

Abstract
Both cyclooxygenase (COX)‐2 and human epidermal growth factor receptor (HER)‐2 promote breast cancer progression; however, the relationship between the two molecules remains unclear. We utilized human breast cancer tissues and cell lines to examine whether COX‐2 and HER‐2 played independent or interdependent roles in vascular endothelial growth factor (VEGF)‐C up‐regulation and lymphangiogenesis. A paired correlation of immunodetectable levels of COX‐2, VEGF‐C, and HER‐2 proteins and lymphovascular density (LVD; D2‐40‐immunolabeled) in 55 breast cancer specimens revealed a positive correlation between COX‐2 and HER‐2 irrespective of clinicopathological status. However COX‐2 alone positively correlated with LVD. In 10 independent specimens, mRNA levels showed a positive correlation between HER‐2 and COX‐2 or VEGF‐C but not LYVE‐1 (lymphovascular endothelial marker). These findings implicate COX‐2, but not HER‐2, in breast cancer–associated lymphangiogenesis. Manipulation of the COX‐2 or HER‐2 genes in breast cancer cell lines varying widely in COX‐2 and HER‐2 expression revealed a direct role of COX‐2 and an indirect COX‐2 dependent role of HER‐2 in VEGF‐C up‐regulation: (i) high VEGF‐C expression in high COX‐2/low HER‐2 expressing MDA‐MB‐231 cells was reduced by siRNA‐mediated down‐regulation of COX‐2, but not HER‐2; (ii) integration of HER‐2 in these cells simultaneously up‐regulated COX‐2 protein as well as VEGF‐C secretion; and (iii) low VEGF‐C secretion by high HER‐2/low COX‐2 expressing SK‐BR‐3 cells was stimulated by COX‐2 overexpression. These findings of the primary role of COX‐2 and the COX‐2‐dependent role of HER‐2, if any, in VEGF‐C up‐regulation and lymphangiogenesis suggest that COX‐2 inhibitors may abrogate lymphatic metastasis in breast cancer irrespective of HER‐2 status. (Cancer Sci 2010)
In spite of rapid advancement in early detection and treatment of breast cancer in recent years, mortality from metastatic disease still remains high. Metastasis by the lymphatic route, often the first mode of spread of this disease, negatively impacts patient survival.( 1 ) However, the underlying mechanisms remain poorly understood. Vascular endothelial growth factors (VEGF)‐C and ‐D were shown to stimulate lymphangiogenesis by binding to VEGF receptor (R)‐3 expressed by lymphatic endothelial cells.( 2 ) Vascular endothelial growth factor (VEGF)‐C transfection into human MCF‐7 breast cancer cells induced lymphangiogenesis and intra‐lymphatic tumor growth in vivo,( 3 ) and VEGF‐C expression was correlated with lymphatic metastasis in a variety of human cancers.( 4 , 5 , 6 )
Cyclooxygenase (COX)‐2 is an important determinant for tumor progression. Its role in tumorigenesis was demonstrated by both overexpression( 7 ) and disruption( 8 ) of COX‐2, and protective effects of COX‐2 inhibitors against colorectal and mammary carcinogenesis.( 9 , 10 ) Indeed, COX‐2 up‐regulation, reported in many aggressive human cancers,( 11 , 12 , 13 ) is also a marker of poor prognosis for breast cancer.( 14 ) We reported that high prostaglandin (PG) E2 production, associated with COX‐2 expression, promotes tumor progression by inactivating antitumor immune cells, stimulating breast cancer cell migration and invasiveness, and tumor‐associated angiogenesis.( 15 , 16 , 17 , 18 )
Besides promoting angiogenesis( 16 , 17 , 18 , 19 ), COX‐2 was shown to stimulate lymphangiogenesis in non‐small‐cell lung cancer( 20 ) and breast cancer( 21 ) by up‐regulating VEGF‐C. We found that the elevated VEGF‐C expression was due to endogenous PGE2‐mediated activation of PGE receptors (EP1, EP4) expressed by breast cancer cells.( 21 ) Tumor‐derived VEGF‐C also directly stimulated breast cancer cell migration by binding to a diverse group of VEGF‐C receptors.( 22 )
Human epidermal growth factor receptor (HER)‐2, another major determinant of breast cancer progression( 23 ), is amplified in 20–30% patients, and often associated with elevated COX‐2 expression.( 24 ) The role of HER‐2 in VEGF‐C regulation and lymphatic metastasis of breast cancer remains at best speculative. A possible role of HER‐2 in VEGF‐C synthesis was suggested by VEGF‐C up‐regulation in HER‐2/VEGF‐C‐negative MCF‐7 cells exposed to heregulin β‐1, an HER‐2 ligand.( 25 ) However, HER‐2‐mediated breast cancer progression in HER‐2 transgenic mice was shown to be dependent on COX‐2.( 26 , 27 , 28 ) Although both COX‐2 and HER‐2 promote human breast cancer progression, the relationship between the two in VEGF‐C up‐regulation and lymphangiogenesis remains unclear. Here, we aimed to determine whether they played independent or interdependent role(s). Such knowledge is fundamental to chemo‐intervention and therapy of lymphatic metastasis.
We utilized human breast cancer tissues and cell lines to meet a number of objectives. First, we aimed to examine the relationship between pairs of the following parameters: the levels of immunohistochemically detectable COX‐2, VEGF‐C, and HER‐2, and relative areas occupied by D2‐40 immuno‐labeled lymphatics. Second, we aimed to compare the level of HER‐2 mRNA expression with the mRNA levels of COX‐2, VEGF‐C, and lymphovascular endothelial hyaluron receptor (LYVE)‐1 in human breast cancer tissues. Third, we aimed to identify the role(s) of COX‐2 and HER‐2 in VEGF‐C production in human breast cancer cell lines varying widely in COX‐2 and HER‐2 expression through genetic manipulations such as: (i) knocking down COX‐2 or HER‐2 or overexpressing HER‐2 in the high COX‐2/low HER‐2 expressing cell line MDA‐MB‐231 capable of high VEGF‐C production; and (ii) knocking down HER‐2 or overexpressing COX‐2 in the low COX‐2/high HER‐2 expressing cell line SK‐BR‐3 which produced little VEGF‐C. Results revealed that COX‐2, not HER‐2, was the primary determinant of VEGF‐C up‐regulation and lymphangiogenesis, and that the roles of HER‐2, if any, were COX‐2‐dependent.
Materials and Methods
Human breast cancer tissues.
Four‐μm‐thick sections of paraffin‐embedded formalin‐fixed breast cancer tissues from 55 patients (group 1) and liquid nitrogen frozen tissues from 10 additional patients (group 2) were obtained from our Department of Pathology, and all patient identifiers were blocked (approval no. TB209) in compliance with Institutional Ethics Review (1995 Declaration of Helsinki, 2004 Tokyo revisions). Group 1 was used for immunohistochemical staining, and group 2 for gene expression. Synopsis of clinicopathological parameters is given in Table 1.
Table 1.
Clinicopathological characteristics of tissue samples
| Histo. status | Group 1 (n = 55) | Group 2 (n = 10) | ||||
|---|---|---|---|---|---|---|
| (−)ve | (+)ve | No rec. | (−)ve | (+)ve | No rec. | |
| 1. LN status | 34 | 21 | 0 | 5 | 2 | 3 |
| 2. ERa | 15 | 26 | 14 | 5 | 5 | 0 |
| 3. PRb | 14 | 25 | 16 | 5 | 5 | 0 |
| 4. SBRc grade | SBR I/II | SBR III | No rec. | SBR I/II | SBR III | No rec. |
| 27 | 27 | 1 | 2 | 3 | 5 | |
aER (estrogen receptor) or bPR (progesterone receptor) status was classified as negative/(−)ve or positive/(+)ve if the respective staining was reported as negative/weak or moderate to strongly positive; or no records (no rec.). Groups 1 and 2 were independent samples, respectively utilized for immunohistochemistry and mRNA analysis. cSBR indicates Scarff‐Bloom‐Richardson grade. LN, lymph node.
Human breast cancer cell lines.
Cell lines with strong disparity in COX‐2 and Her‐2 expression, for example MCF‐7 (low‐COX‐2/low‐HER‐2), MDA‐MB‐231 (high‐COX‐2/low‐HER‐2), and SK‐BR‐3 (high‐HER‐2/low‐COX‐2) were obtained from the ATCC (Manassas, VA, USA) and grown in DMEM (first two) or modified McCoy’s 5A (third line). Human epidermal growth factor receptor (HER)‐2 overexpressing variants of the MDA‐MB‐231 cell line MDA‐MB‐231‐ERbB2 (gift of Dr Moulay Alaoui Jamail, McGill University) were grown like parental cells. Human COX‐2 cDNA cloned into the eukaryotic expression vector pIRES2‐EGFP (from Dr Michael Archer, University of Toronto) was transiently transfected into SK‐BR‐3 cells.
Transfection of cells with siRNAs.
Cells (1.5 × 105) were transfected with 100 nM of either non‐targeting siRNA or SMARTpool VEGF‐C siRNA or HER‐2 siRNAs in presence of 0.2%‐DharmaFECT (Dharmacon, Lafayette, CO, USA) in antibiotic‐free medium. Transfection efficiency was measured by qPCR, and VEGF‐C secretion by ELISA.
Real‐time RT‐PCR.
Primers were as follows: HER‐2 (sense‐5′‐AGACGAAGCATACGTGA‐3′; antisense‐5′‐GTACGAGCCGCACATC‐3′), VEGF‐C (sense‐5′‐CGGGAGGTGTGTATAGATGTG‐3′; antisense‐5′‐ATTGGCTGGGGAAGAGTTTG‐3′), COX‐2 (sense‐5′‐GAATGGGGTGATGAGCAGTT‐3′; antisense‐5′‐CAGAAGGGCAGGATACAGC‐3′), and GAPDH (sense‐5′ACCACAGTCCATGCCATCAC‐3′; antisense‐5′‐TCCACCACCCTGTTGCTGTA‐3′). Quantitative PCR (qPCR) was performed with a LightCycler (Roche‐Canada, Quebec, Canada) and SYBR Green Tag ReadyMix (Sigma, St. Louis, MO, USA), normalized relative to GAPDH.
Cyclooxygenase (COX)‐2 protein expression and VEGF‐C secretion.
Cells were lysed in RIPA buffer (1% NP‐40, 0.1% SDS, 0.5% sodium deoxycholate, 100 ng/mL PMSF, and 70 ng/mL aprotinin). Samples containing 40‐μg proteins were separated on 10% SDS‐PAGE gel. Cyclooxygenase (COX)‐2 protein (72 kDa) was detected with a monoclonal antihuman COX‐2 antibody (Cayman, Ann Arbor, MI, USA). Secreted VEGF‐C was measured with ELISA (IBL, Takasaki, Japan).
Immunohistochemical staining.
A Vestastain ABC kit (Vector, Burlingame, CA, USA) was used to detect VEGF‐C, HER‐2, and D2‐40 antigen in breast cancer tissues. Formalin‐fixed paraffin‐embedded tissue sections (4 μm) were deparaffinized in xylene and rehydrated through decreasing concentrations of ethanol. Except for D2‐40, antigen retrieval was achieved with brief microwaving. To inhibit endogenous peroxidases, rehydrated sections were treated with 0.3% H2O2 in methanol for 30 min. To block non‐specific antibody binding, sections were treated with species‐specific normal sera. The following primary antibodies were applied for 30 min at RT: (i) mouse monoclonal D2‐40 (1:40 dilution; Cedarlane, Hornby, ON, Canada); (ii) rabbit antihuman COX‐2 (1:20; IBL); (iii) rabbit antihuman VEGF‐C (1:20; IBL); and (iv) rabbit antihuman HER2/ErbB2 (1:50; Cell Signaling, Beverley, MA, USA). Biotinylated secondary antibody (Vestastain kit) followed by ABC reagent was applied for 30 min at RT. To visualize immunostaining, 3, 3′‐diaminobenzidine/urea hydrogen peroxide solution was used. Finally, sections were counterstained with Mayer’s hematoxylin. Omission of the primary antibody and treatments with control Ig served as the negative control.
Quantification of immunostaining.
Immunostaining intensities for COX‐2, VEGF‐C, and HER‐2 were scored by matching with a brown color chart in ascending order (0, no staining; 1–6, weakest to strongest staining), the percent of cells in each score category was determined, and then the cumulative score for each tissue sample was calculated on the basis of an independent full scan of 3–5 non‐overlapping sections by a pathologist (P.K.L.) and another operator (J.C.) with <10% deviation. Thus the immunostaining score = Σnp/100, where n is the color grade in ascending order (0–6), and p is the percentage of cells in that particular color grade. Because of a heterogeneity in the presence of marker‐bearing cells (particularly in the case of HER‐2), this scoring method provided a more rigorous way or correlating the expression levels of immunodetectable proteins such as COX‐2, HER‐2, and VEGF‐C in pairs, than the conventional immunoscoring methods.
D2‐40‐stained lymphatics were scored in two different ways, providing two different scores: lymphovascular density (LVD) and lymphovascular spaces (LVS). Lymphovascular density (LVD) was computed as the percent of the total area of the tissue sections (inclusive of tumor cells as well as stroma) occupied by D2‐40‐marked lymphatic endothelial spaces, that may be complete (closed) or incomplete (open), including narrow spaces occupied by endothelial linings in linear or curvilinear orientations. All lymphatics irrespective of sectional plane or degree of opening were scored. Lymphovascular space (LVS) was quantitated as the percent area of the tissue section occupied by lymphatic spaces having complete lumens, outlined by D2‐40‐stained endothelial cells. Identifiable blood vessels served as negative controls for D2‐40 staining.
Statistics.
Statistical analyses were performed using SigmaStat program (Systat Software, Richmond, CA, USA). Spearman’s rank–order correlation tested significant relationships between mRNA expression levels or immunostaining scores of two different markers within the total patient population or different clinicopathological subgroups. The Mann–Whitney test was employed in subgroups where data were not normally distributed. The Student’s t‐test was used to determine differences in comparing two data sets including the effects of siRNA treatments or gene transfection on gene expression and VEGF‐C synthesis in breast cancer cell lines. All tests were two‐tailed with a P‐value of <0.05 for significant differences between the groups.
Results
Immunostaining.
A synopsis of clinicopathological characteristics of breast cancer patients based on pathology reports is given in Table 1. The total number of samples utilized for immunostaining was 55 (Table 1, group 1). Immunoreactivity for COX‐2, HER‐2, and VEGF‐C was confined to cancer cells and not detected in the stroma (Fig. 1a–f). Cyclooxygenase (COX)‐2 (Fig. 1a,c) and VEGF‐C (Fig. 1f) staining was confined to the cytoplasm, whereas HER‐2 staining (Fig. 1b) was noted on cell membranes and to a lesser extent in the cytoplasm as reported.( 23 ) A co‐localization of COX‐2 and HER‐2 in cancer cells (Fig. 1a,b) was evident in some serial sections. Interestingly, in the case of strong COX‐2 positivity of a primary tumor (Fig. 1c), metastatic cells of the same tumor in the lymph node were also strongly COX‐2 positive (Fig. 1e). In some sections there was a distinct heterogeneity of HER‐2‐positive and ‐negative cancer cells side by side (Fig. 1d). Cyclooxygenase (COX)‐2and VEGF‐C staining were also noted in lesions featuring ductal carcinoma in situ (DCIS) (Fig. 2a showing VEGF‐C staining).
Figure 1.
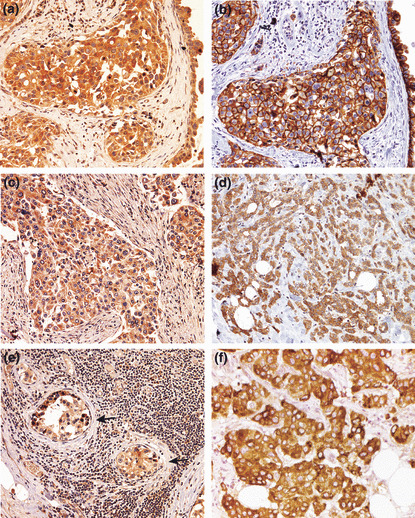
(a–f) Representative images of immunostaining (brown) for cyclooxygenase (COX)‐2 (a, c, e), human epidermal growth factor receptor (HER)‐2 (b,d), and vascular endothelial growth factor (VEGF)‐C (f) in breast cancer tissues, showing immunoreactivity confined to cancer cells and a lack of stromal staining in all cases. Immunostaining is noted in the cytoplasm in each case and also the cell membranes for HER‐2 (evident in b). (e) A metastatic lymph node of the primary tumor (c), showing COX‐2‐positive cancer cells located within an arteriole (arrows). (a,b) Semi‐serial sections of the same tumor showing COX‐2 and HER‐2 positivity, respectively, of cancer cells in the same location, indicating a co‐expression of both markers. (d) Human epidermal growth factor receptor (HER)‐2‐positive (brown) and ‐negative (blue) cancer cells side by side within the cluster. (f) Vascular endothelial growth factor (VEGF)‐C‐positive cancer cells. Original magnifications, ×400.
Figure 2.
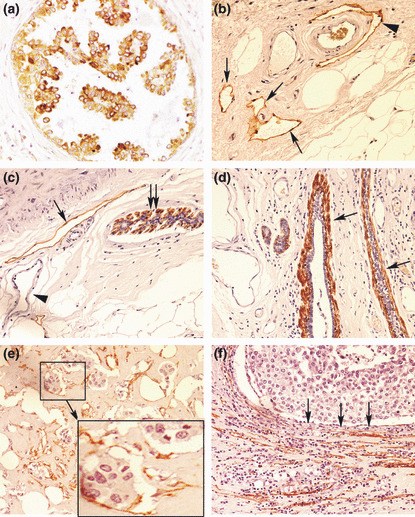
(a) Vascular endothelial growth factor (VEGF)‐C‐positive cancer cells in ductal carcinoma in situ (DCIS). (b–f) Various types of D2‐40 staining (brown): (b) lymphatics (arrows) in the peritumoral stroma, one of them (arrow head) embracing an artery which is unstained; (c) myo‐epithelial cells in the outermost layer of DCIS (double‐arrows); and a lymphatic (single‐arrow) in the stroma, blood capillaries (arrow head) remaining unstained; (d) myo‐epithelial cells (arrows) comprising the outermost layer of mammary ducts; (e) lymphatics with incomplete endothelial linings containing cancer cells indicative of lymphovascular invasion, shown clearly in the magnified inset of the selected area; (f) collapsed/compressed lymphatics in the stroma, adjacent to lobular cancerous tissue (arrows indicate direction of compression). Original magnifications, ×400.
D2‐40‐stained lymphatics were detected primarily in the peritumoral stroma, with the arteries serving as negative controls (Fig. 2b). Myoepithelial cells in DCIS (Fig. 2c) as well as mammary ducts (Fig. 2d) always showed strong D2‐40 immunostaining, indicating that myoepithelial cells express this antigen. They were easily distinguishable from lymphatics on morphological grounds. In certain sections displaying lobular morphology, the cancerous tissue appeared to compress the lymphatics in the surrounding stroma, so that the lymphatic spaces were collapsed and very narrow (Fig. 2f). Finally, some lymphatic spaces, often showing incomplete lining, exhibited the presence of tumor cell clusters within, indicative of lymphovascular invasion (LVI) (Fig. 2e). Lymphovascular invasion (LVI) was never observed in lymphatic spaces with complete lumen scored as LVS (e.g. Fig. 2b,c).
The quantitative score of immunostaining ranged from 0 to 3.75 for COX‐2, 0 to 4.05 for Her2/neu, and 0.01 to 4.60 for VEGF‐C. Paired correlation of immunoscores in all tissue samples and clinical subgroups in which the correlation was significant are presented in Table 2. Significant positive correlations within the entire group of tissue samples were found in three cases, namely between COX‐2 and HER‐2 (n = 47; Table 2, Fig. 3a), between COX‐2 and LVD or LVS (n = 55, Table 2), and between LVD and LVS (n = 55, data not shown). The latter was expected, since LVS was a component of LVD. When we compartmentalized tissue samples into various clinicopathological subgroups (Table 2), a few observations emerged: (i) The positive correlation between COX‐2 and HER‐2 persisted in almost all subgroups irrespective of clinicopathological status; (ii) Cyclooxygenase (COX)‐2 was positively correlated with VEGF‐C only in the estrogen receptor (ER)‐ negative subgroup; (iii) Human epidermal growth factor receptor (HER)‐2 was positively correlated with VEGF‐C in the node‐negative, ER‐negative and progesterone receptor (PR)‐ negative subgroups; (iv) A positive correlation between COX‐2 and LVD but not LVS was retained in almost all subgroups. Interestingly, for LVS, this correlation was significant in subgroups representing less aggressive phenotypes, such as the node‐negative, ER+, PR+, and Scarff‐Bloom‐Richardson (SBR) grade 1/ll subgroups; (v) Human epidermal growth factor receptor (HER)‐2 was correlated with LVS in the node‐negative and low‐SBR subgroups; and (vi) Vascular endothelial growth factor (VEGF)‐C was correlated with LVD only in the SBR‐high subgroup.
Table 2.
Synopsis of correlations among different clinical subgroups of breast cancer patients
| Parameter pair | Clinical subgroup | No. of patients | Spearman correlation | P‐value |
|---|---|---|---|---|
| COX‐2 versus HER‐2 | All cases | 47 | 0.575 | <0.001 |
| − | 28 | 0.626 | <0.001 | |
| ER− | 13 | 0.681 | 0.009 | |
| ER+ | 26 | 0.531 | 0.005 | |
| PR− | 12 | 0.815 | <0.001 | |
| PR+ | 25 | 0.560 | 0.004 | |
| SBR I/II | 23 | 0.547 | 0.007 | |
| SBR III | 23 | 0.660 | <0.001 | |
| COX‐2 versus VEGF‐C | All cases | 54 | 0.147 | 0.287 |
| ER− | 15 | 0.534 | 0.037 | |
| HER‐2 versus VEGF‐C | All cases | 46 | 0.176 | 0.241 |
| − | 27 | 0.453 | 0.018 | |
| ER− | 13 | 0.675 | 0.010 | |
| PR− | 12 | 0.577 | 0.045 | |
| COX‐2 versus LVD | All cases | 55 | 0.481 | <0.001 |
| − | 34 | 0.432 | 0.011 | |
| + | 21 | 0.557 | 0.009 | |
| ER− | 15 | 0.521 | 0.045 | |
| PR+ | 25 | 0.574 | 0.003 | |
| SBR I/II | 27 | 0.575 | 0.002 | |
| SBR III | 27 | 0.384 | 0.047 | |
| HER‐2 versus LVD | All cases | 47 | 0.128 | 0.390 |
| VEGF‐C versus LVD | All cases | 54 | 0.038 | 0.782 |
| SBR III | 27 | 0.413 | 0.032 | |
| COX‐2 versus LVS | All cases | 55 | 0.400 | 0.003 |
| − | 34 | 0.574 | <0.001 | |
| ER+ | 26 | 0.564 | 0.003 | |
| PR+ | 25 | 0.623 | <0.001 | |
| SBR I/II | 27 | 0.554 | 0.003 | |
| HER‐2 versus LVS | All cases | 47 | 0.124 | 0.405 |
| − | 28 | 0.430 | 0.023 | |
| SBR I/II | 23 | 0.433 | 0.039 |
(−) Negative; (+) positive; COX‐2, cyclooxygenase‐2; ER, estrogen receptor; HER‐2, human epidermal growth factor receptor; LVD, lymphovascular density; LVS, lymphovascular space; PR, progesterone receptor; SBR, Scarff‐Bloom‐Richardson grade; VEGF‐C, vascular endothelial growth factor C.
Figure 3.
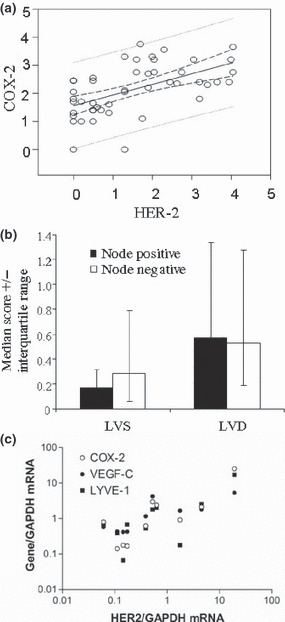
Immunoscores (a,b) and mRNA (c) in breast cancer tissues. (a) Correlation of immunoscores for cyclooxygenase (COX)‐2 versus human epidermal growth factor receptor (HER)‐2 in ungrouped patients. (Spearman r = 0.575, P < 0.0001) (b) Comparison of lymphovascular density (LVD) and lymphovascular spaces (LVS) scores in node‐positive (+ve) versus ‐negative (−ve) tissues. (c) Spearman correlations of mRNA expression levels of human epidermal growth factor receptor (HER)‐2 versus COX‐2, vascular endothelial growth factor (VEGF)‐C, and lymphovascular endothelial hyaluron receptor (LYVE)‐1 in 10 ungrouped breast cancer tissue samples (HER‐2 versus COX‐2, r = 0.758, P = 0.015; HER‐2 versus VEGF‐C, r = 0.782, P = 0.011; HER‐2 versus LYVE‐1, r = 0.576, P = 0.088).
Lymphovascular density (LVD) and LVS were used as respective measures of lymphatic‐occupied areas for all lymphatics and lymphatic spaces with complete lumens. In both cases, the lymphatics were located primarily in the peritumoral stroma. However intratumoral lymphatics, when found, were usually narrow with incomplete lining, and thus scored as part of LVD. The median LVD was numerically higher in the node‐positive than in the node‐negative patients, whereas the LVS was higher in node‐negative patients, although these differences were not significant (Fig. 3b).We interpret these results to suggest that LVS primarily measured open preformed lymphatics that were not engaged in LVI. This suggestion is supported by our findings that such lymphatics were never seen to contain intraluminal tumor cells whereas tumor cell clusters were only found in lymphatics having incomplete lining, included in our LVD scores (e.g. Fig. 2e). Furthermore, LVI was exclusively noted in samples with high tumor grades and high VEGF‐C immunoscores.
Significant correlation of COX‐2 immunoscores with LVD is consistent with our previous findings of a correlation between COX‐ 2 and LYVE‐1 mRNA. 21 In spite of a positive correlation between COX‐2 and HER‐2 immunoscores, a correlation between COX‐2 or HER‐2 with VEGF‐C immunoscores was noted only in certain subgroups. This prompted us to further examine the relationship among the three systems using gene expression analysis in human breast cancer tissues and cell lines.
mRNA expression of HER‐2, COX‐2, VEGF‐C, and LYVE‐1 in breast cancer tissues.
We subjected 10 independent breast cancer tissues to real time qPCR (Table 1, group 2). Human epidermal growth factor receptor (HER)‐2 mRNA expression in these tissues was strongly and significantly correlated with both COX‐2 and VEGF‐C mRNA expression (Fig. 3c). No significant correlation, however, was noticed between HER‐2 and LYVE‐1 mRNAs. Taking into account that COX‐2 mRNA levels correlated significantly with both VEGF‐C and LYVE‐1 mRNAs, these data questioned the contribution of HER‐2 to breast cancer–associated lymphangiogenesis.
Vascular endothelial growth factor (VEGF)‐C expression/secretion is not correlated with HER‐2 expression in human breast cancer cell lines.
Although SK‐BR‐3 is a well‐recognized standard for HER‐2 amplification,( 29 ) other breast cancer cell lines, for example Hs578T and MDA‐MB‐231 which are often considered as HER‐2 negative, also expressed detectable levels of HER‐2 mRNA (Fig. 4a). However, in contrast to the positive association of COX‐2 with VEGF‐C secretion in numerous breast cancer cell lines,( 21 ) no such correlation was found between HER‐2 and VEGF‐C expression in any of the cell lines tested (data not shown). Vascular endothelial growth factor (VEGF)‐C mRNA and protein levels could be significantly reduced by knocking down COX‐2 in the MDA‐MB‐231 cell line. 21 To find out whether HER‐2 had any role in VEGF‐C production in this cell line, we treated the cells with specific siRNAs for HER‐2 and VEGF‐C (as control). Each siRNA efficiently and specifically knocked down the expression of the corresponding gene (Fig. 4b). These effects were sustained at least for 48 h. While VEGF‐C secretion by MDA‐MB‐231 cells was inhibited by VEGF‐C siRNA as expected, HER‐2 siRNA had no effect on VEGF‐C secretion (Fig. 4c).
Figure 4.
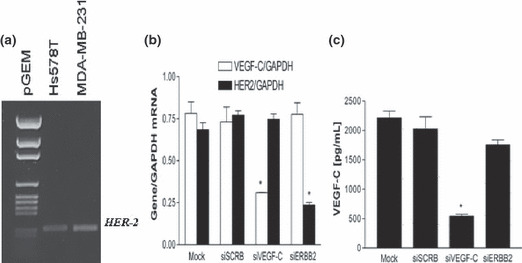
(a) Reverse transcription–PCR detection of human epidermal growth factor receptor (HER)‐2 mRNA in Hs578T and MDA‐MB‐231. (b,c) Effects of HER‐2 and vascular endothelial growth factor (VEGF)‐C siRNA on the expression of these genes (b) and VEGF‐C secretion (c) by MDA‐MB‐231 cells. Cells were treated for 24 h with siRNAs and scramble siRNA (siSCRB) as a control and used either for gene expression or VEGF‐C secretion. Both HER‐2 and VEGF‐C siRNAs efficiently knocked down the cognate genes; however, down‐regulation of HER‐2 did not significantly affect VEGF‐C gene expression or the protein secretion.
Cyclooxygenase (COX)‐2 dependence of VEGF‐C production in HER‐2‐expressing cells.
We found that low the COX‐2, high HER‐2‐expressing SK‐BR‐3 cell line was a poor producer of VEGF‐C, which could not be down‐regulated any further by knocking down HER‐2 (data not shown). However, when COX‐2 was successfully up‐regulated in these cells by transient transfection with a COX‐2 expression vector (Fig. 5a), a stimulation of VEGF‐C secretion was observed (Fig. 5b). Finally, upon stable integration of exogenous HER‐2 into HER‐2 low MDA‐MB‐231 cell line, we noted an up‐regulation of COX‐2 protein in association with a modest increase in VEGF‐C secretion (Fig. 5c,d). These results strongly indicate COX‐2 dependence of VEGF‐C production in HER‐2 expressing cells. Human epidermal growth factor receptor (HER)‐2 on its own, even when expressed at high levels, failed to stimulate VEGF‐C in the absence of COX‐2. On the other hand, such cells became significant producers of VEGF‐C when co‐expressing COX‐2.
Figure 5.
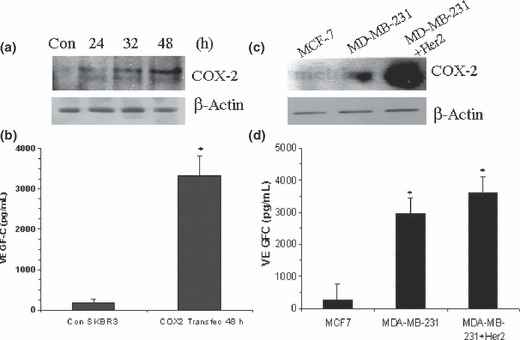
Effects of insertion of cyclooxygenase (COX)‐2 into SK‐BR‐3 (a,b) and human epidermal growth factor receptor (HER)‐2 into MDA‐MB‐231 cells (c,d) on vascular endothelial growth factor (VEGF)‐C secretion. Cyclooxygenase (COX)‐2‐low SK‐BR‐3 cells were transfected with mammalian expression vector containing COX‐2 (4 μg of DNA) for 24, 32, and 48 h. Cell lysates and supernatants were respectively used to monitor COX‐2 expression and VEGF‐C secretion. (a) COX‐2 protein expression (western‐blot) and (b) VEGF‐C secretion (ELISA), 48 h post transfection by control (vector) and COX2‐transected SKBR3 cells (*P < 0.001). Strong up‐regulation of COX‐2 protein at 48 h (a) is associated with higher VEGF‐C secretion (b). (c) Cyclooxygenase (COX)‐2 expression in MCF‐7, MDA‐MB‐231, and MDA‐MB‐231‐HER‐2 cells; (d) VEGF‐C ELISA in the above cells. Note the up‐regulation of both COX‐2 and VEGF‐C upon introduction of HER‐2.
Discussion
The present study was designed to investigate whether COX‐2 and HER‐2, the two major determinants of human breast cancer progression, played independent or interdependent roles in VEGF‐C up‐regulation and lymphangiogenesis. Utilizing correlative studies at the mRNA and protein levels in breast cancer tissues and mechanistic studies with genetic manipulation of breast cancer cell lines, we show that COX‐2 but not HER‐2 is the dominant mediator of VEGF‐C up‐regulation and lymphangiogenesis in human breast cancer. The role of HER‐2, if any, appears to be COX‐2 dependent.
Significant association of COX‐2 and HER‐2 mRNA expression with VEGF‐C mRNA expression in human breast cancer tissues suggest that both COX‐2 and HER‐2 are directly or indirectly associated with VEGF‐C synthesis. In contrast to a significant correlation between COX‐2 and LYVE‐1 mRNA, the correlation between HER‐2 and LYVE‐1 was not significant, suggesting a direct role of COX‐2 rather than HER‐2 in breast cancer–associated lymphangiogenesis.
Immunostaining for COX‐2, VEGF‐C, and HER‐2 in breast cancer tissues was localized to breast cancer cells and absent in stromal cells. A strong correlation between the COX‐2 and HER‐2 immunoscores in nearly every clinicopathological subset of patients is in general agreement with previous reports on DCIS( 30 ) and invasive ductal carcinomas,( 31 ) as well as our current findings at the mRNA level. However, in spite of significant mRNA correlation between COX‐2 and VEGF‐C, such a correlation at the protein level was only found in the ER‐negative tissues. This is consistent with our in vitro observations that ER‐negative/high COX‐2 cell lines (MDA‐MB‐231 and Hs578T) produced high levels of VEGF‐C, whereas ER‐positive/COX‐2‐lacking MCF‐7 cells produced very little.( 21 ) The observed correlation between HER‐2 and VEGF‐C immunoreactivity confined to ER‐negative tissues is possibly explained by co‐expression of COX‐2, rather than a direct role of HER‐2, since high HER‐2/low COX‐2/ER‐negative SK‐BR‐3 cells produced little VEGF‐C.
Whether lymph node metastasis in human breast cancer occurs via newly formed or preformed lymphatics remains a debated issue.( 32 , 33 ) D2‐40 antigen is an epitope of podoplanin expressed by lymphatic and not vascular endothelial cells, making it a reliable immnostainable marker for lymphatics in fixed human tissues in preference to LYVE‐1.( 34 ) Lymphatics were seen primarily in the peritumoral stroma, supporting earlier reports of the predominance of peritumoral lymphatics in non‐inflammatory breast cancers as opposed to inflammatory breast cancers( 33 , 34 ) or head and neck cancer( 35 ) which displayed profuse intratumoral lymphatics. Higher LVS scores in node‐negative than in node‐positive cancers as well as the absence of LVI in open, dilated lymphatics associated with LVS scores suggests that LVS represents either preformed lymphatics or products of lymphangiogenesis earlier in tumor life, not associated with lymphatic metastasis. On the other hand, partially open or collapsed lymphatics nearer to tumor cell clusters, that are included in our LVD scores, are most likely products of neo‐lymphangiogenesis and were also responsible for lymphatic metastasis by lymphovascular invasion as illustrated in Figure 2(f). Lymphovascular density scores (LVD), as expected, were higher (although not significant) in node‐positive than in node‐negative tissues. Significantly positive association of LVD with COX‐2 but not HER‐2 reinforces the notion of a direct role of COX‐2 in lymphangiogenesis and lymphatic metastasis.
A significant correlation between VEGF‐C immunoscores and LVD was only found in tissues with high tumor grade. This finding, combined with a strong correlation between VEGF‐C and LYVE‐1 mRNA levels, reinforces the role of VEGF‐C in lymphangiogenesis. Unexpectedly, however, with regard to VEGF‐C immunoscores, although higher in node‐positive than in node‐negative tissues, the differences were not significant. We did not find additional macrophage‐associated VEGF‐C immunostaining in node‐positive cases that could obscure our scores. This may suggest additional lymphangiogenesis‐independent role(s) of VEGF‐C in breast cancer progression. This hypothesis is supported by two sets of data: our findings that endogenous VEGF‐C promotes breast cancer cell migration, 22 a critical step in metastasis, and our current findings that lymphovascular invasion was limited to high tumor grade samples, in which there was also significant correlation between VEGF‐C immunoscore and LVD.
We had earlier utilized several human breast cancer cell lines showing that the positive correlation between COX‐2 mRNA expression or activity and VEGF‐C production resulted from a cause–effect relationship.( 21 ) In the present study, such a correlation or cause–effect relationship between HER‐2 mRNA and VEGF‐C mRNA or VEGF‐C secretion in breast cancer cell lines was lacking. However, introduction of COX‐2 into low COX‐2/high HER‐2/low VEGF‐C‐producing SK‐BR‐3 cells up‐regulated VEGF‐C production, showing that HER‐2 is permissive to COX‐2‐mediated VEGF‐C up‐regulation or utilizes the intermediary role of COX‐2. The latter suggestion is supported by our observation of a parallel up‐regulation of COX‐2 and VEGF‐C in HER‐2‐transfected MDA‐MB‐231 cells. In support of our human data, an intermediary or obligatory role of COX‐2 in HER‐2‐mediated breast cancer progression has been reported in transgenic murine models, representing multiple mechanisms: an up‐regulation of aromatase,( 28 ) a stimulation of angiogenesis,( 27 ) and resistance to apoptosis.( 36 )
Our findings of the primary role of COX‐2 in VEGF‐C up‐regulation and lymphangiogenesis suggests the potential role of COX‐2 inhibitors in chemo‐intervention of lymphatic metastasis in COX‐2‐expressing breast cancer irrespective of HER‐2 status. This view is supported by the findings that celecoxib treatment of nude mice bearing xenografts of MDA‐MB‐231 cells, as well as a HER‐2‐transfected MCF‐7 cells that expressed COX‐2 in vivo, resulted in reduced tumor growth and lymphangiogenesis.( 37 ) The efficacy of COX‐2 inhibitors in HER‐2/COX‐2 bearing breast cancer remains to be tested adequately. In one small phase II trial of 11 trastuzumab‐resistant patients afflicted with HER‐2‐overexpressing metastatic breast cancer, celecoxib combined with trastuzumab did not produce any additional benefit.( 38 ) However, the authors stated that the COX‐2 status of these patients was undetermined. Since the long‐term safety of high‐dose COX‐2 inhibitors remains under scrutiny,( 39 ) low‐dose COX‐2 inhibitors deserve testing in a combined therapeutic modality, for example with EP4 antagonists because of the primary role of EP4 receptors in COX‐2‐mediated VEGF‐C up‐regulation.( 21 ) Indeed, we found that therapy with an EP4 antagonist inhibited tumor‐associated lymphangiogenesis and lymphatic metastasis in a COX‐2‐expressing syngeneic murine breast cancer model.( 40 )
Acknowledgments
We thank Dr Graham Wagner for RNA from frozen tissues, Dr Michael Archer for COX‐2 vector, Dr Moulay Alaoui‐Jamali for the MDA‐MB‐231‐ERbB2 cell line, Dr Lynne‐Marie Postovit for statistical analysis, and Xiping Xin for tabulating immunoscores. We also thank the Ontario Institute of Cancer Research and the Canadian Breast Cancer Foundation, Ontario Chapter, for grant support to P.K.L.
References
- 1. Truong PT, Vinh‐Hung V, Cserni G, Woodwar WA, Tai P, Vlastos G. The number of positive nodes and the ratio of positive to excised nodes are significant predictors of survival in women with micrometastatic node‐positive breast cancer. Eur J Cancer 2008; 44: 1670–7. [DOI] [PubMed] [Google Scholar]
- 2. Wissmann C, Detmar M. Pathways targeting tumor lymphangiogenesis. Clin Cancer Res 2006; 12: 6865–8. [DOI] [PubMed] [Google Scholar]
- 3. Karpanen T, Egeblad M, Karkkainen MJ et al. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res 2001; 61: 1786–90. [PubMed] [Google Scholar]
- 4. Tsurusaki T, Kanda S, Sakai H et al. Vascular endothelial growth factor‐C expression in human prostatic carcinoma and its relationship to lymph node metastasis. Br J Cancer 1999; 80: 309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yonemura Y, Endo Y, Fujita H et al. Role of vascular endothelial growth factor C expression in the development of lymph node metastasis in gastric cancer. Clin Cancer Res 1999; 7: 1823–9. [PubMed] [Google Scholar]
- 6. Akagi A, Ikeda Y, Miyazaki M et al. Vascular endothelial growth factor‐C (VEGF‐C) expression in human colorectal cancer tissues. Br J Cancer 2000; 83: 887–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu CH, Chang SH, Narko K et al. Overexpression of cyclooxygenase‐2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem 2001; 276: 18563–9. [DOI] [PubMed] [Google Scholar]
- 8. Chulada PC, Thompson MB, Mahler JF et al. Genetic disruption of Ptgs‐1, as well as Ptgs‐2, reduces intestinal tumorigenesis in Min mice. Cancer Res 2000; 60: 4705–8. [PubMed] [Google Scholar]
- 9. Oshima M, Dinchuk JE, Kargman SL et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX‐2). Cell 1996; 87: 803–9. [DOI] [PubMed] [Google Scholar]
- 10. Harris RE. Cyclooxygenase 2 (COX‐2) blockade in the chemoprevention of cancers of the colon, breast, prostate and lung. Immunopharmacology 2009; 17(2): 55–67. [DOI] [PubMed] [Google Scholar]
- 11. Chan G, Boyle JO, Yang EK et al. Cyclooxygenase‐2 expression is up‐regulated in squamous cell carcinoma of the head and neck. Cancer Res 1999; 59: 991–4. [PubMed] [Google Scholar]
- 12. Tucker ON, Dannenberg AJ, Yang EK et al. Cyclooxygenase‐2 expression is up‐regulated in human pancreatic cancer. Cancer Res 1999; 59: 987–90. [PubMed] [Google Scholar]
- 13. Soslow RA, Dannenberg AJ, Rush D et al. COX‐2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 2000; 89: 2637–45. [DOI] [PubMed] [Google Scholar]
- 14. Ristimäki A, Sivula A, Lundin J et al. Prognostic significance of elevated cyclooxygenase‐2 expression in breast cancer. Cancer Res 2002; 62: 632–5. [PubMed] [Google Scholar]
- 15. Lala PK, Parhar RS, Singh P. Indomethacin‐therapy abrogates in prosta‐glandin mediated suppression of natural killer activity in tumor bearing mice and prevents tumor metastasis. Cell Immunol 1986; 99: 108–18. [DOI] [PubMed] [Google Scholar]
- 16. Lala PK, Al‐Mutter N, Orucevic A. Effects of chronic indomethacin therapy on the development and progression of spontaneous mammary tumors in C3H/HeJ mice. Int J Cancer 1997; 173: 371–80. [DOI] [PubMed] [Google Scholar]
- 17. Rozic JG, Chakraborty C, Lala PK. Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int J Cancer 2001; 93: 497–506. [DOI] [PubMed] [Google Scholar]
- 18. Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E(2) receptors in migration of murine and human breast cancer cells. Exp Cell Res 2003; 289: 265–74. [DOI] [PubMed] [Google Scholar]
- 19. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998; 93: 705–16. [DOI] [PubMed] [Google Scholar]
- 20. Su JL, Shih JY, Yen ML et al. Cyclooxygenase‐2 induces EP1‐and HER‐2/Neu‐Dependent vascular endothelial growth factor‐C up‐regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res 2004; 64: 554–64. [DOI] [PubMed] [Google Scholar]
- 21. Timoshenko AV, Chakraborty C, Wagner GF, Lala PK. COX‐2‐mediated upregulation of the lymphangiogenic factor VEGF‐C in human breast cancer. Br J Cancer 2006; 94: 1154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Timoshenko AV, Rastogi S, Lala PK. Migration‐promoting role of VEGF‐C and VEGF‐C binding receptors in human breast cancer cells. Br J Cancer 2007; 97: 1090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wright C, Angus B, Nicholson S et al. Expression of c‐erbB‐2 oncoprotein: a prognostic indicator breast cancer. Cancer Res 1989; 49: 2087–90. [PubMed] [Google Scholar]
- 24. Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase‐2 is overexpressed in HER‐2/neu‐positive breast cancer: evidence for involvement of AP‐1 and PEA3. J Biol Chem 2002; 277: 18649–57. [DOI] [PubMed] [Google Scholar]
- 25. Tsai PW, Shiah SG, Lin MT, Wu CW, Kuo ML. Up‐regulation of vascular endothelial growth factor C in breast cancer cells by heregulin‐beta 1. A critical role of p38/nuclear factor‐kappa B signaling pathway. J Biol Chem 2003; 278: 5750–9. [DOI] [PubMed] [Google Scholar]
- 26. Lanza‐Jacoby S, Miller S, Flynn J et al. The cyclooxygenase‐2 inhibitor, celecoxib, prevents the development of mammary tumors in Her‐2/neu mice. Cancer Epidemiol Biomarkers Prev 2003; 12: 1486–91. [PubMed] [Google Scholar]
- 27. Howe LR, Chang SH, Tolle KC et al. HER2/neu‐induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase‐2 knockout mice. Cancer Res 2005; 65: 10113–9. [DOI] [PubMed] [Google Scholar]
- 28. Subbaramaiah K, Howe LR, Port ER et al. HER‐2/neu status is a determinant of mammary aromatase activity in vivo: evidence for a cyclooxygenase‐2‐dependent mechanism. Cancer Res 2006; 66: 5504–11. [DOI] [PubMed] [Google Scholar]
- 29. Kraus MH, Popescu NC, Amsbaugh SC, King CR. Overexpression of the EGF receptor‐related proto‐oncogene erbB‐2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J 1987; 6: 605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perrone G, Santini D, Vincenzi B et al. COX‐2 expression in DCIS: correlation with VEGF, HER‐2/neu, prognostic molecular markers and clinicopathological features. Histopathology 2005; 46: 561–8. [DOI] [PubMed] [Google Scholar]
- 31. Cho MH, Yoon JH, Jaegal YJ et al. Expression of cyclooxygenase‐2 in breast carcinogenesis and its relation to HER‐2/neu and p53 protein expression in invasive ductal carcinoma. Breast 2006; 15: 390–8. [DOI] [PubMed] [Google Scholar]
- 32. Pepper MS, Tille JC, Nisato R, Skobe M. Lymphangiogenesis and tumor metastasis. Cell Tissue Res 2003; 314: 167–77. [DOI] [PubMed] [Google Scholar]
- 33. Vleugel MM, Bos R, Groep Pvander et al. Lack of lymphangiogenesis during breast carcinogenesis. J Clin Pathol 2004; 57: 746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van der Auwera I, Van den Eynden GG, Colpaert CG et al. Tumor lymphangiogenesis in inflammatory breast carcinoma: a histmorphometric study. Clin Cancer Res 2005; 11: 7637–42. [DOI] [PubMed] [Google Scholar]
- 35. Beasley NJ, Prevo R, Banerji S et al. Intratumoral lymphangiogenesis and lymph node metastasis in head and neck cancer. Cancer Res 2002; 62: 1315–20. [PubMed] [Google Scholar]
- 36. Simeone AM, Li YJ, Broemeling LD, Johnson MM, Tuna M, Tari AM. Cyclooxygenase‐2 is essential for HER2/neu to suppress N‐ (4‐hydroxyphenyl)retinamide apoptotic effects in breast cancer cells. Cancer Res 2004; 64: 1224–8. [DOI] [PubMed] [Google Scholar]
- 37. Barnes NL, Warnberg F, Farnie G et al. Cyclooxygenase‐2 inhibition effects on tumour growth, cell cycling and lymphangiogenesis in a xenograft model of breast cancer. Br J Cancer 2007; 96:575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dang CT, Dannenberg AJ, Subbaramaiah K et al. Phase II study of celecoxib and trastuzumab in metastatic breast cancer patients who have progressed after prior trastuzumab‐based treatments. Clin Cancer Res 2004; 10: 4062–7. [DOI] [PubMed] [Google Scholar]
- 39. Solomon SD, McMurray JJ, Pfeffer MA et al. Adenoma Prevention with Celecoxib (APC) Study Investigators. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 2005; 352: 1071–80. [DOI] [PubMed] [Google Scholar]
- 40. Lala PK, Mohindra V, Xin X, Lieu L, Girish G. COX‐2 and EP4 receptor as targets to ameliorate tumor growth, lymphangiogenesis and metastasis in a murine breast cancer model. Proc Am Assoc Cancer Res 2010; 51: 385–6. # 1601. [Google Scholar]