the NIH (Bethesda, MD, USA).
Counting the number of CD133+/VEGFR2+ cells to estimate EPC number. PBS or VEGF (300 ng) was injected intraperitoneally into 10 nude mice in each group to mobilize EPC from the bone marrow, as previously described.( 36 , 37 ) In each group, EGCG (5 mg/kg) or vehicle (0.05% DMSO) was also injected once a day for 2 days. Injection of EGCG into nude mice had no toxic effects. Forty‐eight hours after the first injection, peripheral blood (700–1000 µL) was collected under anesthesia in each group (n = 5 for each group) before they were killed. Mononuclear cells were isolated by sucrose gradient centrifugation from peripheral blood as previously described with modifications( 38 ) and the number of cells was counted. Next, they were incubated with FITC–anti‐mouse CD133 and PE–anti‐mouse VEGFR2 in order to count the number of CD133+/VEGFR2+ cells by flow cytometry, using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA). A minimum of 10 000 events was counted for each mouse.
RT‐PCR and quantitative real‐time PCR. Total RNA was isolated from EC and Bone Marrow Stromal Cell (BMSC) using the RNeasy Micro Kit (Qiagen, Santa Clarita, CA, USA) with RNase‐Free DNase Set (Qiagen). RNA was quantified by spectrophotometry. Total RNA (5 µg) was then used for first‐strand complementary DNA (cDNA) synthesis in ReverTra‐Plus (Toyobo). The cDNA was amplified by PCR with primer pairs specific for mouse stromal derived factor (SDF)‐1, fibroblast activation protein (FAP), α‐smooth muscle actin (SMA), and Sca‐1. The annealing temperature was 60°C and the reaction proceeded for 30 cycles. PCR products were visualized by ethidium bromide staining and ultraviolet transillumination. NEC were used as a negative control. The primers were as follows: GAPDH, forward 5′‐TCTGACGTGCCGCCTGGAG‐3′, reverse 5′‐TCGCAGGAGACAACCTGGTC‐3′; SDF‐1, forward 5′‐CTCGGTGTCCTCTTGCTGTCC‐3′, reverse 5′‐CGGTATCA GGCTGACTGGTTTACCG‐3′; FAP, forward 5′‐CAAATGTGGCA TAGCAGTGG‐3′, reverse 5′‐TTCTGCTCTTGCCATCACAG‐3′; α‐SMA, forward 5′‐CTGACAGAGGCACCACTGAA‐3′, reverse 5′‐CATCTCCAGAGTCCAGCACA‐3′; and Sca‐1, forward 5′‐GAAGAGGCAGAATTCCAAGG‐3′, reverse 5′‐ATGTGGGA ACATTGCAGGAC‐3′. In addition, total RNA (5 µg) was used and real‐time PCR was conducted using SYBR Green Real‐time PCR Master Mix Plus (Toyobo) in BMSC with or without EGCG (0, 25, 50, 100 µM) and EC and EPC. Cycling conditions were according to the manufacturer's instructions based on the use of Opticon Monitor version 3.0 (Bio‐Rad, Hercules, CA, USA). The expression levels of MMP‐9 mRNA in BMSC with or without EGCG treatment and VEGFR2 mRNA, Sca‐1 and VEGFR2 mRNA in EC and EPC were normalized to GAPDH. The primers were as follows:
MMP‐9, forward, 5′‐CTCTACAGAGTCTTTGAGTCCGGCAG‐3′, reverse, 5′‐TCAGGAACTTCCAGTACCAACCGTC‐3′; and VEGFR2, forward, 5′‐GGCAAATGTGTCAGCTTTGTACA‐3′; reverse, 5′‐GGTCACGTGGAAGGAGATCAC‐3′.
Primary culture and characterization of Bone Marrow Stromal Cell (BMSC). BMSC were isolated from the femoral bone marrow of nude mice. The cells were triturated with a 20‐gauge needle and passed through a 70‐µm nylon mesh cell strainer (Becton‐Dickinson, Franklin Lakes, NJ, USA) to obtain a single‐cell suspension in DMEM (Sigma Chemical Co.) supplemented with 20% inactivated FBS, as described previously with modifications.( 39 ) We characterized BMSC that had been grown under the control conditions by RT‐PCR to analyze the expression of stromal cell (SDF‐1, α‐SMA, FAP) and stem cell marker (Sca‐1).
In vivo tumor growth experiments. Seven‐week‐old KSN nude mice were purchased from Sankyo Labo and housed in pathogen‐free conditions. Melanoma was obtained by s.c. injection of A375SM (1 × 106 cells) in the flanks of nude mice as described previously.( 23 ) Controls were injected with 0.05% DMSO and one group was injected with EGCG (20 mg/kg) i.p. (n = 5, for each group). EGCG was injected every day. Animals were weighed, and tumor growth was monitored for 36 days by measuring two tumor diameters every 3 days with calipers and calculating the tumor volumes with the formula: (shortest diameter)2× (longest diameter) × 0.5. On day 36, the animals were killed and the tumors were removed. Each sample was snap frozen. Eight‐µm cryosections of tumors were stained with anti‐CD31 antibody for histological study. The microvessel density (MVD) was measured using a computer‐aided image analysis system (Metamorph Software, Universal Imaging Corporation, West Chester, PA, USA).
Statistical analysis. Differences between groups were evaluated using Student's t‐test, two‐way ANOVA test.
Results
TEC and peripheral blood‐derived endothelial cells expressed Sca‐1. Endothelial cells and peripheral blood‐derived endothelial cells (EPC) were characterized by flow cytometry. All EC and peripheral blood‐derived endothelial cells were positive for BS1‐B4, which binds specifically to mouse endothelial cells (Fig. 1A: a–d). Peripheral blood‐derived endothelial cells expressed the stem cell marker Sca‐1. Furthermore, Sca‐1 was overexpressed also in TEC from both melanoma and oral carcinoma whereas it was expressed at low‐level in NEC (Fig. 1A: e–h). Consistently, mRNA expression of Sca‐1 was higher in TEC and peripheral blood‐derived endothelial cells, than in NEC by real‐time RT‐PCR (*P < 0.05 vs NEC) (Fig. 1B). The cryosections of xenografted melanoma were processed for immunohistochemistry using FITC–antimouse Sca‐1 and PE–antimouse CD31. Representative images demonstrate CD31+ cells (red), Sca‐1+ cells (green), and CD31+/Sca‐1+ (yellow). Nuclei were stained with DAPI (blue). CD31+/Sca‐1+ cells (yellow) localized to some tumor vessels. These data suggest that TEC and peripheral blood‐derived endothelial cells contain EPC‐derived endothelial cells. As peripheral blood‐derived endothelial cells were cultured from PBMC, we assumed that they can be used to investigate the effects of EGCG on EPC.
Figure 1.
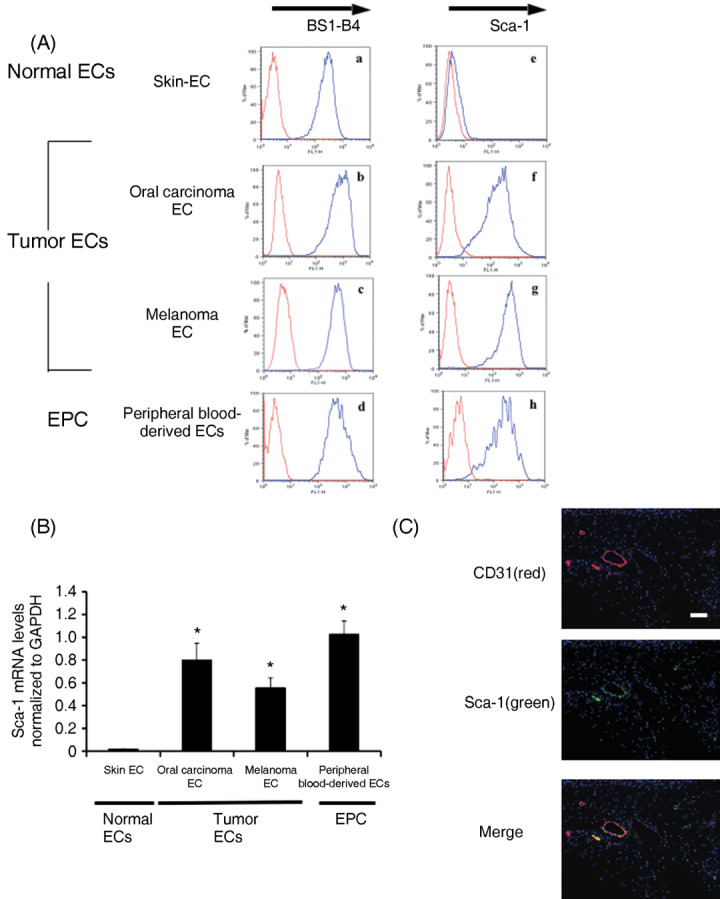
Peripheral blood‐derived endothelial cells (EPC) and tumor‐associated endothelial cells (TEC) bind BS1‐B4 lectin (mouse endothelial marker) and express stem‐cell antigen (Sca)‐1 (stem cell marker). (A) Normal endothelial cells (NEC), TEC, and EPC were characterized by flow cytometry. (a–d) NEC, TEC, and EPC were positive for BS1‐B4. (e–h) EPC expressed the stem cell marker Sca‐1. Sca‐1 was overexpressed also in TEC from both melanoma and oral carcinoma whereas it was expressed at a low level in NEC. (B) The mRNA expression of Sca‐1 was higher in TEC and EPC compared to NEC in real‐time PCR (*P < 0.05). (C) The cryosections of xenografted melanoma tumors were processed for immunohistochemistry using FITC‐conjugated antimouse Sca‐1 and phycoerythin‐conjugated antimouse CD31. Representative images are shown demonstrating CD31‐positive cells (red), Sca‐1‐positive cells (green), and CD31+/Sca‐1+ cells (yellow). Nuclei were stained with DAPI (blue). CD31+/Sca‐1+ cells existed in tumor vessels. Scale bar = 100 µm. EC, endothelial cells.
EGCG inhibited the migration of TEC and EPC. Because the PI3K–Akt signaling pathway has recently been shown to promote survival of TEC and migration( 22 , 40 ) that subsequently leads to angiogenesis, we analyzed the effect of the PI3K inhibitor LY294002 on VEGF‐induced migration in EC and EPC. LY294002 (20 µM) suppressed the VEGF‐induced cell migration of EC and EPC. These results suggest that cell migration of EC and EPC is stimulated by VEGF through activation of the PI3K signaling pathway. Then, the cell migration toward VEGF was analyzed in vitro with or without EGCG. EGCG significantly inhibited the migration of TEC and EPC toward VEGF compared with NEC (*P < 0.05) (Fig. 2).
Figure 2.
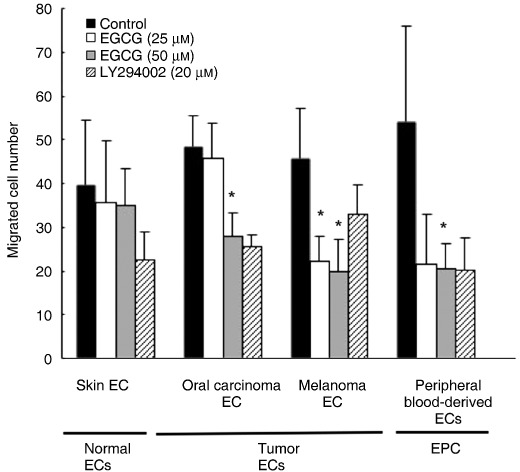
Epigallocatechin‐3 gallate (EGCG) inhibited tumor‐associated endothelial cells (TEC) and peripheral blood‐derived ECs (EPC) migration. Endothelial cells (EC) and EPC were pretreated with EGCG (25, 50 µM) for 16 h and the phosphoinositide 3‐kinase (PI3K) inhibitor LY294002 (20 µM) for 2 h. Migration of EC and EPC toward vascular endothelial growth factor (VEGF) was analyzed on fibronectin‐coated membranes using a Boyden chamber. Cells that migrated to the lower side of the membrane were counted in seven different high‐power fields. Migration of TEC and EPC, compared to normal endothelial cells, toward VEGF was inhibited significantly by EGCG (*P < 0.05 vs controls). Results represent the average of three experiments.
EGCG inhibited Akt phosphorylation in TEC. During angiogenesis, VEGF signaling is mediated by ligand‐dependent signaling through the PI3K–Akt pathway.( 41 ) The protein kinase Akt plays a central role in mature endothelial cells. Activation of Akt promotes survival by inhibiting apoptosis( 42 , 43 ) and mediates VEGF‐induced migration in endothelial cell.( 44 , 45 ) Because it was shown that the PI3K–Akt pathway is involved in cell migration of EC and EPC, we analyzed the level of Akt phosphorylation in these cells, using LY294002 or EGCG. Pretreatment with LY294002 (20 µM) decreased VEGF‐induced Akt activation in TEC but not in NEC. EGCG also reduced the level of phosphorylated Akt stimulated by VEGF in TEC (Fig. 3a,b). Specific inhibition of the PI3K–Akt pathway by EGCG in TEC is a possible explanation for the differential effect of EGCG on cell migration between TEC and NEC, as shown in Figure 2.
Figure 3.
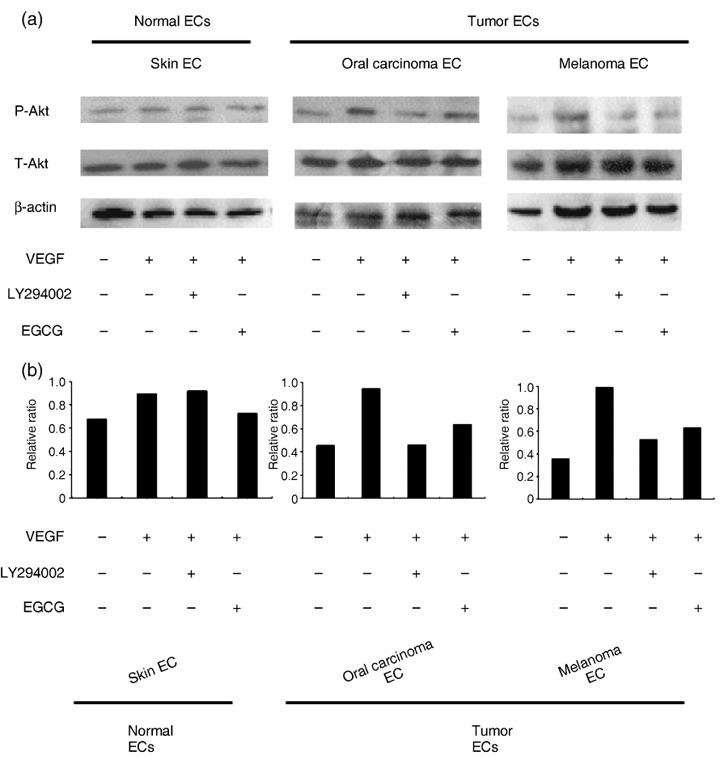
Epigallocatechin‐3 gallate (EGCG) inhibited Akt phosphorylation in tumor‐associated endothelial cells (TEC) but not in normal endothelial cells (NEC). (a) Serum‐starved endothelial cells (EC) were pretreated with EGCG (100 µM) for 16 h or the phosphoinositide 3‐kinase (PI3K) inhibitor LY294002 (20 µM) for 2 h before addition of vascular endothelial growth factor (VEGF) (10 ng/mL). EGCG was not toxic to EC. EC were stimulated with VEGF for 30 min. The levels of phosphorylated Akt (P‐Akt) were determined by western blotting using an antiphospho‐Akt antibody. After western blot analysis with antiphospho‐Akt (Ser473) antibody (top), the membrane was stripped and reincubated with anti‐total Akt (T‐Akt) antibody (middle) and β‐actin antibody (bottom) to detect the amount of total‐Akt (T‐Akt) and β‐actin protein. (b) Levels of P‐Akt were normalized for total Akt by scanning densitometry using computed image analysis. The graph shows the relative ratio of P‐Akt (Ser473)/T‐Akt by densitometric analysis. The experiment was carried out three times with similar results.
VEGFR2 was upregulated in TEC and EPC. VEGFR2 plays a major role in tumor angiogenesis. VEGFR2 is responsible for most of the mitogenic and chemotactic effects of VEGF in tumor angiogenesis. Some studies have provided evidence that EGCG inhibits VEGF‐induced phosphorylation of VEGFR2.( 15 ) To address the mechanism of differential response to EGCG between TEC, EPC, and NEC under VEGF treatment, the expression level of mRNA for VEGFR2 was analyzed by real‐time RT‐PCR. TEC and EPC upregulated VEGFR2 compared to NEC (Fig. 4). It was suggested that the different levels of VEGFR2 expression may be one reason why EGCG causes inhibitory effects more in TEC, and EPC in the presence of VEGF, compared with NEC.
Figure 4.
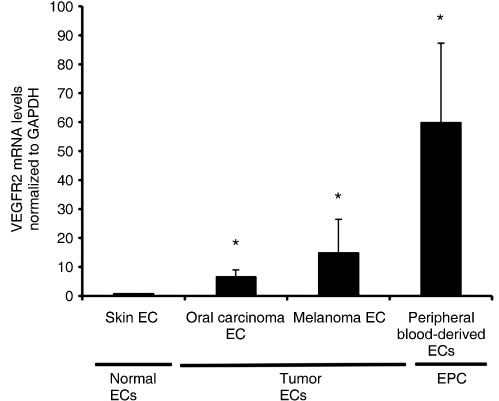
VEGF receptor (VEGFR) 2 is upregulated in tumor‐associated endothelial cells (TEC) and peripheral blood‐derived ECs (EPC). TEC and EPC showed higher levels of VEGFR2 mRNA compared to normal endothelial cells (NEC) by real‐time RT‐PCR. Expression levels of VEGFR2 mRNA were normalized to GAPDH (*P < 0.05 vs NEC).
EGCG suppressed the VEGF‐stimulated mobilization of CD133+/VEGFR2+ cells into peripheral blood. To evaluate the effect of EGCG on EPC mobilization, the number of CD133+/VEGFR2+ cells in circulation was analyzed in mice with or without EGCG treatment. VEGF was injected i.p. into mice to mobilize EPC and then a low dose of EGCG or vehicle was injected once a day for 2 days. Forty‐eight hours after the first injection, PBMC were isolated and counted. PBMC were incubated with anti‐CD133 and anti‐VEGFR2 antibodies, and the CD133+/VEGFR2+ cells in circulation were counted by flow cytometry. EGCG alone did not affect the number of CD133+/VEGFR2+ cells. The number of CD133+/VEGFR2+ cells in the control group was 11.86 ± 2.09/µL, whereas the number of CD133+/VEGFR2+ cells in the EGCG‐treated group was 9.71 ± 8.33/µL. However, in the VEGF‐treated group, the number of circulating CD133+/VEGFR2+ cells was lower after treatment with EGCG. The number of CD133+/VEGFR2+ cells in the VEGF‐treated group was 19.66 ± 19.12/µL, whereas the number of CD133+/VEGFR2+ cells in the VEGF + EGCG‐treated group was 8.8 ± 3.49/µL (Fig. 5). These results suggest that EGCG inhibits the VEGF‐induced mobilization of EPC into circulation. There was no significant difference between the VEGF group and the VEGF + EGCG‐treated group.
Figure 5.
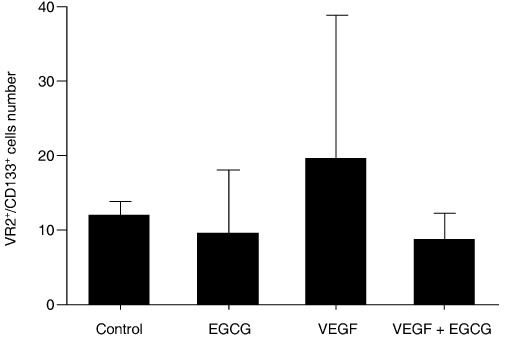
Epigallocatechin‐3 gallate (EGCG) suppressed VEGF‐stimulated mobilization of CD133+/VEGF receptor (VEGFR) 2+ cells into circulation. Ten nude mice in each group were injected intraperitoneally with PBS or a high dose of VEGF (300 ng) to mobilize EPC from the bone marrow. Next, in each group, EGCG (5 mg/kg) or vehicle was injected once a day for 2 days. Forty‐eight hours later, peripheral blood was collected from each mouse in each group (n = 5 for each group) before they were killed. Peripheral blood mononuclear cells (PBMC) were collected and incubated with FITC–anti‐mouse CD133 and PE–anti‐mouse VEGFR2, and the number of CD133+/VEGFR2+ cells was counted by flow cytometry. Although there was no significant difference between the VEGF group and the VEGF + EGCG group, the low dose of EGCG (5 mg/kg) moderately decreased the number of VEGF‐mobilized CD133+/VEGFR2+ cells that migrated into peripheral circulation.
EGCG suppressed the expression of MMP‐9 in BMSC from oral carcinoma‐bearing mice. MMP‐9 in the bone marrow plays an important role in the mobilization of bone marrow‐derived VEGFR2+ cells into peripheral circulation.( 27 ) In order to test the effects of EGCG on MMP‐9 expression in BMSC, we isolated BMSC from oral carcinoma‐bearing mice. Expression of mRNA for Sca‐1, SDF‐1, α‐SMA, and FAP was detected in BMSC but not in skin endothelial cells by RT‐PCR (Fig. 6a). To investigate whether EGCG affects the expression of MMP‐9 mRNA in BMSC, these cells were treated with or without EGCG (25, 50, 100 µM) for 24 h and the expression levels of MMP‐9 mRNA, normalized to GAPDH, were analyzed by real‐time PCR. MMP‐9 mRNA expression in BMSC was downregulated by EGCG. The relative expression level of MMP‐9 after 25, 50, and 100 µM of EGCG treatment was 0.49, 0.46, and 0.37, respectively (P < 0.05) (Fig. 6b).
Figure 6.
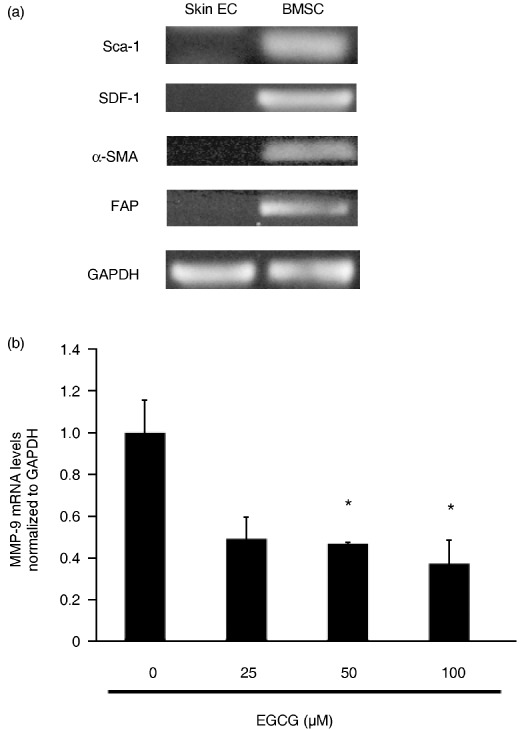
Epigallocatechin‐3 gallate (EGCG) suppressed the mRNA expression of MMP‐9 in bone marrow stromal cells (BMSC) of oral carcinoma‐bearing mice. (a) RT‐PCR characterization of BMSC for expression of the stem cell marker stem‐cell antigen (Sca)‐1, and the stromal cell markers stromal derived factor (SDF)‐1, fibroblast activation protein (FAP), and α‐smooth muscle actin (SMA). Sca‐1, SDF‐1, α‐SMA, and FAP were expressed in BMSC but not skin endothelial cells. (b) The possibility that EGCG affects mRNA expression of MMP‐9 in BMSC was assessed by real‐time PCR. BMSC were pretreated with or without EGCG (25, 50, 100 µM) for 24 h. Expression levels of MMP‐9 mRNA were normalized to GAPDH. EGCG significantly reduced MMP‐9 mRNA levels in BMSC. The experiment was carried out three times with similar results (*P < 0.05 vs 0 µM). EC, endothelial cells.
EGCG inhibited tumor growth in vivo. To address the question of whether EGCG inhibits tumor growth in vivo, either EGCG or vehicle as a control was administered i.p. into the melanoma xenograft mice. After 36 days, EGCG suppressed melanoma growth (689.8 ± 276.6 mm3 in vivo) compared with controls (354.2 ± 188.2 mm3). There was a significant difference between EGCG‐treated and control tumors (P < 0.05) (Fig. 7a). Drug treatment was well tolerated without apparent toxicity or bodyweight changes throughout the study. Cryosections of each tumor were stained with an anti‐CD31 antibody (Fig. 7b). Mice treated with EGCG showed a significant reduction in MVD (control 3.11 ± 1.02% vs EGCG 1.83 ± 0.35, mean ± SD, P < 0.05) (Fig. 7c). These results suggest that EGCG inhibits tumor growth with anti‐angiogenic activity.
Figure 7.
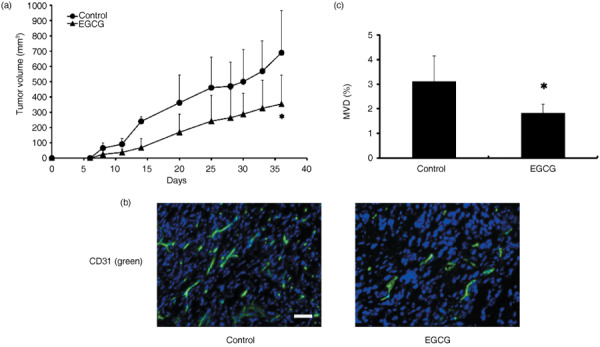
Epigallocatechin‐3 gallate (EGCG) repressed melanoma tumor growth. (a) The melanoma tumor cells were inoculated subcutaneously into nude mice. Day 0 is defined as the first day of treatment. Two groups of animals were then treated with the vehicle (0.05% DMSO, n = 5) or EGCG (20 mg/kg, n = 5) intraperitoneally every day. Tumor growth was significantly suppressed by EGCG treatment compared with the controls (*P < 0.05). Means ± SD are shown. (b) Immunohistochemical analysis of tumors. Snap‐frozen tumor tissue specimens were processed for immunohistochemical study when mice were killed. Cryosections were stained with FITC‐labeled anti‐CD31 antibody (green). The sections were counterstained with DAPI (blue). The CD31‐positive vessel area was decreased in EGCG‐treated tumors compared to controls. Scale bar = 100 µm. (c) Microvessel density (MVD) was analyzed quantitatively. Immunohistochemical analysis of tumors indicated a significant reduction in MVD in tumors after treatment with EGCG (*P < 0.05).
Discussion
We provide new evidence about the mechanism by which EGCG inhibits tumor angiogenesis. First, EGCG inhibited cell migration toward VEGF in TEC and EPC, but not in NEC. Second, EGCG inhibited Akt phosphorylation in TEC. Third, EGCG suppressed the VEGF‐induced mobilization of CD133+/VEGFR2+ cells into peripheral circulation in vivo. EGCG downregulated the expression level of MMP‐9 in BMSC, which is related to the mobilization of EPC into peripheral circulation.( 26 , 27 ) Finally, EGCG suppressed in vivo tumor growth in nude mice with anti‐angiogenic activity.
We demonstrate for the first time that EGCG specifically targets TEC and EPC. Although we did not identify the molecules responsible for this specificity, the specific inhibition of Akt phosphorylation in TEC suggests that the PI3K–Akt pathway mediates targeted signaling. In our previous studies with isolated TEC, we found that they were different from NEC in many respects, including sensitivity to growth factors and to certain drugs such as an EGF receptor kinase inhibitor.( 23 , 24 , 46 ) Activation of Akt mediates VEGF‐induced endothelial cell migration.( 44 , 45 ) In our study, the PI3K inhibitor LY294002 significantly reduced cell migration in TEC and EPC and Akt phosphorylation was inhibited by LY294002 in TEC. These results suggest that a PI3K‐dependent pathway modulates cell migration of TEC and EPC, which we isolated, consistent with other reports on microvascular endothelial cells.( 45 ) Furthermore, our results show that EGCG inhibited VEGF‐induced Akt phosphorylation in TEC, but not in NEC. These results suggest that inactivation of Akt signaling is involved in the anti‐angiogenic effects of EGCG on TEC and EPC causing the inhibition of migration. It was speculated that selective inhibition by EGCG on TEC and EPC may have resulted from their upregulation of VEGFR2, which stimulates the PI3K–Akt pathway.
EPC are currently considered to be a novel target for anti‐angiogenic therapy, as are TEC because they play important roles in tumor metastasis.( 26 ) It has been shown that tumors mobilize bone marrow‐derived EPC, apart from recruiting neighboring blood vessels or EC, and that EPC migrate to tumors and become incorporated into their developing vasculature.( 47 ) The contribution of EPC to tumor angiogenesis in mouse models ranges from 2 to 50%.( 28 , 48 ) Several anti‐angiogenic drugs have already been reported to produce inhibitory effects on EPC.( 28 ) EPC are considered to express CD133 and VEGFR2.( 36 , 37 , 49 ) Here we showed for the first time, using a mouse model, that EGCG inhibits mobilization of CD133+/VEGFR2+ cells into peripheral circulation in vivo. It is expected that EGCG will target EPC besides TEC when used for cancer therapy.
The expression of MMP‐9 mRNA was significantly downregulated by EGCG treatment in BMSC. MMP play key roles in cancer invasion and metastasis. Upregulated MMP‐9 activity in bone marrow cleaves the membrane‐bound stem cell cytokine mKitL expressed by BMSC to liberate soluble sKitL. The survival activity of sKitL then stimulates the mobilization of bone marrow‐derived VEGFR2+ cells to peripheral circulation. In MMP‐9−/– mice, release of sKitL, hematopoietic cell motility, and the mobilization of bone marrow‐derived VEGFR2+ cells are impaired.( 27 ) EGCG has been reported to inhibit the activity of MMP‐2, MMP‐3, and MMP‐9 in cancer cells.( 50 ) In our study, suppression of MMP‐9 expression by EGCG in BMSC may be one explanation for reduction of EPC mobilization.
Finally, we demonstrated that EGCG inhibited the growth of tumors in nude mice with impaired angiogenesis. It was suggested that the antitumor effects of EGCG resulted from anti‐angiogenic activity on TEC and EPC.
We conclude that EGCG inhibits tumor angiogenesis by inhibiting PI3K–Akt signaling specifically in TEC and peripheral blood‐derived endothelial cells, but not in NEC. It might be possible to use EGCG as an anti‐angiogenic drug, either as a single agent or for cancer treatment in adjuvant settings for relatively long periods, as it is not toxic to normal cells.
Acknowledgments
We thank Dr I. J. Fidler for providing the A375SM super‐metastatic human malignant melanoma cell line, Ms Tomoko Takahashi and Ms Midai Muranaka for technical assistance. This work was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan (K. Hida and M. Shindoh), The Haraguchi Memorial Foundation for Cancer Research, The Akiyama Foundation, and The Takeda Science Foundation (K. Hida), and a research grant from the Tea Association of Kyoto Prefecture (Y. Hida).
References
- 1. Yang CS, Wang ZY. Tea and cancer. J Natl Cancer Inst 1993; 85: 1038–49. [DOI] [PubMed] [Google Scholar]
- 2. Wang ZY, Wang LD, Lee MJ et al . Inhibition of N‐nitrosomethylbenzylamine‐induced esophageal tumorigenesis in rats by green and black tea. Carcinogenesis 1995; 16: 2143–8. [DOI] [PubMed] [Google Scholar]
- 3. Yang CS, Chung JY, Yang G, Chhabra SK, Lee MJ. Tea and tea polyphenols in cancer prevention. J Nutr 2000; 130: 472S–8S. [DOI] [PubMed] [Google Scholar]
- 4. Fujiki H, Suganuma M, Imai K, Nakachi K. Green tea: cancer preventive beverage and/or drug. Cancer Lett 2002; 188: 9–13. [DOI] [PubMed] [Google Scholar]
- 5. Mukhtar H, Katiyar SK, Agarwal R. Cancer chemoprevention by green tea components. Adv Exp Med Biol 1994; 354: 123–34. [DOI] [PubMed] [Google Scholar]
- 6. Mukhtar H, Katiyar SK, Agarwal R. Green tea and skin – anticarcinogenic effects. J Invest Dermatol 1994; 102: 3–7. [DOI] [PubMed] [Google Scholar]
- 7. Jankun J, Selman SH, Swiercz R, Skrzypczak‐Jankun E. Why drinking green tea could prevent cancer. Nature 1997; 387: 561. [DOI] [PubMed] [Google Scholar]
- 8. Cao Y, Cao R, Brakenhielm E. Antiangiogenic mechanisms of diet‐derived polyphenols. J Nutr Biochem 2002; 13: 380–90. [DOI] [PubMed] [Google Scholar]
- 9. Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer 2007; 7: 139–47. [DOI] [PubMed] [Google Scholar]
- 10. Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science 2006; 312: 1171–5. [DOI] [PubMed] [Google Scholar]
- 11. Ferrara N, Gerber HP. The role of vascular endothelial growth factor in angiogenesis. Acta Haematol 2001; 106: 148–56. [DOI] [PubMed] [Google Scholar]
- 12. Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol 2001; 280: C1358–66. [DOI] [PubMed] [Google Scholar]
- 13. Fassina G, Vene R, Morini M et al . Mechanisms of inhibition of tumor angiogenesis and vascular tumor growth by epigallocatechin‐3‐gallate. Clin Cancer Res 2004; 10: 4865–73. [DOI] [PubMed] [Google Scholar]
- 14. Kondo T, Ohta T, Igura K, Hara Y, Kaji K. Tea catechins inhibit angiogenesis in vitro, measured by human endothelial cell growth, migration and tube formation, through inhibition of VEGF receptor binding. Cancer Lett 2002; 180: 139–44. [DOI] [PubMed] [Google Scholar]
- 15. Lamy S, Gingras D, Beliveau R. Green tea catechins inhibit vascular endothelial growth factor receptor phosphorylation. Cancer Res 2002; 62: 381–5. [PubMed] [Google Scholar]
- 16. Tang FY, Meydani M. Green tea catechins and vitamin E inhibit angiogenesis of human microvascular endothelial cells through suppression of IL‐8 production. Nutr Cancer 2001; 41: 119–25. [DOI] [PubMed] [Google Scholar]
- 17. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–6. [DOI] [PubMed] [Google Scholar]
- 18. Allinen M, Beroukhim R, Cai L et al . Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004; 6: 17–32. [DOI] [PubMed] [Google Scholar]
- 19. McDonald DM, Choyke PL. Imaging of angiogenesis: from microscope to clinic. Nat Med 2003; 9: 713–25. [DOI] [PubMed] [Google Scholar]
- 20. St Croix B, Rago C, Velculescu V et al . Genes expressed in human tumor endothelium. Science 2000; 289: 1197–202. [DOI] [PubMed] [Google Scholar]
- 21. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002; 160: 985–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor‐derived endothelial cells. Faseb J 2003; 17: 1159–61. [DOI] [PubMed] [Google Scholar]
- 23. Hida K, Hida Y, Amin DN et al . Tumor‐associated endothelial cells with cytogenetic abnormalities. Cancer Res 2004; 64: 8249–55. [DOI] [PubMed] [Google Scholar]
- 24. Hida K, Klagsbrun M. A new perspective on tumor endothelial cells: unexpected chromosome and centrosome abnormalities. Cancer Res 2005; 65: 2507–10. [DOI] [PubMed] [Google Scholar]
- 25. Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res 2006; 66: 2173–80. [DOI] [PubMed] [Google Scholar]
- 26. Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti‐angiogenesis therapy? Nat Rev Cancer 2002; 2: 826–35. [DOI] [PubMed] [Google Scholar]
- 27. Heissig B, Werb Z, Rafii S, Hattori K. Role of c‐kit/Kit ligand signaling in regulating vasculogenesis. Thromb Haemost 2003; 90: 570–6. [DOI] [PubMed] [Google Scholar]
- 28. Bertolini F, Shaked Y, Mancuso P, Kerbel RS. The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer 2006; 6: 835–45. [DOI] [PubMed] [Google Scholar]
- 29. Nolan DJ, Ciarrocchi A, Mellick AS et al . Bone marrow‐derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev 2007; 21: 1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng XW, Kuzuya M, Nakamura K et al . Mechanisms underlying the impairment of ischemia‐induced neovascularization in matrix metalloproteinase 2‐deficient mice. Circ Res 2007; 100: 904–13. [DOI] [PubMed] [Google Scholar]
- 31. Arbiser JL, Raab G, Rohan RM et al . Isolation of mouse stromal cells associated with a human tumor using differential diphtheria toxin sensitivity. Am J Pathol 1999; 155: 723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beaudry P, Hida Y, Udagawa T et al . Endothelial progenitor cells contribute to accelerated liver regeneration. J Pediatr Surg 2007; 42: 1190–8. [DOI] [PubMed] [Google Scholar]
- 33. Dormond O, Foletti A, Paroz C, Ruegg C. NSAIDs inhibit αVβ3 integrin‐mediated and Cdc42/Rac‐dependent endothelial‐cell spreading, migration and angiogenesis. Nat Med 2001; 7: 1041–7. [DOI] [PubMed] [Google Scholar]
- 34. Bielenberg DR, Hida Y, Shimizu A et al . Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest 2004; 114: 1260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murphy DA, Makonnen S, Lassoued W, Feldman MD, Carter C, Lee WM. Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43‐9006). Am J Pathol 2006; 169: 1875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asahara T, Murohara T, Sullivan A et al . Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997; 275: 964–7. [DOI] [PubMed] [Google Scholar]
- 37. Asahara T, Takahashi T, Masuda H et al . VEGF contributes to postnatal neovascularization by mobilizing bone marrow‐derived endothelial progenitor cells. EMBO J 1999; 18: 3964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharpe EE 3rd, Teleron AA, Li B et al . The origin and in vivo significance of murine and human culture‐expanded endothelial progenitor cells. Am J Pathol 2006; 168: 1710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramanadham S, Yarasheski KE, Silva MJ et al . Age‐related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2beta)‐null mice. Am J Pathol 2008; 172: 868–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bussolati B, Assenzio B, Deregibus MC, Camussi G. The proangiogenic phenotype of human tumor‐derived endothelial cells depends on thrombospondin‐1 downregulation via phosphatidylinositol 3‐kinase/Akt pathway. J Mol Med 2006; 84: 852–63. [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez SK, Guo W, Liu L, Band MA, Paulson EK, Meydani M. Green tea catechin, epigallocatechin‐3‐gallate, inhibits vascular endothelial growth factor angiogenic signaling by disrupting the formation of a receptor complex. Int J Cancer 2006; 118: 1635–44. [DOI] [PubMed] [Google Scholar]
- 42. Gerber HP, McMurtrey A, Kowalski J et al . Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′‐kinase/Akt signal transduction pathway. Requirement for Flk‐1/KDR activation. J Biol Chem 1998; 273: 30 336–43. [DOI] [PubMed] [Google Scholar]
- 43. Dimmeler S, Hermann C, Zeiher AM. Apoptosis of endothelial cells. Contribution to the pathophysiology of atherosclerosis? Eur Cytokine Netw 1998; 9: 697–8. [PubMed] [Google Scholar]
- 44. Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser‐1177 is required for VEGF‐induced endothelial cell migration. FEBS Lett 2000; 477: 258–62. [DOI] [PubMed] [Google Scholar]
- 45. Morales‐Ruiz M, Fulton D, Sowa G et al . Vascular endothelial growth factor‐stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res 2000; 86: 892–6. [DOI] [PubMed] [Google Scholar]
- 46. Hida K, Hida Y, Shindoh M. Understanding tumor endothelial cell abnormalities to develop ideal anti‐angiogenic therapies. Cancer Sci 2008; 99: 459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lyden D, Hattori K, Dias S et al . Impaired recruitment of bone‐marrow‐derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med 2001; 7: 1194–201. [DOI] [PubMed] [Google Scholar]
- 48. Shaked Y, Bertolini F, Man S et al . Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis: Implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell 2005; 7: 101–11. [DOI] [PubMed] [Google Scholar]
- 49. Gehling UM, Ergun S, Schumacher U et al . In vitro differentiation of endothelial cells from AC133‐positive progenitor cells. Blood 2000; 95: 3106–12. [PubMed] [Google Scholar]
- 50. Siddiqui IA, Malik A, Adhami VM et al . Green tea polyphenol EGCG sensitizes human prostate carcinoma LNCaP cells to TRAIL‐mediated apoptosis and synergistically inhibits biomarkers associated with angiogenesis and metastasis. Oncogene 2007; 27: 2055–63. [DOI] [PubMed] [Google Scholar]