

Abstract
Although heat shock proteins (HSP) are well known to contribute to thermotolerance, they only play a supporting role in the phenomenon. Recently, it has been reported that heat sensitivity depends on heat‐induced DNA double‐strand breaks (DSB), and that thermotolerance also depends on the suppression of DSB formation. However the critical elements involved in thermotolerance have not yet been fully identified. Heat produces DSB and leads to cell death through denaturation and dysfunction of heat‐labile repair proteins such as DNA polymerase‐β (Polβ). Here the authors show that thermotolerance was partially suppressed in Polβ−/– mouse embryonic fibroblasts (MEF) when compared to the wild‐type MEF, and was also suppressed in the presence of the HSP inhibitor, KNK437, in both cell lines. Moreover, the authors found that heat‐induced γH2AX was suppressed in the thermotolerant cells. These results suggest that Polβ at least contributes to thermotolerance through its reactivation and stimulation by Hsp27 and Hsp70. In addition, it appears possible that fewer DSB were formed after a challenging heat exposure because preheat‐induced Hsp27 and Hsp70 can rescue or restore other, as yet unidentified, heat‐labile proteins besides Polβ. The present novel findings provide strong evidence that Polβ functions as a critical element involved in thermotolerance and exerts an important role in heat‐induced DSB. (Cancer Sci 2008; 99: 973–978)
Hyperthermia is widely used to treat patients with various types of cancers, and is performed in combination with radiation and/or anticancer agents.( 1 ) The enhancement of radiation‐induced cell killing by heat,( 2 ) and the existence of an inflection point in Arrhenius plots of 1/T0 (the mean lethal heating period [min]) versus temperature,( 3 ) indicate that heat‐induced protein inactivation is critically involved in heat‐induced cell killing. Heat‐induced protein denaturation results in the disruption of centrosome‐dependent mitosis,( 4 ) and of multiple nuclear matrix‐dependent functions.( 5 )
Exposure of cells to a transient, non‐lethal elevation in temperature results in the activation of cellular stress responses and induces a state of thermotolerance in cells that renders them resistant to subsequent lethal insults.( 6 ) Thermotolerant cells are less sensitive to hyperthermia‐induced cytotoxicity, growth factor withdrawal, heavy metals, radiation, and anticancer drugs.( 7 ) Thermotolerance is associated with the synthesis and cellular accumulation of a family of highly conserved proteins referred to as heat shock proteins (HSP). It has been reported that Hsp27 and Hsp70 mainly contribute to thermotolerance( 8 , 9 , 10 ) through molecular chaperone activity.( 11 , 12 ) However, little is known about their partners in the mechanism of thermotolerance. Therefore, the authors focused on just Hsp27 and Hsp70.
Among the variety of possible DNA‐damaging events that can occur, DNA double‐strand breaks (DSB) are the most serious. Genome integrity is maintained by dynamic responses to the presence of DNA damage, in which damage sensing, cell‐cycle arrest, and repair factors are involved.( 13 ) Therefore, the existence of DSB repair systems is a critical component in cellular radiation sensitivity and may be important in the mechanisms involved in heat sensitivity of cancer cells. Recently, heat‐induced DSB have been detected and shown to be involved in heat‐induced cell killing.( 14 ) A positive correlation was seen between thermotolerance for heat killing and the heat‐induced loss of DNA polymerase‐β (Polβ) activity in cells.( 15 ) Moreover, recent work has indicated that heat‐induced γH2AX (histone H2AX phosphorylated at serine 139) foci formation was suppressed in thermotolerance development.( 14 ) However, the physiological function of Polβin vivo has not been investigated. It is hypothesized that heat induces DSB formation through the denaturation and dysfunction of heat‐labile repair enzymes such as Polβ because DNA alone is quite resistant to heat, and because the in vitro polymerase chain reaction (PCR) assay results in almost no DNA lesions.( 13 ) In the present study, the role of Polβ in thermotolerance development was investigated using Polβ‐knockout mouse embryonic fibroblasts (Polβ−/– MEF).( 16 )
Materials and Methods
Cells. H1299 (human non‐small lung carcinoma p53 deficient cells) cells were provided by Dr M. Oren (Department of Molecular Cell Biology, Weizmann Institute of Science, Israel).( 14 ) Polβ−/– MEF was established as previously described.( 16 ) These cells were cultured in Dulbecco's modified Eagle's medium‐10 (DMEM‐10; MP Biomedicals Inc., Illkirch, France) containing 10% (v/v) fetal bovine serum (FBS; MP Biomedicals Inc.), 20 mmol/L 2‐[4‐(2‐hydroxyethyl)‐1‐piperazinyl] ethanesulfonic acid (Nacalai Tesque, Kyoto, Japan), 30 µg/mL penicillin (Meiji Seika Kaisha Ltd, Tokyo, Japan), 50 µg/mL streptomycin (Meiji Seika Kaisha Ltd), and 50 µg/mL kanamycin (Nacalai Tesque). The cells were cultured at 37°C in a humidified CO2 incubator.
Hyperthermia. Exponentially growing cells were immersed in a water bath (Thermominder EX; Taitec Co., Ltd, Koshigaya, Japan) maintained at 45.5°C. After a preheating treatment and a challenging heat treatment, the cells were cool down immediately and then incubated at 37°C in a humidified CO2 incubator. Under the present experimental conditions, no marked change in pH values was detected in the medium during the treatment.
Co‐immunoprecipitation and Western blot analysis. Exponentially growing H1299 cells were cultured in 25‐cm2 flasks. For the immunoprecipitation studies, the medium was removed, and the cells were washed with phosphate‐buffered saline (PBS) buffer. Nuclear extracts from cells were obtained using a NE‐PER Nuclear and Cytoplasmic Extraction Reagents Kit and Slide‐A‐Lyzer 3.5K MWCO Dialysis Cassettes (Pierce Biotechnology, Inc., Rockford, IL, USA). 10 ng of antimouse IgG1 antibody (IHC, BD Biosciences Pharmingen, San Diego, CA, USA) was added to 100 µg of nuclear proteins, and the mixture was gently rotated at 4°C for 1 h. MagaCell Protein G beads (150 µL; Bio‐Nobile, Turku, Finland) were then added, and the incubation was continued for an additional 1 h. The beads were removed using a PickPen (Bio‐Nobile). This step was repeated twice. Anti‐polymerase‐β monoclonal antibodies (18S; NeoMarkers, Fremont, CA, USA) and MagaCell Protein G beads (300 µL) were incubated together and gently rotated at 4°C for 1 h, and the mixture was then added to nuclear proteins. This preparation was then gently rotated at 4°C for 1 h. The MagaCell protein G beads were collected with a PickPen, and the beads were then suspended and washed three times with IP buffer (50 mmol tris‐HCl, pH 8.0, containing 5 mmol ethylene diamine tetra‐acetic acid [EDTA], 150 mmol NaCl, 1 mmol phenylmethylsulfonyl fluoride [PMSF], 50 mmol sodium fluoride, 0.1 mmol sodium orthovanadate, 1%[v/v] Nonidet P‐40). The beads were then suspended in 50 µL of sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) loading buffer and heated to 95°C for 5 min. The beads were removed with a PickPen. Immunoprecipitated proteins (20 µL) were subjected to Western blot analysis. After electrophoresis on 15% polyacrylamide gels containing 0.1% SDS, the proteins were transferred electrophoretically onto Poly Screen polyvinylidene difluoride (PVDF) membranes (DuPont/Biotechnology Systems, New England Nuclear Research Products, Boston, MA, USA). The membranes were then incubated with anti‐Hsp27 monoclonal antibody (Ab1; Calbiochem, San Diego, CA, USA), anti‐Hsp70 monoclonal antibody (C92F3 A‐5; StressGen Biotechnologies Corp., Victoria, Canada), Polβ mouse monoclonal antibody, and antiactin polyclonal antibody (I‐19; Santa Cruz Biotechnology, Santa Cruz, CA, USA). For visualization of the bands, horseradish peroxidase (HP)‐conjugated antimouse IgG antibody (Zymed Laboratories, Inc., San Francisco, CA, USA) for Hsp27, Hsp70 and Polβ, and HP‐conjugated antirabbit IgG antibody (Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA) for actin were used with the BLAST: Blotting Amplification System (DuPont/Biotechnology Systems).
Colony forming assays. Cell survival was measured using a standard colony forming assay. Three flasks were used for each data point, and three independent experiments were performed for each survival point. The colonies obtained after 10 days were fixed with methanol and stained with 2% Giemsa solution. Microscopic colonies composed of more than approximately 50 cells were counted as surviving cells. Surviving fraction values were fitted using the single‐hit multitarget model:
where S/S0 is the surviving fraction, T is the heating period (min), T0 is the mean lethal heating period (min) required to reduce the fraction of cells to 37% indicative of single‐event killing and n is the number of targets in the cell, all of which must be inactivated to kill the cell. The parameters n and T0 were calculated using KaleidaGraph (Synergy Software, PA, USA). The thermotolerance ratio (TTR) was calculated according the formula; TTR = Tx/T0 , where Tx is the mean lethal heating period (min) with preheating, and T0 is the heating period without preheating.
KNK437 treatment. N‐formyl‐3,4‐methylenedioxy‐γ‐butyrolactam (KNK437; synthesized by the Kaneka Co., Osaka, Japan),( 17 ) was dissolved in dimethyl sulfoxide (DMSO) and added to culture medium at a final concentration of 0, 100 or 300 µmol 1 h before preheating. For cell colony forming assays, the medium containing KNK437 was exchanged for KNK437‐free medium 16 h after the start of preheating. For flow cytometry, the medium containing KNK437 was not changed until sampling. The same concentration of DMSO was used as a control.
Flow cytometry. The heat treated Polβ+/+ and Polβ−/– MEF were cultured for 30 min. Then the cells were fixed with cold 70% methanol and kept at –20°C. Cells were centrifuged and rinsed with TPBS (PBS containing 0.05% Tween 20). The cells were blocked with rabbit serum for 15 min at room temperature and rinsed with TPBS. The cells were then incubated with antiphospho‐H2AX monoclonal antibody (JBW301; Upstate Biotechnology, Lake Placid, NY, USA) at a 300‐fold dilution for 60 min at room temperature; rinsed with TPBS; incubated with AlexaFluor 488‐conjugated antimouse IgG second antibody (Molecular Probes, Eugene, OR, USA) at a 400‐fold dilution for 60 min at room temperature; and rinsed in TPBS. The cell cycle distribution was measured by determining the cellular DNA content. For the determination of DNA content, cells were fixed with cold 70% methanol. The cells were then incubated for 30 min at room temperature with 1 mg/mL RNase and 50 µg/mL propidium iodide. Before flow cytometric analysis, samples were filtered through a 35‐µm nylon mesh. Samples were analyzed using a flow cytometer (Becton Dickinson, San Jose, CA, USA).
Statistical analysis. Levels of significance were calculated using an unpaired Student's t‐test. P < 0.05 was considered significant.
Results
HSP accumulate and bind to Polβ in the nucleus during the development of thermotolerance. To clarify the relationship between thermotolerance and heat‐induced HSP, control H1299 cells were conditioned by preheating (45.5°C, 5 min), incubating at 37°C for various periods up to 24 h, and then exposing to a challenging heat treatment at 45.5°C. The thermotolerance ratio increased when H1299 cells were incubated for 3–6 h after a conditioning preheat treatment before the challenging heat treatment (Fig. 1a). The results of Western blot analysis on nuclear extracts indicated that Hsp27 and Hsp70 levels increased substantially in heated cells when compared with control cells (Fig. 1b). Quantitatively, Hsp27 and Hsp70 levels in the heated cells increased 1.1–1.7‐fold at 3–6 h after heat treatment (Fig. 1c).
Figure 1.
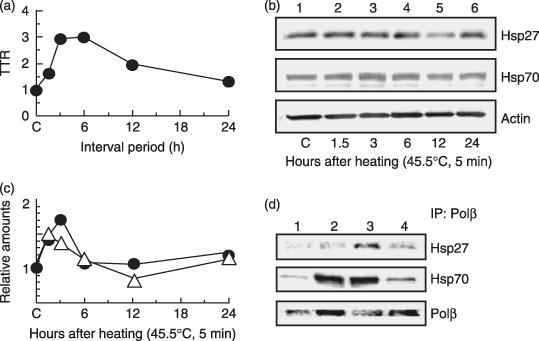
Heat shock protein (HSP) accumulation and binding to polymerase‐β (Polβ) in the nucleus. H1299 cells were treated at various intervals after a preheating treatment (45.5°C, 5 min). (a) Thermotolerance ratio (TTR). (b) HSP bands in Western blot analysis. Lane 1, untreated control samples. Lanes 2–6, samples at various periods after a heat treatment alone (45.5°C, 5 min): lane 2, 1.5 h; lane 3, 3 h; lane 4, 6 h; lane 5, 12 h; lane 6, 24 h. (c) Density of HSP bands with Western blot analysis. (j) ( ) Hsp27. (
) Hsp27. ( ) Hsp70. (d) Co‐immunoprecipitation experiments with nuclear extracts. Lane 1, non‐heating treatment; lane 2, 3 h after a preheating treatment alone (45.5°C, 5 min); lane 3, challenging heat treatment (45.5°C, 20 min) at 3 h after a preheating treatment (45.5°C, 5 min); lane 4, challenging heat treatment alone (45.5°C, 20 min).
) Hsp70. (d) Co‐immunoprecipitation experiments with nuclear extracts. Lane 1, non‐heating treatment; lane 2, 3 h after a preheating treatment alone (45.5°C, 5 min); lane 3, challenging heat treatment (45.5°C, 20 min) at 3 h after a preheating treatment (45.5°C, 5 min); lane 4, challenging heat treatment alone (45.5°C, 20 min).
Any protective effect of HSP on Polβ function during heat stress probably would be derived from a direct physical association between the two proteins. However, evidence for any direct binding between HSP and Polβ has not been reported to date. A co‐immunoprecipitation technique was applied to detect any physical association between them. Evidence for co‐immunoprecipitation of HSP with Polβ was sought using an anti‐Polβ antibody (Fig. 1d). Fig. 1d shows that Hsp27 and Hsp70 did co‐immunoprecipitate with Polβ after challenging heat treatment with preheating treatment, but not after non‐heating treatment and challenging heat treatment alone. In addition, Hsp70 did co‐immunoprecipitate with Polβ after preheating treatment alone.
Thermotolerance was suppressed in Polβ−/– MEF and KNK437‐treated cells. To examine whether the loss of Polβ induction impaired thermotolerance, Polβ+/+ and Polβ−/– MEF were heated at 45.5°C (with or without preheating at 45.5°C for 5 min), and cell viability was measured (Fig. 2). Both types of MEF cells showed similar levels of heat sensitivity without preheating, but preheating markedly increased cell viability in Polβ+/+ MEF. Although thermotolerance reached its peak at 12 h after preheating treatment in both types of cells, thermotolerance was suppressed in Polβ−/– MEF. Thermotolerance in Polβ−/– MEF was reduced approximately one‐half to one‐third when compared with thermotolerance in Polβ+/+ MEF (Fig. 2b).
Figure 2.
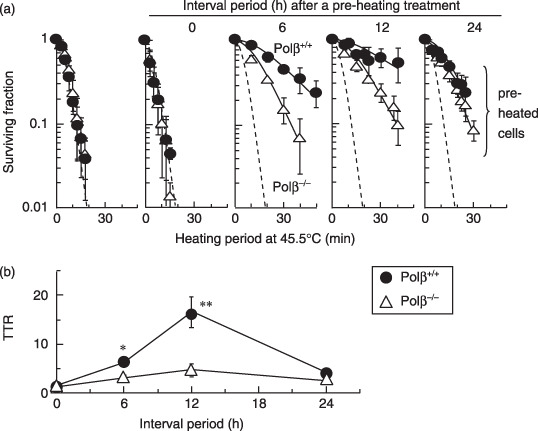
The effect of polymerase‐β (Polβ) on thermotolerance. Polβ+/+ and Polβ−/– mouse embryonic fibroblasts (MEF) were heated (45.5°C) after various times at intervals (0, 6, 12, or 24 h) after a preheating treatment (45.5°C, 5 min). (a) Survival curves. Dotted lines show survival curves at 45.5°C for different heating periods without preheating. Error bars represent ±SD. (b) Thermotolerance ratio (TTR). () Polβ+/+ MEFs. () Polβ−/– MEFs. *P < 0.05 and **P < 0.01, by Student's t‐test between Polβ+/+ and Polβ−/– MEF, respectively.
To demonstrate the effect of HSP on thermotolerance, the cells were exposed to KNK437 (Fig. 3), an inhibitor of HSP. There was no significant effect of KNK437 on heat sensitivity in either type of MEF. However, thermotolerance was markedly suppressed by KNK437, not only in Polβ+/+ MEF but also in Polβ−/– MEF.
Figure 3.
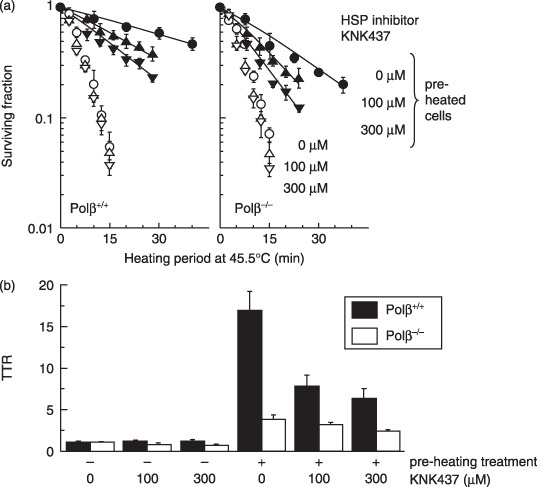
The effect of the heat shock protein (HSP) inhibitor, KNK437 on thermotolerance in polymerase‐β (Polβ)+/+ and Polβ−/– mouse embryonic fibroblasts (MEF). Polβ+/+ and Polβ−/– MEF were heated (45.5°C) at 12 h after a preheating treatment (45.5°C, 5 min) with KNK437. (a) Survival curves. Error bars represent ±SD. ( ,,
,, ) Cells were not conditioned by a preheating treatment. (,
) Cells were not conditioned by a preheating treatment. (, ,
, ) Cells were conditioned by a preheating treatment. Left panel: Polβ+/+ MEF. Right panel: Polβ−/– MEF. (,) 0 µM KNK437. (,) 100 µM KNK437. (,) 300 µM KNK437. (b) Thermotolerance ratios. (
) Cells were conditioned by a preheating treatment. Left panel: Polβ+/+ MEF. Right panel: Polβ−/– MEF. (,) 0 µM KNK437. (,) 100 µM KNK437. (,) 300 µM KNK437. (b) Thermotolerance ratios. ( ) Polβ+/+ MEF. (
) Polβ+/+ MEF. ( ) Polβ−/– MEF.
) Polβ−/– MEF.
Heat treatment increases γH2AX induction in Polβ−/– MEF and KNK437‐treated cells with preheating. To clarify the effects of Polβ and HSP on the phosphorylation of histone H2AX, flow cytometric analysis was performed on Polβ+/+ and Polβ−/– MEF exposed to KNK437 (Fig. 4). The relative γH2AX intensity was increased in Polβ−/– MEF when compared with parental MEF, and was also increased in the presence of KNK437 in both cell lines. In Polβ+/+ cells, γH2AX frequency almost doubled, while in Polβ−/– cells the γH2AX frequency also increased significantly.
Figure 4.
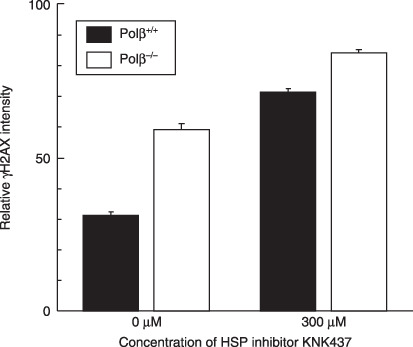
Relative γH2AX intensity. Polymerase‐β (Polβ)+/+ and Polβ−/– mouse embryonic fibroblasts (MEF) were heated (45.5°C, 20 min) at 12 h after a preheating exposure (45.5°C, 5 min) with or without KNK437. () Polβ+/+ MEF. () Polβ−/– MEF.
Discussion
Polβ is a key enzyme involved in the protection of the genome from DNA damage through its role in base excision repair (BER), and in mammalian cells most BER synthesis is carried out by Polβ.( 18 , 19 ) Polβ not only functions as a DNA polymerase, but also catalyzes the excision of deoxyribose phosphate.( 20 ) A relationship has been reported between thermotolerance against heat killing and the heat‐induced loss of Polβ activity in cells.( 15 ) It has also been shown that the number of cellular γH2AX foci was reduced in preheated cells that were subsequently treated with a challenging heat exposure.( 14 ) In the present study, thermotolerance was shown to be suppressed in Polβ‐deficient cells as compared with the parental cells. Therefore, it was strongly suggested that Polβ has an important role in thermotolerance.
HSP interact with Polβ. HSP have been implicated in the induction of radiation resistance via the adaptive response.( 21 ) Moreover, recent studies support an important role for BER in radiosensitivity.( 22 ) Hsp70 associates with Polβ and stimulates this activity.( 23 ) In addition, uracil DNA glycosylase and apurinic–apyrimidinic endonuclease (APE) have been associated with Hsp27 and Hsp70,( 24 ) lending support for the idea that Hsp27 and Hsp70 have a role in the BER of DNA damage. The widespread conservation of HSP in nature may be the result of its selection, because it can protect the genomes of cells from oxidation and radiation damage through the stimulation of DNA repair enzymes. KNK437 inhibits the acquisition of thermotolerance in a dose‐dependent manner, and the induction of various other HSP (including those with approximate molecular weights of 25, 40, 70, 90 and 110 kDa).( 10 , 17 ) Hsp70.1 and Hsp70.3, which are stress‐induced HSP, have an essential role in maintaining genomic stability under stress conditions.( 25 ) Although the authors were not able to get a remarkable result about Hsp40 and Hsp90 (data not shown), Hsp27 and Hsp70 were found to accumulate and bind with Polβ in the nucleus during the period of thermotolerance development. Furthermore, thermotolerance was suppressed in cells treated with the HSP inhibitor KNK437. These results indicate that Hsp27 and Hsp70, which accumulate in response to preheating treatments, are able to: (i) reactivate heat‐denatured Polβ via chaperone activity; and (ii) stimulate Polβ activity along with its role in the repair of DNA damage during the period when thermotolerance is being established.
Involvement of Polβ in heat‐induced γH2AX. An immunocytochemical assay that recognizes γH2AX foci is an extremely sensitive indicator for the existence of a DSB that is induced by heat.( 14 ) There is an inflection point at 42.5°C in the Arrhenius plot of cell killing and Polβ inactivation.( 26 ) The activation enthalpy for cell killing is also similar to that of protein denaturation,( 3 ) such as for Polβ.( 27 ) There is an inflection point at 42.5°C in the Arrhenius plot of cell killing and γH2AX foci formation, and the thermal activation energies of both cell killing and foci formation are almost the same above and below this inflection point.( 14 ) Heat stress activates ATM,( 28 ) and heat‐induced H2AX phosphorylation is mediated by ATM and DNA‐PK.( 29 ) It was proposed that the inhibition of BER of base damage is induced by heat stress through the production of reactive oxygen species,( 30 ) and leads to an increase in the number of existing DSB. Recently, reports have shown that the DSB repair component NBS1 is phosphorylated and involved in cellular responses to DNA damage that are induced by heat exposure.( 31 ) In the present study, an increased level of heat‐induced γH2AX after a preheating treatment was detected in Polβ‐deficient cells when compared with Polβ‐proficient cells, and was also detected in the presence of KNK437 in both cells. In other words, heat‐induced γH2AX was suppressed in thermotolerant cells. These findings support the concept that heat‐induced DSB contribute to heat‐induced cell killing, and are a part of the process leading to heat‐induced DSB formation.
A model for involvement of Polβ in heat‐induced DSB. Polβ is more sensitive to heat than are incision enzymes such as APE. This theoretically provides a mechanism that could account for the increased numbers of DNA breaks observed in heat‐treated cells. Therefore, it appears that there is a possible mechanism that can explain how heat induces nick formation through enzymatic repair processes. DSB could then be generated where nicks form in close proximity to each other on opposite DNA strands (Fig. 5). In fact, the inhibition of poly(ADP‐ribose) polymerase (PARP), which is involved with BER and single‐strand break (SSB) repair, induces γH2AX foci,( 32 ) providing support for the above hypothesis. Although Polβ, XRCC1, PARP‐1, and DNA ligase III are considered to contribute predominantly to BER and SSB repair,( 33 , 34 , 35 , 36 , 37 ) DNA ligase III and PARP‐1 have been reported to be a candidate component of backup pathways for non‐homologous end joining,( 38 , 39 , 40 ) and presumably Polβ and XRCC1 could also participate in this pathway. If so, then it might be a possibility that elevated temperatures could also produce DSB by inactivating these alternative components of repair. The present results suggest the involvement of Polβ in conjunction with HSP (Hsp27 and Hsp70) in the protecting cells from elevated temperatures, but a specific contribution to heat‐induced DSB has not been completely defined.
Figure 5.
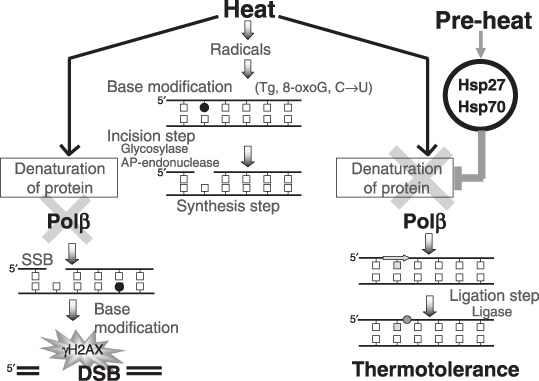
A model for involvement of polymerase‐β (Polβ) in heat‐induced double‐strand breaks (DSB). Heat induces base modifications through free radical species. Polβ are heat sensitive compared with incision enzymes for excision repair. Heat indirectly induces nicks through inhibition of base excision repair. DSB appears when nicks form in close proximity to each other on both strands through a cell cycle, and a nick is converted into DSB at a DNA replication fork during the S‐phase. When cells are preconditioned, Polβ is protected or reactivated through the interaction with heat shock protein (HSP) and fewer DSB are generated.
Together, this data demonstrates clear in vivo relevance for the interaction between Polβ and HSP (Hsp27 and Hsp70) and has important biological implications for the understanding of cellular responses to elevated temperatures. Although HSP can rescue or restore many other heat‐labile proteins besides Polβ, these findings suggest that Polβ contributes to thermotolerance through reactivation and stimulation from HSP, and leads to fewer DSB forming. These observations provide support for the concept that heat‐induced DSB contribute to heat‐induced cell killing. Further investigations are still required to address the exact mechanism leading to heat‐induced DSB formation. Such studies could contribute to new concepts and further understanding of hyperthermic biology and oncology.
Acknowledgments
This work was supported in part by grants from the Ministry of Education, Science, Sports, Culture and Technology of Japan.
References
- 1. Ohnishi K, Ohnishi T. Heat‐induced p53‐dependent signal transduction and its role in hyperthermic cancer therapy. Int J Hyperthermia 2001; 17: 415–27. [DOI] [PubMed] [Google Scholar]
- 2. Kampinga HH, Dikomey E. Hyperthermic radiosensitization: mode of action and clinical relevance. Int J Radiat Biol 2001; 77: 399–408. [DOI] [PubMed] [Google Scholar]
- 3. Dewey WC. Arrhenius relationships from the molecule and cell to the clinic. Int J Hyperthermia 1994; 10: 457–83. [DOI] [PubMed] [Google Scholar]
- 4. Nakahata K, Miyakoda M, Suzuki K, Kodama S, Watanabe M. Heat shock induces centrosomal dysfunction, and causes non‐apoptotic mitotic catastrophe in human tumour cells. Int J Hyperthermia 2002; 18: 332–43. [DOI] [PubMed] [Google Scholar]
- 5. Roti Roti JL, Kampinga HH, Malyapa RS, Wright WD, Van Der Waal RP, Xu M. Nuclear matrix as a target for hyperthermic killing of cancer cells. Cell Stress Chaperones 1998; 3: 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li GC, Mivechi NF, Weitzel G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int J Hyperthermia 1995; 11: 459–88. [DOI] [PubMed] [Google Scholar]
- 7. Takayama S, Reed JC, Homma S. Heat‐shock proteins as regulators of apoptosis. Oncogene 2003; 22: 9041–7. [DOI] [PubMed] [Google Scholar]
- 8. Li GC, Werb Z. Correlation between synthesis of heat shock proteins and development of thermotolerance in Chinese hamster fibroblasts. Proc Natl Acad Sci USA 1982; 79: 3218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crête P, Landry J. Induction of HSP27 phosphorylation and thermoresistance in Chinese hamster cells by arsenite, cycloheximide, A23187, and EGTA. Radiat Res 1990; 121: 320–7. [PubMed] [Google Scholar]
- 10. Ohnishi K, Takahashi A, Yokota S, Ohnishi T. Effects of a heat shock protein inhibitor KNK437 on heat sensitivity and heat tolerance in human squamous cell carcinoma cell lines differing in p53 status. Int J Radiat Biol 2004; 80: 607–14. [DOI] [PubMed] [Google Scholar]
- 11. Ehrnsperger M, Graber S, Gaestel M, Buchner J. Binding of non‐native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 1997; 16: 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hartl FU, Hayer‐Hartl M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002; 295: 1852–8. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi A, Ohnishi T. Does γH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett 2005; 229: 171–9. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi A, Matsumoto H, Nagayama K et al . Evidence for the involvement of double‐strand breaks in heat‐induced cell killing. Cancer Res 2004; 64: 8839–45. [DOI] [PubMed] [Google Scholar]
- 15. Dewey WC, Esch JL. Transient thermal tolerance: cell killing and polymerase activities. Radiat Res 1982; 92: 611–14. [PubMed] [Google Scholar]
- 16. Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase β. EMBO J 2000; 19: 1397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yokota S, Kitahara M, Nagata K. Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res 2000; 60: 2942–8. [PubMed] [Google Scholar]
- 18. Sobol RW, Horton JK, Kuhn R et al . Requirement of mammalian DNA polymerase‐β in base‐excision repair. Nature 1996; 379: 183–6. [DOI] [PubMed] [Google Scholar]
- 19. Sobol RW, Prasad R, Evenski A et al . The lyase activity of the DNA repair protein β‐polymerase protects from DNA‐damage‐induced cytotoxicity. Nature 2000; 405: 807–10. [DOI] [PubMed] [Google Scholar]
- 20. Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science 1995; 269: 699–702. [DOI] [PubMed] [Google Scholar]
- 21. Lee YJ, Park GH, Cho HN et al . Induction of adaptive response by low‐dose radiation in RIF cells transfected with Hspb1 (Hsp25) or inducible Hspa (Hsp70). Radiat Res 2002; 157: 371–7. [DOI] [PubMed] [Google Scholar]
- 22. Vens C, Dahmen‐Mooren E, Verwijs‐Janssen M et al . The role of DNA polymerase β in determining sensitivity to ionizing radiation in human tumor cells. Nucleic Acids Res 2002; 30: 2995–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mendez F, Kozin E, Bases R. Heat shock protein 70 stimulation of the deoxyribonucleic acid base excision repair enzyme polymerase β. Cell Stress Chaperones 2003; 8: 153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mendez F, Sandigursky M, Franklin WA, Kenny MK, Kureekattil R, Bases R. Heat‐shock proteins associated with base excision repair enzymes in HeLa cells. Radiat Res 2000; 153: 186–95. [DOI] [PubMed] [Google Scholar]
- 25. Hunt CR, Dix DJ, Sharma GG et al . Genomic instability and enhanced radiosensitivity in Hsp70.1‐ and Hsp70.3‐deficient mice. Mol Cell Biol 2004; 24: 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spiro IJ, Denman DL, Dewey WC. Effect of hyperthermia on isolated DNA polymerase‐β. Radiat Res 1983; 95: 68–77. [PubMed] [Google Scholar]
- 27. Spiro IJ, Denman DL, Dewey WC. Effect of hyperthermia on CHO DNA polymerases α and β. Radiat Res 1982; 89: 134–49. [PubMed] [Google Scholar]
- 28. Miyakoda M, Suzuki K, Kodama S, Watanabe M. Activation of ATM and phosphorylation of p53 by heat shock. Oncogene 2002; 21: 1090–6. [DOI] [PubMed] [Google Scholar]
- 29. Kaneko H, Igarashi K, Kataoka K, Miura M. Heat shock induces phosphorylation of histone H2AX in mammalian cells. Biochem Biophys Res Commun 2005; 328: 1101–6. [DOI] [PubMed] [Google Scholar]
- 30. Bruskov VI, Malakhova LV, Masalimov ZK, Chernikov AV. Heat‐induced formation of reactive oxygen species and 8‐oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res 2002; 30: 1354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohnishi K, Scuric Z, Yau D et al . Heat‐induced phosphorylation of NBS1 in human skin fibroblast cells. J Cell Biochem 2006; 99: 1642–50. [DOI] [PubMed] [Google Scholar]
- 32. Bryant HE, Schultz N, Thomas HD et al . Specific killing of BRCA2‐deficient tumours with inhibitors of poly (ADP‐ribose) polymerase. Nature 2005; 434: 913–17. [DOI] [PubMed] [Google Scholar]
- 33. Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol 1994; 14: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caldecott KW, Tucker JD, Stanker LH, Thompson LH. Characterization of the XRCC1‐DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res 1995; 23: 4836–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, Frosina G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J Biol Chem 1997; 272: 23 970–5. [DOI] [PubMed] [Google Scholar]
- 36. Mackey ZB, Niedergang C, Murcia JM et al . DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly (ADP‐ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J Biol Chem 1999; 274: 21 679–87. [DOI] [PubMed] [Google Scholar]
- 37. Wong HK, Wilson DM 3rd. XRCC1 and DNA polymerase β interaction contributes to cellular alkylating‐agent resistance and single‐strand break repair. J Cell Biochem 2005; 95: 794–804. [DOI] [PubMed] [Google Scholar]
- 38. Audebert M, Salles B, Calsou P. Involvement of poly (ADP‐ribose) polymerase‐1 and XRCC1/DNA ligase III in an alternative route for DNA double‐strand breaks rejoining. J Biol Chem 2004; 279: 55 117–26. [DOI] [PubMed] [Google Scholar]
- 39. Audebert M, Salles B, Weinfeld M, Calsou P. Involvement of polynucleotide kinase in a poly (ADP‐ribose) polymerase‐1‐dependent DNA double‐strand breaks rejoining pathway. J Mol Biol 2006; 356: 257–65. [DOI] [PubMed] [Google Scholar]
- 40. Wang H, Rosidi B, Perrault R et al . DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res 2005; 65: 4020–30. [DOI] [PubMed] [Google Scholar]