

Abstract
Recent developments in isolation and characterization of tumor stem cells (TSCs) have opened new possibilities for developing TSC‐targeted therapies. Extensive efforts have been made to ascertain markers of TSCs, including cell surface, enzymatic, gene expression profile, and functional markers. These markers and the technologies used to identify and isolate TSCs are discussed in this review. TSC characteristics, such as quiescence, multidrug resistance, enhanced DNA repair ability, and anti‐apoptotic mechanisms, and various features of the in vivo niche, which may make them resistant to conventional therapy, are also discussed here. The increasing understanding of aberrantly expressed molecules and signaling pathways in TSCs may provide the foundation for design of therapeutic strategies for TSC ablation. (Cancer Sci 2009)
The idea of targeting TSCs with chemotherapy first became prominent in the 1970s with the introduction of the ‘human tumor stem cell assay’ by Anne Hamburger and Sydney Salmon.( 1 ) Although the initial orientation of this assay was for customization of therapy to individual patients’ TSCs, it was subsequently extended to use in drug discovery. A multicenter NCI contract drug screening effort( 2 ) concluded that the technology was not suitable for large‐scale drug screening, but did identify two novel agents, which were evaluated in clinical trials. One of the compounds identified in new drug screening with the human tumor stem cell assay, chloroquinoxaline sulfonamide (NSC 339004), showed some evidence of activity on the first schedule used in Phase I trials, but none on a second schedule. Some closely related sulphonamide compounds, whose mechanisms have not been fully elucidated either, were subsequently identified by Eisai Pharmaceuticals (E7974 and E7820) and are currently in development. Approximately 30 years after the Hamburger and Salmon report, interest in TSCs has once again become prominent in published cancer research (Fig. 1). This has grown out of the increasing understanding of normal human embryonic stem cell biology and work documenting the existence of human leukemic stem cells bearing characteristic cell surface markers.( 3 ) Numerous investigators have probed various solid tumor types and cell lines for the presence of TSCs using cell surface and other putative TSC markers. Among the markers that have been studied, expression of ABC transporters and ALDH have been considered to confer resistance to chemotherapy with transporter substrates and cyclophosphamide, respectively. Sequestration of TSCs in particular anatomic niches might further contribute to therapeutic resistance in vivo and in the clinic. However, expression of stem cell signaling pathways and cell surface markers might provide opportunities for targeted therapy. Interest in targeting TSCs has clearly been based on the notion that these cells resist currently available therapies and contribute to the re‐growth of tumors following chemotherapy. In the case of solid tumors, experimental proof of this rather compelling hypothesis has been difficult to approach. Very recently, it has been shown that elimination of LIC in promyelocytic leukemia by retinoic acid and/or arsenic trioxide treatment, rather than induction of differentiation in the bulk of the leukemic cell population, is key to curative treatment.( 4 ) Mechanistic studies in this case have been facilitated by insight into the role of specific chromosomal translocations and resulting fusion proteins in the pathogenesis of the disease. Other malignancies may present substantially different biology and therapeutic challenges. Recently, it has been shown that human melanoma might contain a very high percentage of cells with tumor initiating ability.( 5 ) This observation was made possible by the use of xenotransplantation models with enhanced sensitivity for detection of tumor initiating cells (NOD/SCID mice deficient for IL‐2 gamma receptors and the use of Matrigel for support of engrafted cells). Regardless of the absolute fraction of solid tumor cells constituted by TSCs, development of therapeutic approaches capable of eliminating these cells would seem to be an important goal. In this review, we discuss means of identifying and isolating putative TSCs and potential approaches to therapeutic targeting.
Figure 1.
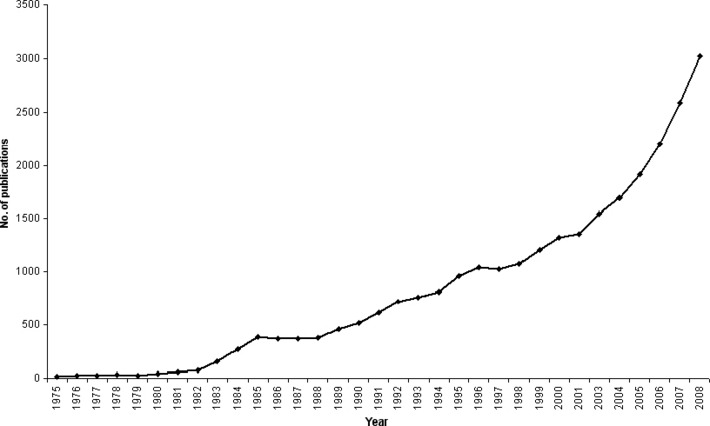
Tumor stem cell (TSC) published reports, 1975–2008. The search term ‘tumor stem cell’ was fed into ISI Web of Knowledge (http://isiwebofknowledge.com/). It generated approximately 27 000 hits for publications from 1959 to 2008. The number of publications ranged between 1 and 6 from 1959 to 1974 (data not shown), then it started picking up in 1975 with 20 publications. This number spiked to 388 in 1985 and maintained a plateau until 1988. Subsequently, the number of publications has increased steadily with more than 3000 publications in 2008. This increase in publications corresponds to an increase in the interest in TSC biology and the realization of the importance of TSCs for achieving successful tumor treatment.
Identification and Isolation of TSCs
Tumor stem cells were first isolated as clones based on the soft agar cloning technique. However, this technique is time and labor intensive and only a small fraction of primary tumors yield adequate numbers of colonies.( 2 ) An alternative is to culture TSCs as floating spheres. Historically, bulk tumor cells have been cultured as spheroids since the 1970s. Mouse EMT6/Ro( 6 ) and various other cells were cultured as structured spheroids in the presence of serum. More recently, relatively undifferentiated TSC sphere cultures have been derived from primary tumors and cell lines using serum‐free medium that is supplemented with growth factors, such as epidermal growth factor and basic fibroblast growth factor. Serum replacement results in the morphological change of adherent monolayer cultures to TSC‐enriched floating spheres, such as neurospheres (Fig. 2).( 7 )
Figure 2.
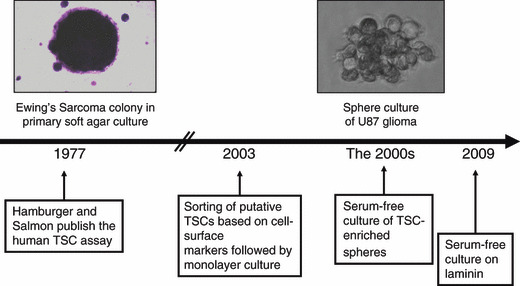
Evolution of cell culture technologies relevant to tumor stem cell (TSC) culture. To understand TSC biology, it is important to identify and isolate these cells from the bulk population. Initially, TSCs were isolated as colonies on soft agar (1971).( 1 ) This labor‐intensive method remained as the state‐of‐the‐art for decades. In 2003, breast TSCs were isolated using a complex cell surface marker signature and validated based on the functional tumorigenicity ability of TSCs.( 15 ) In the 2000s, TSC‐enriched spheres have been grown, such as neurospheres, under serum‐free conditions.( 7 ) These spheres contain multiple cell types, including dead cells, differentiated cells, and TSCs. To obtain a highly TSC‐enriched more homogeneous culture, these have recently been grown as monolayers on a modified surface.( 8 )
Tumor stem cell culture as floating spheres presents certain problems: (i) as the sphere size increases, it restricts the core cells’ access to nutrients; and (ii) in addition to TSCs, spheres also contain dead and differentiated cells. To overcome these limitations, recently, glioma TSCs have been adapted to grow as adherent cells using a laminin‐coated culture surface.( 8 )
Cell surface markers. Cell surface proteins, such as CD24, CD44, CD133, and integrins, to name a few, have been used as TSC markers. CD133 is discussed briefly as it will be used several times to highlight various themes in TSC research.
CD133 is a transmembrane glycoprotein that has been reported as a TSC marker for breast, lung, liver, colon, prostate, and brain tumors.( 9 , 10 , 11 , 12 ) Although CD133 expression might point to a possible TSC, it is not a reliable marker due to the technical limitations of its detection and due to inherent TSC heterogeneity resulting in cellular subsets within a TSC population. CD133 expressing cells have been identified and isolated using the monoclonal antibodies AC133 and AC141. However, these antibodies recognize poorly characterized epitopes. Furthermore, CD133 expression might be regulated by biological variables, such as oxygen concentration.( 13 ) TSC heterogeneity within a particular tumor appears to hinder the use of a single marker for reliable TSC identification. TSCs have been isolated from gliomas based on the expression of CD133 and A2B5 (a glial progenitor marker found to be expressed in human gliomas). When these TSCs were tested in nude mice for their tumorigenicity potential, it was observed that TSCs that were either A2B5+CD133+, or A2B5+CD133−, or A2B5−CD133− were able to form tumors.( 14 ) This indicates that multiple TSC types exist within gliomas. TSCs have recently been shown to vary from patient to patient.( 8 ) Thus, cell surface markers might be useful in the identification of potential TSCs, but additional markers need to be considered when defining a specific TSC type.
A combination of various cell surface proteins is increasingly used as a complex TSC signature. Breast TSCs have been identified based on the cell surface marker signature CD44+CD24−/low coupled to functional tumorigenicity capability.( 15 ) For a list of these complex signatures, please refer to another review.( 16 )
Enzymatic markers. Aldehyde dehydrogenase has been reported as a TSC marker. ALDHs catalyze the oxidation of a wide variety of aldehydes to carboxylic acids, and are known to play an important role in endobiotic and xenobiotic metabolism. Accordingly, ALDHs have been known to provide resistance to hematopoietic stem cells against alkylating agents of the oxazaphosphorines family, such as cyclophosphamide and its derivatives. Based on the broad utility of ALDHs, methods to detect ALDH activity have been commercially developed, such as the Aldefluor assay (Stem Cell Technologies, Vancouver, Canada), and have been used to sort cells with variant ALDH activity. ALDH isozymes have been reported as TSC markers in pancreatic cancer, breast cancer, prostate cancer, lung cancer, multiple myeloma, and leukemia.( 10 , 17 , 18 ) Although ALDH activity may indicate potential TSCs, there are drawbacks to its utility as a marker. For example, although putative TSCs in lung cancer can be sorted based on high ALDH activity,( 18 ) it has been reported that smoking can also elevate ALDH levels. This may complicate the identification of TSCs and may also result in false positives.
Gene expression profiles. Cell surface and enzymatic molecules offer practically useful TSCs markers. However, they do not provide a comprehensive view of the molecular pathways operating in TSCs. To gather this information, genetic profiling has been used. Human prostate TSCs were isolated based on the cell surface signature CD133+/α2β1 hi. Subsequently, these were subjected to expression analysis and compared to their normal and differentiated (CD133−/α2β1 low) counterparts. The gene expression profile of these TSCs provided a signature consisting of significantly variable expression of 581 genes. This variable gene expression was used for functional annotation of the signature. This pointed to various pathways involved in TSC biology: JAK‐STAT signaling; cell adhesion and extracellular matrix interactions, focal adhesion signaling, and Wnt signaling. These are some of the pathways that could be targeted to deplete TSCs (Table 2).( 19 )
Table 2.
Molecular targets in tumor stem cells and prototype targeted agents
| Targets in tumor stem cells | Therapeutic molecules |
| Multidrug resistance | Thiosemicarbazone derivative (NSC 73306) |
| Telomerase | RHPS4, BIBR 1532, BIBR 1591 |
| PML‐RARA | Retinoic acid and arsenic trioxide |
| Wnt/β‐catenin pathway | Planomycin, sulindac sulfide |
| Hedgehog pathway | Cyclopamine, purmorphamine |
| Notch pathway | γ‐Secretase inhibitors (LY‐411575, DAPT) |
BIBR 1532, {2‐[(E)‐3‐naphtalen‐2‐yl‐but‐2‐enoylamino]‐benzoic acid}; BIBR 1591, {5‐morpholin‐4‐yl‐2‐[(E)‐3‐naphtalen‐2‐yl‐but‐2‐enoylamino]‐benzoic acid}; DAPT, N‐[N‐(3,5‐difluorophenacetyl)‐l‐alanyl]‐S‐phenylglycine t‐butyl ester; LY‐411575, N2‐[(2S)‐2‐(3,5‐difluorophenyl)‐2‐hydroxyethanoyl]‐N1‐[(7S)‐5‐methyl‐6‐oxo‐6,7‐dihydro‐5H‐dibenzo(b,d)azepin‐7‐yl]‐l‐alaninamide; PML‐RARA, promyelocytic leukemia–retinoic acid receptor‐α fusion; RHPS4, 3,11‐difluoro‐6,8,13‐trimethyl‐8H‐quino[4,3,2‐kl] acridinium methosulfate.
Functional markers. Although cell surface molecules, enzymes, and genetic profiles have been used to identify potential TSCs, a more reliable proof may be based on functional TSC markers. These include in vitro (proliferation, colony‐forming capability, adhesion, migration, and invasion) and in vivo (tumorigenicity, metastatic capacity, and ability to recapitulate the morphological features of a specific tumor).( 7 , 17 )
In vivo tumorigenicity of TSCs is often assessed in NOD/SCID mice. Based on this assay, the percentage of TSCs has been estimated to be between 0.1% and 0.0001%. Recently, it has been reported that these earlier studies may have underestimated the TSC percentage. The use of a more highly immunocompromised variant of NOD/SCID mice and Matrigel for tumor implantation indicated one in four tumor initiating cells in malignant melanoma. Thus, altering the criteria for analyzing tumorigenicity can result in profoundly different estimates of TSC tumorigenic ability.( 5 ) However, the existence of a higher TSC population might be a tumor specific feature, that is, melanoma is known to be a highly aggressive tumor, and thus, might have a higher percentage of TSCs.
There are many properties of TSCs that can be used as markers for identification, but more research is required to understand the relationships among these markers. For example, when CD133 expression was used to isolate melanoma TSCs, the frequency of CD133+ cells was observed to be lower than the frequency of tumorigenic cells in melanoma patients. Also, CD133+ cells did not demonstrate enrichment for tumorigenic melanoma cells. Moreover, both CD133+ and CD133− melanoma cells were observed to contain a high frequency of tumorigenic cells.( 5 ) Thus, different markers point to different facets of TSCs, and there is a discrepancy in the total number of TSCs measured using these various markers. Therefore, it is important to consider combinations of markers to define TSCs, which after further research could aid in more reliable TSC identification and isolation.
Addressing TSC Heterogeneity
To isolate TSCs, they need to be identified. Various markers, discussed above, point towards the complexity of such an endeavor. It is apparent that no single marker can be used for TSC identification across the spectrum of tumors. Moreover, there are discrepancies among various markers for validation of TSCs. This results from heterogeneity associated with tumor biology at various levels: different tumors may express different molecules; different patients suffering from the same tumor type may have variant expression patterns because of variant genetic, epigenetic, and environmental factors; and in a single patient, distinct populations of tumor cells may co‐exist in primary and metastatic tumors, including a heterogeneous population of TSCs.
Currently, most researchers are studying TSCs in a single tumor type using only a few established cancer cell lines. Screening models, like the NCI‐60,( 20 ) could be used to address this issue. The NCI‐60 contains multiple cells lines representing central nervous system, pulmonary, colon, breast, ovarian, and prostate tumors, leukemia, and melanoma. To help put the tumor and TSC heterogeneity in perspective, we have analyzed the expression of putative TSC markers (CD15, CD24, CD44, CD133, CD166, CD326, ABCB1, and ALDH) in the NCI‐60 panel.( 20 ) As expected, the expression pattern for individual markers varied widely across the panel. However, multidimensional analysis showed that marker co‐expression patterns correlated with tumor types (C.H. Stuelten, unpubl. data, 2009). This study was carried out in tumor cell lines cultured in the presence of serum. Various methods, discussed above, have been developed for TSC‐enriched culture of tumor cell lines. We are in the process of establishing an NCI‐60 screen of TSC‐enriched cell populations. Data generated in this project may help in identifying tumor specific and general TSC trends and result in a collection of TSCs useful for new drug screening.
Drug Resistance in TSCs
After initial response to chemotherapy, relapse is often observed. This has been attributed to the existence of drug‐resistant TSCs in a variety of tumors, such as breast tumor and osteosarcoma.( 21 , 22 ) Various mechanisms known to confer longer lifespan to stem cells have been speculated to contribute to drug resistance in TSCs. These mechanisms might involve relative quiescence, expression of multidrug‐resistance proteins (discussed in another section), robust DNA repair capability, and effective strategies to avoid apoptosis.( 23 , 24 )
Quiescence. The TSC niche has been envisioned as a modulator of TSC quiescence. It has been suggested that there might be two types of TSC niches: one that maintains quiescence and another that maintains proliferative cells.( 25 ) Tumors are known to grow in hypoxic environments. Hypoxia induces production of HIF1α and HIF2α. These HIFs can exert opposite effects on proliferation.( 26 ) For example, HIF1α inhibits c‐myc and mTOR and activates p53; thus, decreases proliferation. HIF2α activates c‐myc and mTOR and inhibits p53; thus, increases proliferation.( 27 ) Therefore, a regulated balance between HIF1α and HIF2α, among other molecules, could be expected to regulate TSC quiescence and proliferation.
Further understanding of the molecular regulation of quiescence has been gained from gene expression profiling of TSCs, enriched based on higher β1‐integrin expression in squamous cell carcinoma.( 28 ) Among various markers that were examined, the ones that correlated with diminished differentiation status and increased proliferation were: leucine‐rich repeats and immunoglobulin‐like domains 1 (Lrig1) and microtubule‐associated protein 4 (MAP4) (downregulated), and melanoma chondroitin sulphate proteoglycan (MCSP) (upregulated).
Lrig1 negatively regulates epidermal growth factor receptor signaling, thus it maintains epidermal stem cells in a quiescent state.( 28 , 29 , 30 , 31 ) Its downregulation would perturb the quiescence and aid in proliferation of TSCs. However, although it was observed to be downregulated in unfractionated squamous cell carcinoma lines and primary tumors, its levels were comparable in TSC‐enriched fractions and normal epidermal and oral stem cells.( 28 ) This complicated scenario might explain quiescence observed in TSCs. There is a fraction of TSCs that remains quiescent to maintain its viability when non‐quiescent and proliferating TSCs are susceptible to therapy.( 28 )
MAP4 has been reported to be involved in the regulation of cell cycle progression and cytokinesis.( 28 , 32 ) Thus, although requiring further proof, it has been suggested that downregulation of MAP4 could enhance cell cycle progression of TSCs.( 28 )
MCSP has been known to stimulate integrin‐mediated adhesion and spreading of tumor cells by activating the small GTPases CDC42 and Rac1.( 28 , 33 , 34 ) Thus, upregulation of MCSP would favor metastatic potential of TSCs.( 28 )
Various aspects governing the TSC niche and new therapeutic strategies against the quiescent cells in the niche have recently been reviewed.( 35 ) It is difficult to recapitulate the various factors operating in the in vivo TSC niche in an in vitro culture system (Table 1). However, it is important to decipher the molecular cues that govern quiescence, proliferation, and metastasis as these may modulate TSC longevity and tumor relapse.
Table 1.
In vivo tumor stem cell (TSC) niche factors relevant to therapy†
| Factors in TSC niche | Role/effect |
|---|---|
| Vascular factors | Nutritional and oxygen gradients with effects on survival and death pathways, the targeted drug delivery system has to navigate through the bloodstream and cross the vasculature surrounding the TSC niche |
| Stromal cell interactions | Cytokine and growth factor effects on potential cellular targets |
| Extracellular matrix | Barrier functions, effects on cells mediated by receptors |
| TSC signaling | Molecular pathways modulated in response to the microenvironment |
†Cell culture models can be engineered to address some of these by inclusion of growth factors, extracellular matrix, and oxygen gradients.
Enhanced DNA repair. DNA damage can be fatal for rapidly dividing tumor cells. In this regard, alkylating agents, such as temozolomide and carmustine, have been used to induce DNA damage for glioma chemotherapy. However, TSCs that initiate and sustain tumors might have enhanced DNA repair mechanisms, which can resolve the alkylation damage to DNA.( 36 ) Furthermore, TSCs might have a higher tolerance limit for mutations due to defects in apoptosis machinery.
Anti‐apoptotic characteristics. Apoptosis can effectively remove damaged or potentially harmful cells by the extrinsic and intrinsic pathways. As noted above, CD44 has been used as a TSC marker. CD44 perturbs the Fas‐based extrinsic apoptotic pathway, thus aiding in longer TSC life span. Also, TSCs have been observed to express higher levels of anti‐apoptotic genes, such as FLIP, BCL‐2, BCL‐XL, and IAP family members (XIAP, cIAP1, and survivin).( 37 , 38 )
Taken together, experimental evidence suggests that TSCs have evolved strategies to evade death mechanisms. The understanding of the underlying molecular pathways may provide potential therapeutic opportunities to achieve targeted TSC ablation.
Therapies against TSCs
To achieve tumor eradication, it is thought to be essential to target TSCs. A variety of molecules and pathways operating in TSCs could potentially be targeted with therapeutic molecules (Table 2).
Tumor stem cells may display multidrug resistance that is conferred by ABC transporters.( 39 ) These ABC transporters have been reported as TSC markers in melanoma and osteosarcoma, among others.( 40 , 41 ) Targeted inactivation of ABC transporters could reinstate the drug sensitivity in TSCs resulting in TSC killing. For example, ABCB5 has been reported as a marker for a subset of CD133+ melanoma stem cells. ABCB5 provides resistance to doxorubicin by functioning as an efflux pump. When it was blocked by anti‐ABCB5 monoclonal antibody, doxorubicin sensitivity was restored in these TSCs (Fig. 3).( 42 ) TSC heterogeneity, discussed above, is likely to limit the effectiveness of individual maneuvers of this type.
Figure 3.
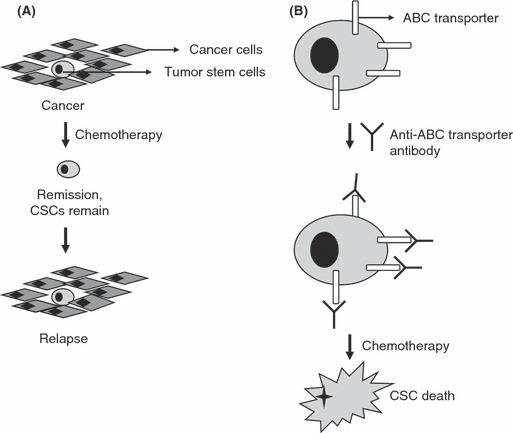
Tumor stem cell (TSC) chemotherapy. (A) A tumor contains tumor cells and TSCs. Conventional therapy, such as chemotherapy, removes tumor cells, but is ineffective against TSCs. After remission, the remaining TSCs re‐form the tumor, resulting in relapse. (B) TSCs are resistant to chemotherapy because they express drug efflux proteins, such as ATP binding cassette (ABC) transporters. Recently, these ABC transporters have been inactivated using anti‐ABC transporter antibodies. Once the transporter has been inactivated, TSCs cannot remove the chemotherapeutic drug and can be killed.
Tumor cells have been observed to reactivate telomerase to achieve immortality.( 43 ) Increased telomerase activity has been reported in approximately 90% of malignant tumors.( 44 ) Sustained telomerase function is necessary for TSCs that have the potential to proliferate indefinitely. Thus, anti‐telomerase agents are expected to target tumor cells as well as TSCs.
Aberrant proteins expressed in TSCs can be targeted to achieve TSC killing. LIC with promyelocytic leukemia–retinoic acid receptor‐α fusion in acute promyelocytic leukemia were selectively depleted by a combination of retinoic acid and arsenic trioxide. These chemicals triggered the catabolism of promyelocytic leukemia–retinoic acid receptor‐α fusion oncoprotein, resulting in LIC eradication.( 4 )
Tumor stem cells can be targeted based on their stem cell‐like properties. For example, breast cancer cells were modified to underexpress E‐cadherin that caused epithelial to mesenchymal transition and resulted in enrichment for TSCs. These TSC‐enriched populations were used for high throughput screening, which identified salinomycin as a targeting agent for cells that have undergone epithelial to mesenchymal transition.( 45 )
The increasing knowledge about molecular pathways governing various aspects of TSCs could be exploited to tailor drug‐based therapies for TSC ablation. A variety of signaling pathways, previously reported to be differentially regulated in tumor cells, have been observed to be aberrantly modulated in TSCs. These include Wnt/β‐catenin, hedgehog, notch, JAK/STAT, PTEN/PI3K/Akt, and TGF‐ β pathways.( 19 , 46 , 47 ) These pathways in TSCs may be targeted with agents, many of which have been advanced to translational applications against tumor cells (Table 2). Thus, there is a possibility of using drugs already available for TSC eradication.
Future Perspectives
Tumor stem cells are poised to play an important role in the effort to achieve successful tumor ablation. The research to understand the biology of TSCs is progressing rapidly. As this knowledge becomes more concrete, it will pave the way for reliable TSC identification and isolation. As discussed above, currently used TSC markers suffer from the limitation of non‐specificity. Also, there is a lack of reagents, such as targeting ligands, against these markers. Moreover, TSCs even from a single type of tumor may contain subsets. Therefore, using multiple markers to identify TSCs may be more reliable than using a single type of marker. Complex combinations are already being used to define TSC populations. For example, in the human breast cancer cell line MDA‐MB‐468, TSCs have been identified based on cell surface (CD133+/CD44+), enzymatic (high activity of ALDH), and functional (proliferation, colony formation, adhesion, migration, invasion, tumorigenicity, and metastasis) markers.( 17 ) It may be beneficial to overlap genetic signatures on these markers to further define these TSCs. For this, bioinformatics‐based tools can be used to generate complex fingerprints based on a combination of markers. Although such a scenario holds promise, no clear and general path has emerged for marker‐based TSC characterization.
Once TSC‐specific molecules have been identified, targeted therapies can be developed. However, various factors may complicate TSC targeting. First, TSCs appear to represent heterogeneous populations. Potential sources of this heterogeneity are genetic, epigenetic, and environmental factors. The constitutional genomic features of individual patients (including gene copy number differences and single nucleotide polymorphisms, among others) as well as somatic mutations/genetic instability features contribute to TSC heterogeneity. The differentiated features of tumor stem cells may reflect the epigenetic status of cells from which the tumor arose. In addition, environmental factors, such as those operating in the TSC niche (Table 1) can contribute to heterogeneity. Therefore, it is unlikely that one particular targeting molecule could be used to deliver therapeutic agents to all TSCs within a single tumor. Second, TSCs reside in niches, and the targeting agent will have to traverse the bloodstream and penetrate through cells and tissues surrounding the niche. Finally, the biology of TSCs is still being elucidated. In the TSC niche, TSCs have complicated interactions with the surrounding stromal cells, which may modulate TSCs at the molecular level. These molecular changes may manifest as epigenetic, genetic, and/or proteomic changes (Table 1). The dynamic TSC changes and adaptations modulated in response to their microenvironment will complicate the efforts to develop a TSC‐targeted therapeutic agent.
Tumor stem cell targeting is essential, but recently it has been speculated that non‐TSCs in a tumor need to be targeted as well. These non‐TSCs could form TSCs and might even sustain the tumor even after TSCs have been destroyed.( 45 )
After resolving the multitude of issues related to TSC culture, identification, isolation, and targeting, better treatment options may be developed. Along with the conventional treatments, new technologies, such as gene therapy and nanotechnology,( 48 , 49 , 50 ) could provide sophisticated multifunctional agents for simultaneous targeting, imaging, and therapy of TSCs.
Abbreviations
- ABC
ATP binding cassette
- ALDH
aldehyde dehydrogenase
- HIF
hypoxia inducible factors
- IAP
inhibitor of apoptosis
- LIC
leukemia initiating cells
- MAP4
microtubule‐associated protein 4
- MCSP
melanoma chondroitin sulphate proteoglycan
- NCI
National Cancer Institute (USA)
- TSC
tumor stem cell
References
- 1. Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science 1977; 197: 461–3. [DOI] [PubMed] [Google Scholar]
- 2. Shoemaker RH, Wolpert‐DeFilippes MK, Kern DH et al. Application of a human tumor colony‐forming assay to new drug screening. Cancer Res 1985; 45: 2145–53. [PubMed] [Google Scholar]
- 3. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–7. [DOI] [PubMed] [Google Scholar]
- 4. Nasr R, Guillemin MC, Ferhi O et al. Eradication of acute promyelocytic leukemia‐initiating cells through PML‐RARA degradation. Nat Med 2008; 14: 1333–42. [DOI] [PubMed] [Google Scholar]
- 5. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 2008; 456: 593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Freyer JP, Sutherland RM. Selective dissociation and characterization of cells from different regions of multicell tumor spheroids. Cancer Res 1980; 40: 3956–65. [PubMed] [Google Scholar]
- 7. Yu SC, Ping YF, Yi L et al. Isolation and characterization of cancer stem cells from a human glioblastoma cell line U87. Cancer Lett 2008; 265 (1): 124–34. [DOI] [PubMed] [Google Scholar]
- 8. Pollard SM, Yoshikawa K, Clarke ID et al. Glioma stem cell lines expanded in adherent culture have tumor‐specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009; 4: 568–80. [DOI] [PubMed] [Google Scholar]
- 9. Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44 + /CD24− and CD133 + cells with cancer stem cell characteristics. Breast Cancer Res 2008; 10 (1): R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boman BM, Wicha MS. Cancer stem cells: a step toward the cure. J Clin Oncol 2008; 26: 2795–9. [DOI] [PubMed] [Google Scholar]
- 11. Vermeulen L, Todaro M, De Sousa Mello F et al. Single‐cell cloning of colon cancer stem cells reveals a multi‐lineage differentiation capacity. Proc Natl Acad Sci USA 2008; 105: 13427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh SK, Clarke ID, Terasaki M et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003; 63: 5821–8. [PubMed] [Google Scholar]
- 13. Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J Mol Med 2008; 86: 1025–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogden AT, Waziri AE, Lochhead RA et al. Identification of A2B5 + CD133‐ tumor‐initiating cells in adult human gliomas. Neurosurgery 2008; 62: 505–14; discussion 14–5. [DOI] [PubMed] [Google Scholar]
- 15. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang YM, Chang JW. Current status and issues in cancer stem cell study. Cancer Invest 2008; 26: 741–55. [DOI] [PubMed] [Google Scholar]
- 17. Croker AK, Goodale D, Chu J et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med 2008. doi: DOI: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ucar D, Cogle CR, Zucali JR et al. Aldehyde dehydrogenase activity as a functional marker for lung cancer. Chem Biol Interact 2009; 178: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Birnie R, Bryce SD, Roome C et al. Gene expression profiling of human prostate cancer stem cells reveals a pro‐inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol 2008; 9: R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006; 6: 813–23. [DOI] [PubMed] [Google Scholar]
- 21. Li X, Lewis MT, Huang J et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100: 672–9. [DOI] [PubMed] [Google Scholar]
- 22. Gillette JM, Nielsen‐Preiss SM. Cancer stem cells: seeds of growth in osteosarcoma. Cancer Biol Ther 2009; 8: 553–554. [DOI] [PubMed] [Google Scholar]
- 23. Kvinlaug BT, Huntly BJ. Targeting cancer stem cells. Expert Opin Ther Targets 2007; 11: 915–27. [DOI] [PubMed] [Google Scholar]
- 24. Raguz S, Yague E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer 2008; 99: 387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sneddon JB, Werb Z. Location, location, location: the cancer stem cell niche. Cell Stem Cell 2007; 1: 607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gordan JD, Thompson CB, Simon MC. HIF and c‐Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007; 12: 108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keith B, Simon MC. Hypoxia‐inducible factors, stem cells, and cancer. Cell 2007. 129: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jensen KB, Jones J, Watt FM. A stem cell gene expression profile of human squamous cell carcinomas. Cancer Lett 2008; 272: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jensen KB, Watt FM. Single‐cell expression profiling of human epidermal stem and transit‐amplifying cells: Lrig1 is a regulator of stem cell quiescence. Proc Natl Acad Sci USA 2006; 103: 11958–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gur G, Rubin C, Katz M et al. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J 2004; 23: 3270–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Laederich MB, Funes‐Duran M, Yen L et al. The leucine‐rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J Biol Chem 2004; 279: 47050–6. [DOI] [PubMed] [Google Scholar]
- 32. Andersen SS. Spindle assembly and the art of regulating microtubule dynamics by MAPs and Stathmin/Op18. Trends Cell Biol 2000; 10: 261–7. [DOI] [PubMed] [Google Scholar]
- 33. Eisenmann KM, McCarthy JB, Simpson MA et al. Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack‐1 and p130cas. Nat Cell Biol 1999; 1: 507–13. [DOI] [PubMed] [Google Scholar]
- 34. Majumdar M, Vuori K, Stallcup WB. Engagement of the NG2 proteoglycan triggers cell spreading via rac and p130cas. Cell Signal 2003; 15: 79–84. [DOI] [PubMed] [Google Scholar]
- 35. Iwasaki H, Suda T. Cancer stem cells and their niche. Cancer Sci 2009; 100: 1166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johannessen TC, Bjerkvig R, Tysnes BB. DNA repair and cancer stem‐like cells – potential partners in glioma drug resistance? Cancer Treat Rev 2008; 34: 558–67. [DOI] [PubMed] [Google Scholar]
- 37. Liu G, Yuan X, Zeng Z et al. Analysis of gene expression and chemoresistance of CD133 + cancer stem cells in glioblastoma. Mol Cancer 2006; 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jin F, Zhao L, Zhao HY et al. Comparison between cells and cancer stem‐like cells isolated from glioblastoma and astrocytoma on expression of anti‐apoptotic and multidrug resistance‐associated protein genes. Neuroscience 2008; 154: 541–50. [DOI] [PubMed] [Google Scholar]
- 39. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 2009; 14 (1): 3–9. [DOI] [PubMed] [Google Scholar]
- 40. Schatton T, Murphy GF, Frank NY et al. Identification of cells initiating human melanomas. Nature 2008; 451 (7176): 345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tirino V, Desiderio V, D’Aquino R et al. Detection and characterization of CD133 + cancer stem cells in human solid tumours. PLoS ONE 2008; 3: e3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frank NY, Margaryan A, Huang Y et al. ABCB5‐mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005; 65: 4320–33. [DOI] [PubMed] [Google Scholar]
- 43. Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer 2008; 8: 167–79. [DOI] [PubMed] [Google Scholar]
- 44. Shay JW, Keith WN. Targeting telomerase for cancer therapeutics. Br J Cancer 2008; 98: 677–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gupta PB, Onder TT, Jiang G et al. Identification of selective inhibitors of cancer stem cells by high‐throughput screening. Cell 2009; 138: 645–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bleau AM, Hambardzumyan D, Ozawa T et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem‐like cells. Cell Stem Cell 2009; 4: 226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peacock CD, Wang Q, Gesell GS et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA 2007; 104: 4048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saini V, Martyshkin DV, Mirov SB et al. An adenoviral platform for selective self‐assembly and targeted delivery of nanoparticles. Small 2008; 4: 262–9. [DOI] [PubMed] [Google Scholar]
- 49. Saini V, Roth JC, Pereboeva L, Everts M. Importance of viruses and cells in cancer gene therapy. Adv Gene Mol Cell Ther 2007; 1: 30–43. [Google Scholar]
- 50. Saini V, Zharov VP, Brazel CS, Nikles DE, Johnson DT, Everts M. Combination of viral biology and nanotechnology: new applications in nanomedicine. Nanomedicine 2006; 2: 200–6. [DOI] [PubMed] [Google Scholar]