

Abstract
Lapatinib and bortezomib are highly active against breast cancer cells. Breast cancer patients who initially respond to lapatinib may eventually manifest acquired resistance to this treatment. Thus, the identification of novel agents that may prevent or delay the development of acquired resistance to lapatinib is critical. In the current study, we show that the combination of lapatinib and bortezomib results in a synergistic growth inhibition in human epidermal receptor 2 (HER2)‐overexpressing breast cancer cells and that the combination enhances apoptosis of SK‐BR‐3 cells. Importantly, we found that the combination of lapatinib plus bortezomib more effectively blocked activation of the HER2 pathway in SK‐BR‐3 cells, compared with monotherapy. In addition, we established a model of acquired resistance to lapatinib by chronically challenging SK‐BR‐3 breast cancer cells with increasing concentrations of lapatinib. Here, we showed that bortezomib notably induced apoptosis of lapatinib‐resistant SK‐BR‐3 pools and further inhibited HER2 signaling in the resistant cells. Taken together, the current data indicate a synergistic interaction between lapatinib and bortezomib in HER2‐overexpressing breast cancer cells and provide the rationale for the clinical evaluation of these two noncross‐resistant targeted therapies. The combination of lapatinib and bortezomib may be a potentially novel approach to prevent or delay the onset of acquired resistance to lapatinib in HER2‐overxpressing/estrogen receptor (ER)‐negative breast cancers. (Cancer Sci 2010); 00: 000–000
Overexpression or amplification of human epidermal receptor 2 (HER2) (ErbB2/neu) occurs in 25–30% of invasive breast carcinomas and is associated with aggressive disease and significantly decreased disease‐free survival and overall survival.( 1 ) Trastuzumab, a recombinant humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor, has proven to be active in HER2‐overexpressing breast cancers, both alone and in combination with chemotherapy.( 2 , 3 ) Lapatinib (GW572016, GlaxoSmithKline, Brentford, UK), a small molecule inhibitor of epidermal growth factor receptor (EGFR) and HER2 tyrosine kinases, is also a promising targeted therapy drug that was recently approved by the US Food and Drug Administration (FDA) in combination with capecitabine for treating HER2‐positive advanced or metastatic breast cancer patients who had progressed on prior trastuzumab‐based therapies.( 4 , 5 , 6 ) This compound has been shown to inhibit phosphatidylinositol‐3‐kinase (PI3K)/Akt and mitogen‐activated protein kinase (MAPK)/extracellular signal‐regulated kinase (ERK) signaling pathways in HER2‐overexpressing breast cancer cell lines, tumor xenografts and in tumor biopsies from patients with HER2‐positive breast cancer.( 7 , 8 , 9 ) In addition, lapatinib has been reported to restore tamoxifen sensitivity to tamoxifen‐resistant breast cancer models, have activity against trastuzumab‐resistant breast cancer cells and to inhibit the formation of brain metastases in a well‐established preclinical metastatic breast cancer xenograft model that happens to be resistant to trastuzumab.( 10 , 11 , 12 ) Importantly, it has been reported that lapatinib treatment of patients with HER2‐positive breast cancer did not lead to an increase in the percentage of tumorigenic cells, which might be resistant to chemotherapy and responsible for cancer relapse.( 13 )
Bortezomib (formerly known as PS‐341) is a selective and reversible proteasome inhibitor that induces cell cycle arrest and apoptosis through inhibition of nuclear factor kappa B (NF‐κB) activity and upregulation of various apoptotic pathways. This drug has been approved in many countries for the treatment of relapsed and refractory multiple myeloma and is currently in clinical trials for a variety of tumor types, including breast cancer.( 14 , 15 , 16 ) Although the exact mechanisms by which bortezomib induces apoptosis of breast cancer cells are unknown, preclinical investigations suggest that bortezomib is active against human breast cancer cell lines and show a modest growth‐inhibitory treatment effect in the trastuzumab‐resistant, HER2‐positive human breast cancer xenograft model.( 17 , 18 ) However, several clinical trials have not found it effective against breast cancer patients when used as a single agent.( 19 , 20 ) Given that preclinical and clinical data have already proven its efficacy in combination with chemotherapeutic agents and a potential role in overcoming tumor cell chemo‐resistance, the future development of bortezomib in breast cancer should focus on combination therapeutic strategies with agents such as trastuzumab or docetaxel.( 21 , 22 )
Although lapatinib has been shown to significantly improve survival in patients with metastatic HER2‐positive breast cancer, recent evidence suggests that patients who initially respond to lapatinib may eventually manifest acquired resistance to this treatment.( 23 ) Thus, it will be important to elucidate the underlying molecular mechanisms of acquired resistance to lapatinib. Moreover, identification of novel agents that may prevent or delay the development of acquired‐resistance to lapatinib is critical to improving the survival of breast cancer patients. In the current study, we characterized the effect of the combination of lapatinib and bortezomib in HER2‐overexpressing breast cancer cells using the combination index (CI) method for multiple drug effect analysis.( 24 ) Importantly, we explored the underling mechanisms of their combined effect on cell growth and apoptosis by focusing on the HER2 signaling transduction pathway. In addition, we established a model of acquired resistance to lapatinib by continuously exposing SK‐BR‐3 breast cancer cells to lapatinib with increasing concentrations. Whether or not bortezomib‐induced apoptosis of lapatinib‐resistant SK‐BR‐3 cells was assayed and the effect of bortezomib on the HER2 signaling pathway in the resistant cells were investigated.
Materials and Methods
Cell lines and cell culture. The human breast cancer cell lines, SK‐BR‐3 and BT‐474, were obtained from the American Type Culture Collection (Manassas, VA, USA). SK‐BR‐3 and BT‐474 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum at 37°C with 5% CO2 in a humidified incubator.
Compounds and antibodies. Lapatinib was kindly provided by GlaxoSmithKline. Bortezomib was purchased from LC Laboratories (Woburn, MA, USA). In both cases, 10 mM aliquots of drug in dimethyl sulfoxide (DMSO) were stored at −20°C and diluted just before use. All antibodies were purchased from commercial sources as indicated below: anti‐Akt1/2, anti‐Neu (F‐11), anti‐phospho Neu (Tyr1248)‐R, anti‐phospho‐Akt1/2/3 (Ser 473)‐R, anti‐phospho‐Erk (E‐4), anti‐Erk1 (K‐23) and anti‐P27 (C‐19) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti‐phospho‐4E‐BP1 (Ser65) antibody was obtained from Cell Signaling Technology (Beverly, MA, USA). Anti‐GAPDH antibody was obtained from ProteinTech Group, Inc. (Chicago, IL, USA). HRP‐conjugated goat‐anti‐rabbit IgG, goat‐anti‐mouse IgG and donkey‐anti‐goat IgG antibodies were purchased from Santa Cruz Biotechnology, Inc.
Establishment of lapatinib‐acquired resistant breast cancer cells. Lapatinib‐resistant pooled SK‐BR‐3 cells were developed by continuously exposing cells to increasing concentrations of lapatinib (0.25∼1.0 μmol/L) for at least 6 months. The resistant pools were cultured by collecting all viable cells on one plate. Pools were then maintained in RPMI 1640 supplemented with 1.0 μmol/L lapatinib, a concentration which is difficult for parental cells to survive in. Resistance to lapatinib was confirmed by cell viability assay as described below. The resistant pools were maintained in medium without lapatinib for 2 or 3 days before each experiment.
Cell viability assay. The cell viability was determined using the CCK‐8 (Cell Counting Kit‐8, Dojindo, Kumamoto, Japan) assay. Briefly, cells were seeded at a density of 3 × 103–5 × 103 cells into a 96‐well microplate. The cells were then treated with various concentrations of bortezomib and/or lapatinib as a single agent or in combination. CCK‐8 solution (Dojindo) was added to each well. The absorbance at 450 nm was measured with an Absorbance Microplate Reader (SUNRISE, Grödig, Austria). Cell survival for all experiments was expressed as the percentage of viable cells compared with the untreated cells.
Annexin V binding assay. Cell apoptosis was detected using Annexin V‐FITC Apoptosis Detection Kit I (Becton, Dickinson, Franklin Lakes, NJ, USA). Ten microliters of Annexin V‐FITC and 5 μL of propidium iodide were added to the cells. The stained cells were analyzed using flow cytometry.
Western blot analysis. The cells were lysed in RIPA buffer. The protein concentration of supernatants was determined using a modification of the Bradford method (Bio‐Rad Labs, Hercules, CA, USA). Equal amounts of proteins were resolved by SDS‐PAGE. Proteins were transferred to Immobilon‐P or nitrocellulose membranes. The membranes were incubated overnight with primary antibodies. The membranes were then incubated with a secondary antibody (Santa Cruz). Target proteins were visualized with the Super‐Signal West Femto Maximum sensitivity substrate kit (Pierce, Rockford, IL, USA) and subsequent exposure to X‐OMAT X‐ray film (Sigma, St Louis, MO, USA) according to the manufacturer’s instructions. Immunoblotting with GAPDH mouse monoclonal antibody (Santa Cruz) was done to confirm equal protein loading.
Multiple drug effect analysis: combination index. Aliquots of 3 × 103–5 × 103 BT‐474 or SK‐BR‐3 cells were seeded into a 96‐well microplate and incubated overnight. Experimental medium containing either control, lapatinib, bortezomib, or the combination of lapatinib and bortezomib was added to the appropriate wells. Multiple drug effect analysis was done using the combination index (CI), as described previously by Chou and Talalay.( 24 ) A statistical test was performed to determine whether the mean CI value at multiple levels was statistically significantly different from 1.0. A CI equal to 1.0 indicates additive interactions between the two agents; CI values statistically significantly <1 indicate synergy; and, conversely, CI values statistically significantly >1 indicate antagonism. Multiple drug‐effect analysis was performed using CalcuSyn software from Biosoft (Cambridge, United Kingdom).
Statistical analysis. Statistical analysis was carried out with SPSS version 11.5 (SPSS Inc., Chicago, IL, USA). To analyze multiple drug effects, the one‐sample Student’s t‐test was used to determine the statistical differences. An unpaired two‐tailed t‐test or anova was used to determine the statistical difference for cell viability and apoptosis. All statistical tests were two‐sided, and P‐values <0.05 were considered statistically significant.
Results
Synergistic growth inhibition of bortezomib and lapatinib in HER2 over‐expressing breast cancer cell lines. The growth‐inhibitory effects of bortezomib and lapatinib were assessed by CCK‐8 assays in breast cancer cell lines (SK‐BR‐3, BT‐474) with increasing concentrations of bortezomib or lapatinib for periods of 24, 48 or 72 h. Lapatinib resulted in time‐dependent and dose‐dependent inhibition growth in SK‐BR‐3 and BT‐474 cells. Slightly different from lapatinib, bortezomib was pro‐proliferative at lower concentrations, especially in SK‐BR‐3 cells (data not shown). To determine the combined effects of bortezomib and lapatinib on cell viability in the two cell lines, we then exposed the cells to the two agents simultaneously for 48 h. The concentrations of lapatinib used for these experiments ranged between 0.05 μmol/L and 12.15 μmol/L (serial 1:3 dilutions), and the concentrations of bortezomib ranged between 1 nmol/L and 243 nmol/L (serial 1:3 dilutions) for SK‐BR‐3 cells, and between 10 nmol/L and 2430 nmol/L for BT‐474 cells, respectively. The concentrations of the two agents spanned clinically relevant concentration ranges, including the 50% inhibitory concentration (IC50). The combination index of the two drugs was calculated using CalcuSyn software (Great Shelford, UK), as described by Chou and Talalay,( 24 ) which provides information on the nature and extent of drug interaction at the different effect levels of the two drugs used in combination. The CI values for both SK‐BR‐3 and BT‐474 cells are summarized in Table 1. The combination of bortezomib and lapatinib yielded strongly synergistic effects in SK‐BR‐3 cells (Fig. 1A), and the average CI value from IC30 to IC90 was 0.63 (P < 0.05). However, the two‐drug combination yielded relatively weak synergistic effects against the BT‐474 cells (Fig. 1B), and the average CI value from IC30 to IC90 was 0.91 (P < 0.05, Table 1). Thus, it appeared that the SK‐BR‐3 cells might be more susceptible to the combination lapatinib and bortezomib compared with the BT‐474 cells.
Table 1.
Combination effects of bortezomib and lapatinib against SK‐BR‐3 and BT‐474 cells
| Combination potency | CI values for SK‐BR‐3 cells* (mean ± SD) | CI values for BT474 cells** (mean ± SD) |
|---|---|---|
| IC30 | 0.33 ± 0.19 | 0.86 ± 0.88 |
| IC50 | 0.43 ± 0.24 | 0.88 ± 0.72 |
| IC70 | 0.59 ± 0.38 | 0.91 ± 0.69 |
| IC90 | 1.04 ± 0.96 | 0.96 ± 0.96 |
CI < 1, CI = 1 and CI > 1 indicate synergism, additive effect and antagonism, respectively. In each experiment, six concentrations were used for each drug and each combination drug. Approximated IC50 equipotency ratios were used as the drug combination ratios. IC30, IC50, IC70 and IC90 = concentration required to inhibit 30%, 50%, 70% or 90%, respectively. Representative results from three experiments. The CI values were derived from the median effect plots. The one‐sample Student’s t‐test was used to determine whether the CI values at multiple effect levels (IC30∼ IC90) were statistically significantly different from 1. * and ** indicate P < 0.05. CI, combination index; IC, inhibitory concentration.
Figure 1.
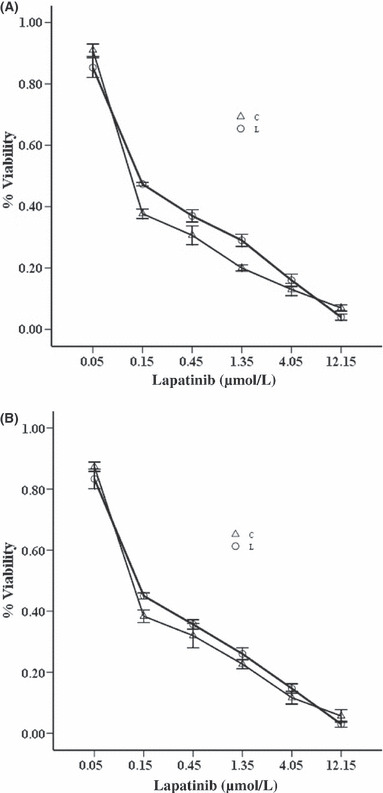
SK‐BR‐3 (A) and BT‐474 (B) cells were treated with threefold serial dilutions of lapatinib ranging from 0.05 to 12.15 μmol/L for 72 h or bortezomib ranging from 1 to 243 nmol/L for 72 h or the combination ranging from 0.05 μmol/L lapatinib and 1 nmol/L bortezomib to 12.15 μmol/L lapatinib and 243 nmol/L bortezomib for 72 h. Cell viability for the lapatinib arm (L) and the combination arm (C) was assayed using Cell Counting Kit‐8 and expressed as relative to the untreated control cells. Representative results are from at least three independent occasions.
Combining lapatinib with bortezomib enhances apoptosis in SK‐BR‐3 cells. We then exposed cells with lapatinib (0.1 μmol/L, 1 μmol/L), bortezomib (1 nmol/L, 10 nmol/L), or lapatinib in combination with bortezomib at different doses for 48 h. Apoptosis of cells was analysed by flow cytometry and measured by Annexin V staining to detect early apoptosis, and propidium iodide to detect the late stage of apoptosis. As shown, lapatinib induced 9% and 17% apoptosis at 0.1 μmol/L and 1 μmol/L for 48 h, respectively, whereas apoptosis of the SK‐BR‐3 cells treated with DMSO was only 6% (P < 0.05, Fig. 2). Similarly, treatment with bortezomib resulted in a significant increase in apoptosis, compared with the controls (Fig. 2). Importantly, combining lapatinib with bortezomib enhances apoptosis compared with either treatment alone. For example, the combination of lapatinib (1 μmol/L) and bortezomib (10 nmol/L) elicited 38% apoptosis, while lapatinib (1 μmol/L) or bortezomib (10 nmol/L) only elicited 17% or 13% apoptosis, respectively, in the SK‐BR‐3 cells (P < 0.05).
Figure 2.
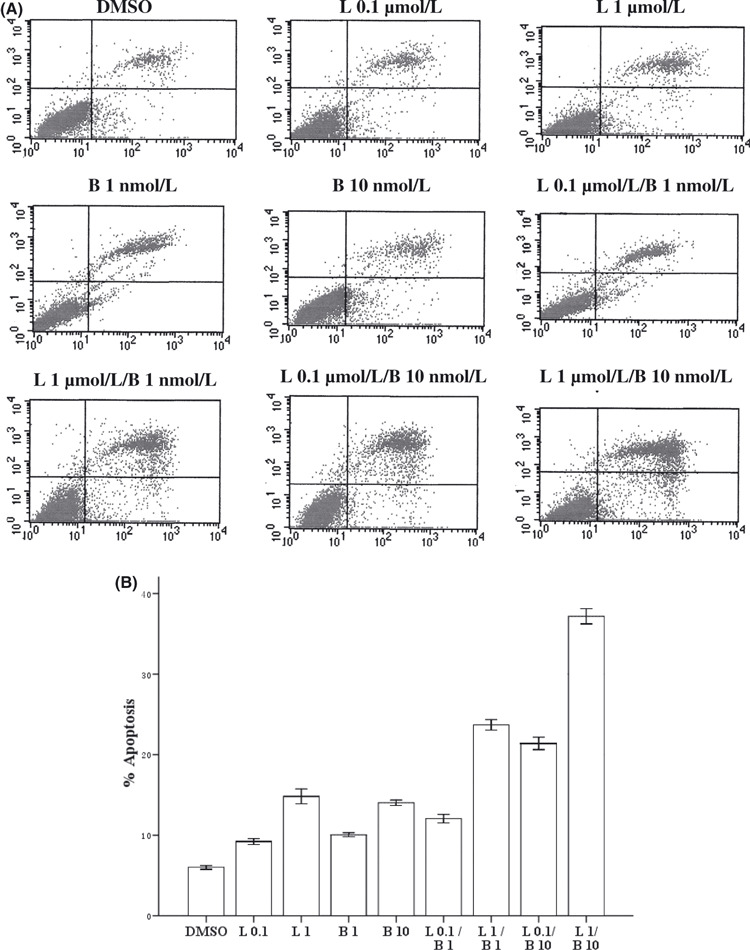
Cells were treated with lapatinib 0.1 μmol/L (L 0.1), lapatinib l μmol/L (L 1), bortezomib (1 nmol/L, B 1), bortezomib (10 nmol/L, B 10) or lapatinib combined with bortezomib for 24 h. Cells were stained with Annexin V and propidium iodide, and analyzed using flow cytometry. (A) Dot plots are displayed with Annexin V (x‐axis) and propidium iodide (y‐axis) staining. Annexin V‐positive cells are in the upper right (late apoptotic cells) and lower right quadrants (early apoptotic cells). (B) The percentage of cells staining positive for Annexin V are indicated. anova indicated a significant difference among the groups (P < 0.05).
Combining lapatinib with bortezomib reduced phosphorylation of HER2 and AKT and upregulated P27. At a concentration of 1 μmol/L for 72 h, lapatinib markedly reduced phosphorylation of HER2 protein in the SK‐BR‐3 cells, without affecting the total HER2 protein (Fig. 3A); when exposed to bortezomib for 72 h, the phosphorylated HER2 protein level also modestly decreased in a concentration‐dependent manner (Fig. 3B). Importantly, combining lapatinib with bortezomib resulted in further inhibition of phosphorylated HER2, compared with lapatinib or bortezomib alone (Fig. 3). Downstream signaling of the HER2 receptor pathway was also blocked by lapatinib and bortezomib. For instance, phosphorylation of AKT dramatically decreased in SK‐BR‐3 cells, especially when exposed to the two‐drug combination.
Figure 3.
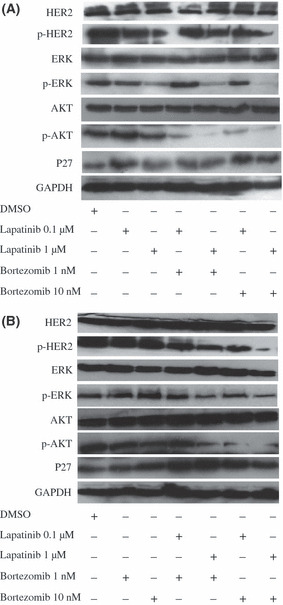
Cells were treated with lapatinib (0.1, l μmol/L; A), bortezomib (1, 10 nmol/L; B) or lapatinib (0.1, l μmol/L) combined with bortezomib (1, 10 nmol/L) for 72 h. Western blot analysis was done for phosphorylated HER2 (p‐HER2) and total HER2, AKT and p‐AKT, ERK and p‐ERK, p‐4E‐BP1 and total P27. The GAPDH served as a loading control.
We then studied the effect of lapatinib and/or bortezomib on the ERK1/2 pathway, and lapatinib reduced phosphorylation of ERK1/2 in the SK‐BR‐3 cells in a concentration‐dependent manner (Fig. 3A). In contrast, as the concentration of bortezomib increased, phosphorylation of ERK1/2 was markedly induced (Fig. 3B). To determine the interaction of lapatinib and bortezomib with ERK1/2, we exposed the SK‐BR‐3 cells to the two drugs simultaneously for 72 h. Interestingly, lapatinib, especially at a higher concentration, blocked phosphorylation of ERK1/2 induced by bortezomib (Fig. 3A). As expected, p27 was significantly induced upon treatment with lapatinib or bortezomib alone. In addition, increased levels of p27 protein were detected in the SK‐BR‐3 cells treated with the combination of lapatinib and bortezomib, compared with either treatment alone.
Bortezomib induced apoptosis in lapatinib‐resistant SK‐BR‐3 cells. To examine whether our cells were resistant to lapatinib, parental SK‐BR‐3 cells and lapatinib‐resistant pool cells derived from parental cells were treated with threefold serial dilutions of lapatinib ranging from 0.05 μmol/L to 12.15 μmol/L for 72 h. The relative resistance of pool cells to lapatinib was determined by CCK‐8 assays, compared with parental cells. Pool cells were resistant to almost any dose of lapatinib as indicated above, especially to lower doses of lapatinib (Fig. 4A). The 50% inhibitory concentration of lapatinib for parental and resistant SK‐BR‐3 cells was approximately 0.13–0.45 μmol/L, respectively. We then exposed parental and resistant cells with threefold serial dilutions of bortezomib ranging from 1 nmol/L to 243 nmol/L for 72 h. The sensitivity of lapatinib‐resistant cells to bortezomib was similar to that of parental cells (P > 0.05, Fig. 4B). To ascertain whether bortezomib would lead to apoptosis in lapatinib‐resistant SK‐BR‐3 cells, cells were treated with bortezomib and then stained with Annexin V‐FITC and propidium iodide to detect apoptosis. Bortezomib induced significant apoptosis in resistant cells (P < 0.05, Fig. 5). For example, at the concentration of 10 nmol/L for 48 h, bortezomib elicited 24% apoptosis in resistant SK‐BR‐3 cells.
Figure 4.
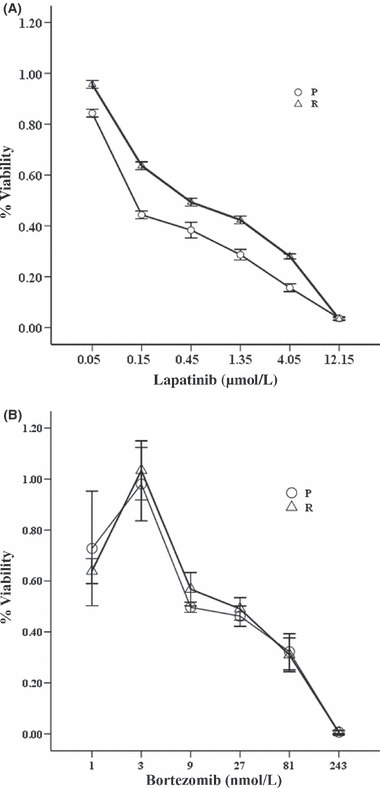
Parental (○) and resistant (△) SK‐BR‐3 cells were treated with threefold serial dilutions of (A) lapatinib ranging from 0.05 to 12.15 μmol/L for 72 h or (B) bortezomib ranging from 1 to 243 nmol/L for 72 h. Cell viability for parental and resistant SK‐BR‐3 cells was assayed using Cell Counting Kit‐8 and expressed as relative to the untreated control cells. Bortezomib reduced cell survival to a similar degree in the parental and resistant SK‐BR‐3 cells (P > 0.05). Representative results are from at least three independent occasions.
Figure 5.
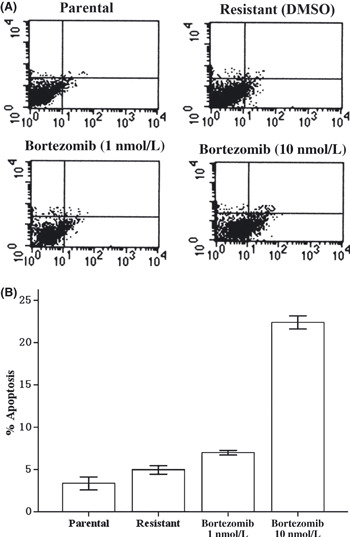
Resistant cells were treated with bortezomib at 1 or 10 nmol/L for 24 h. Cells were stained with Annexin V and propidium iodide, and analyzed by flow cytometry. (A) Dot plots are displayed with Annexin V (x‐axis) and propidium iodide (y‐axis) staining. Annexin V‐positive cells are in the upper right (late apoptotic cells) and lower right quadrants (early apoptotic cells). (B) The percentage of cells staining positive for Annexin V are indicated. Bortezomib induces dramatic apoptosis in resistant cells (P < 0.05).
Bortezomib blocked HER2 signaling and upregulated P27 in lapatinib‐resistant SK‐BR‐3 cells. In pooled SK‐BR‐3 cells resistant to 1 μmol/L lapatinib (Fig. 6B), decreased basal levels of HER2 receptor phosphorylation were observed; the total HER2 protein level was comparable between resistant and parental cells (Fig. 6A). Hence, resistance to lapatinib in our model was not associated with loss of HER2 expression. Immunoblotting demonstrated that bortezomib further reduced phosphorylation of HER2, 4E‐BP1 and AKT in a concentration‐dependent manner. Total HER2 protein receptor modestly decreased in resistant cells, and the total level of AKT remained unchanged (Fig. 6A). We were surprised to find that phospho‐ERK1/2 levels did not increase, but decreased in resistant SK‐BR‐3 cells exposed to bortezomib (Fig. 6B). As expected, p27 was markedly induced upon treatment with bortezomib in resistant cells as the concentration of bortezomib increased.
Figure 6.
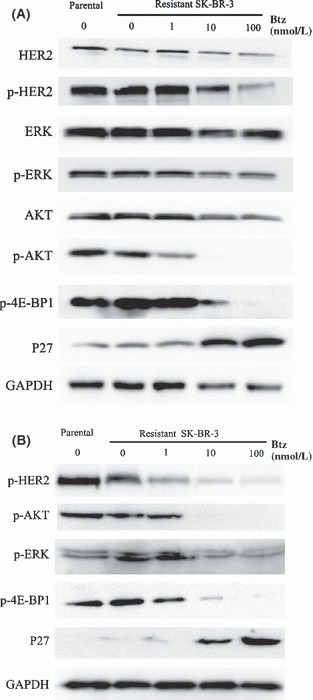
Resistant cells were treated with bortezomib at 1, 10 or 100 nmol/L for 48 h. (A) Immunoblotting was done for phosphorylated and total human epidermal receptor 2 (HER2), AKT and p‐AKT, ERK1/2 and p‐ERK1/2, p‐4E‐BP1 and total P27 in lapatinib‐resistant (0.5 μmol/L) cells. (B) Western blot analysis was done for p‐HER2, p‐AKT, p‐ERK1/2, p‐4E‐BP1 and P27 in lapatinib‐resistant (1 μmol/L) cells. GAPDH served as a loading control.
Discussion
The data presented herein suggest that there consistently was a synergistic interaction between lapatinib and bortezomib in SK‐BR‐3 and BT‐474 cells, when the CI value ranged from 0.30 to 0.90. However, at lower concentrations of bortezomib, for instance, when the CI values were <0.25, the combination was antagonistic in the breast cancer cells tested (Fig. 1). One potential explanation for this phenomenon is that lower concentrations of bortezomib might be pro‐proliferative in breast cancer cells, because several in vitro studies consistently show that lapatinib leads to growth inhibition in HER2‐overexpressing breast cancer cells in a concentration‐dependent manner.( 7 , 11 , 25 ) Recently Codony‐Servat et al. ( 17 ) reported that bortezomib promoted cell proliferation in a panel of six breast cancer cell lines at less than IC50 concentrations, which is in agreement with our observations (Fig. 6) and supports our view. More importantly, we found that bortezomib at lower doses, such as 3 nmol/L, induced apoptosis in parental and resistant SK‐BR‐3 cells (data not shown), whereas this concentration of bortezomib was obviously not growth‐inhibitory (Fig. 4). As mentioned above, the ERK MAPK signaling is a key signaling pathway involved in many cellular responses such as cell proliferation, survival and differentiation.( 26 ) Enhanced phosphorylation of the Raf/MEK/ERK1/2 pathway through bortezomib treatment might be responsible for the observed pro‐proliferative response in breast cancer cells.( 17 , 18 ) Although bortezomib does not have a growth‐inhibitory impact on breast cancer cells at lower concentrations, our results indicate that combining lapatinib with bortezomib enhanced apoptosis of SK‐BR‐3 cells. These findings suggest that each agent might be acting on independent but complementary growth‐regulating mechanisms.
The HER2 signaling transduction pathway plays a significant role in growth and survival of HER2‐overexpressing breast cancer cell lines, such as SK‐BR‐3 and BT474 cells( 27 , 28 ) Thus, we explored the underlying mechanisms of their combined effect on apoptosis by mainly focusing on the HER2 signaling pathway. Although the exact mechanisms by which lapatinib and/or bortezomib induce apoptosis of breast cancer cells is unknown, the two agents inactivate HER2 through distinct and complementary ways. Lapatinib inhibits HER2 activation by binding directly with the HER2 receptor and blocking its kinase activity, thereby preventing subsequent downstream signaling events, whereas bortezomib inactivates HER2 through the buildup of ubiquitinated and Hsp70‐associated HER2 receptor, degradation and loss of HER2 function.( 18 ) We observed that lapatinib or bortezomib inhibited HER2 phosphorylation in SK‐BR‐3 cells when administered individually (Fig. 3), which is consistent with results reported by other investigators.( 8 , 17 , 18 ) Induction of apoptosis in HER2‐overexpressing breast cancer cells depends mainly on the inhibition of HER2 phosphorylation.( 7 , 8 , 11 ) Hence, the reduction of HER2 phosphorylation might be responsible for the pro‐apoptotic effect of bortezomib in SK‐BR‐3 cells. Although bortezomib, not directed specifically against HER2, is a proteasome inhibitor in essence, the present study and work of others( 21 , 29 ) suggest that the combination of HER2‐targeted agents, such as lapatinib and bortezomib, most likely accomplishes a complete blockage of HER2 signaling for cancer therapy.
The mechanisms of acquired resistance to small molecule tyrosine kinase inhibitors are multifactorial, which might all contribute partially to acquired lapatinib resistance in breast cancer treatment.( 30 , 31 ) Xia et al. ( 30 ) developed a cell‐based model of lapatinib resistance by chronically exposing HER2‐overexpressing/estrogen receptor (ER)‐positive breast cancer cells to this agent, and confirmed that acquired resistance to lapatinib was mediated by a switch in cell survival dependence from the HER2 pathway alone to codependence on the ER and HER2 pathways. Here, we established a model of acquired resistance to lapatinib in HER2‐overexpressing/ER‐negative breast cancer cells. In terms of the clinical relevance of our resistant SK‐BR‐3 cell subline, a recent report suggests that, at the FDA‐approved dose of 1250 mg/day, the steady‐state Cmax of lapatinib found in the plasma of patients treated with this compound is 2.43 μg/mL.( 32 ) Additionally, the steady‐state Cmin of the majority of the responders ranges from 0.3 to 0.6 μg/mL.( 23 )
Increasing evidence from experimental models suggests that HER2‐overexpressing breast cancers are HER2 driven and HER2 dependent.( 33 , 34 ) A recent study by Ritter et al. ( 35 ) indicates that BT‐474 breast cancer cells resistant to trastuzumab retained HER2 gene amplification and exhibited elevated levels of EGFR/HER2 heterodimers. Furthermore, HER2 has been shown to interact with and cross‐talk to insulin‐like growth factor‐I receptor in trastuzumab‐resistant SK‐BR‐3 cells.( 36 , 37 ) These findings suggest that HER2 signaling still plays an important role in HER2‐overexpressing breast cancer cells resistant to trastuzumab. Hence, it is tempting to speculate that cells chronically challenged with lapatinib may be, at least in part, also dependent on HER2 kinase function. In our model, decreased basal levels of HER2 receptor phosphorylation were observed. Lapatinib, however, did not completely inactivate HER2 in the resistant cells. Consequently, if HER2 signaling can be effectively blocked by other agents in lapatinib‐resistant SK‐BR‐3 cells, this would produce significant antitumor effects. We found that bortezomib dramatically reduced phosphorylation of HER2, AKT and 4E‐BP1 with subsequent marked apoptosis in our resistant SK‐BR‐3 cells. Although other factors such as inhibition of NF‐κB activation can not be ruled out,( 38 ) inactivation of HER2 through inhibition of HER2 phosphorylation may also contribute to the pro‐apoptotic response of bortezomib in lapatinib‐resistant SK‐BR‐3 cells.
Taken together, the current data presented here indicate a synergistic interaction between lapatinib and bortezomib in HER2‐overexpressing breast cancer cells and provide the rationale for the clinical evaluation of these two noncross‐resistant HER2 targeted therapies. These findings also suggest that the combination of lapatinib and bortezomib may be a potentially novel approach to prevent or delay the onset of acquired resistance to lapatinib in HER2‐overxpressing/ER‐negative breast cancers.
Disclosure Statement
The authors indicate no potential conflict of interest.
Acknowledgments
The authors thank GlaxoSmithKline for providing lapatinib (GW572016 and GSK572016).
References
- 1. Slamon DJ, Clark GM, Wong SG et al. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science 1987; 235: 177–82. [DOI] [PubMed] [Google Scholar]
- 2. Vogel CL, Cobleigh MA, Tripathy D et al. Efficacy and safety of trastuzumab as a single agent in first‐line treatment of HER2‐overexpressing metastatic breast cancer. J Clin Oncol 2002; 20: 719–26. [DOI] [PubMed] [Google Scholar]
- 3. Slamon DJ, Leyland‐Jones B, Shak S et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–92. [DOI] [PubMed] [Google Scholar]
- 4. Moy B, Goss PE. Lapatinib: current status and future directions in breast cancer. Oncologist 2006; 11: 1047–57. [DOI] [PubMed] [Google Scholar]
- 5. Nelson MH, Dolder CR. Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors. Ann Pharmacother 2006; 40: 261–9. [DOI] [PubMed] [Google Scholar]
- 6. Geyer CE, Forster J, Lindquist D et al. Lapatinib plus capecitabine for HER2‐positive advanced breast cancer. N Engl J Med 2006; 355: 2733–43. [DOI] [PubMed] [Google Scholar]
- 7. Rusnak DW, Lackey K, Affleck K et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB‐2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor‐derived cell lines in vitro and in vivo . Mol Cancer Ther 2001; 1: 85–94. [PubMed] [Google Scholar]
- 8. Xia W, Mullin RJ, Keith BR et al. Anti‐tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002; 21: 6255–63. [DOI] [PubMed] [Google Scholar]
- 9. Spector NL, Xia W, Burris H et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005; 23: 2502–12. [DOI] [PubMed] [Google Scholar]
- 10. Chu I, Blackwell K, Chen S et al. The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation‐ and estrogen‐dependent gene expression in antiestrogen‐resistant breast cancer. Cancer Res 2005; 65: 18–25. [PubMed] [Google Scholar]
- 11. Konecny GE, Pegram MD, Venkatesan N et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER‐2‐overexpressing and trastuzumab‐treated breast cancer cells. Cancer Res 2006; 66: 1630–9. [DOI] [PubMed] [Google Scholar]
- 12. Gril B, Palmieri D, Bronder JL et al. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst 2008; 100: 1092–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X, Lewis MT, Huang J et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100: 672–9. [DOI] [PubMed] [Google Scholar]
- 14. Bross PF, Kane R, Farrell AT et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res 2004; 10: 3954–64. [DOI] [PubMed] [Google Scholar]
- 15. Rajkumar SV, Richardson PG, Hideshima T et al. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol 2005; 23: 630–9. [DOI] [PubMed] [Google Scholar]
- 16. Cardoso F, Ross JS, Picart MJ et al. Targeting the ubiquitinproteasome pathway in breast cancer. Clin Breast Cancer 2004; 5: 148–57. [DOI] [PubMed] [Google Scholar]
- 17. Codony‐Servat J, Tapia MA, Bosch M et al. Differential cellular and molecular effects of bortezomib, a proteasome inhibitor, in human breast cancer cells. Mol Cancer Ther 2006; 5: 665–75. [DOI] [PubMed] [Google Scholar]
- 18. Marx C, Yau C, Banwait S et al. Proteasome‐regulated ERBB2 and estrogen receptor pathways in breast cancer. Mol Pharmacol 2007; 71: 1525–34. [DOI] [PubMed] [Google Scholar]
- 19. Brown J, Von Roenn J, O’Regan RM et al. A phase II study of the proteasome inhibitor PS‐341 in patients (pts) with metastatic breast cancer (MBC). J Clin Oncol 2004; 22: abstract 546. [Google Scholar]
- 20. Yang CH, Gonzalez‐Angulo AM, Reuben JM et al. Bortezomib (VELCADE) in metastatic breast cancer: pharmacodynamics, biological effects, and prediction of clinical benefits. Ann Oncol 2006; 17: 813–17. [DOI] [PubMed] [Google Scholar]
- 21. Cardoso F, Durbecq V, Laes JF et al. Bortezomib (PS341, Velcade) increases the efficacy of trastuzumab (Herceptin) in HER2‐positive breast cancer cells in a synergistic manner. Mol Cancer Ther 2006; 5: 3042–51. [DOI] [PubMed] [Google Scholar]
- 22. Awada A, Albanell J, Canney PA et al. Bortezomib/docetaxel combination therapy in patients with anthracycline‐pretreated advanced/metastatic breast cancer: a phase I/II dose‐escalation study. Br J Cancer 2008; 98: 1500–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burris HA 3rd, Hurwitz HI, Dees EC et al. Phase I safety, pharmacokinetics, and clinical activity study of patinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol 2005; 23: 5305–13. [DOI] [PubMed] [Google Scholar]
- 24. Chou T‐C, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 25. Rusnak DW, Alligood KJ, Mullin RJ et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb®, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif 2007; 40: 580–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roberts PJ, Der CJ. Targeting the Raf‐MEK‐ERK mitogen‐activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26: 3291–310. [DOI] [PubMed] [Google Scholar]
- 27. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 28. Badache A, Gonçalves A. The ErbB2 signaling network as a target for breast cancer therapy. J Mammary Gland Biol Neoplasia 2006; 11: 13–25. [DOI] [PubMed] [Google Scholar]
- 29. Xia W, Gerard CM, Liu L et al. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti‐ErbB2 antibodies enhances apoptosis of ErbB2‐overexpressing breast cancer cells. Oncogene 2005; 24: 6213–21. [DOI] [PubMed] [Google Scholar]
- 30. Xia W, Bascus S, Hegde P et al. A model of required autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA 2006; 103: 7795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen FL, Xia W, Spector NL. Acquired resistance to small molecular ErbB2 tyrosine kinase inhibitors. Clin Cancer Res 2008; 14: 6730–4. [DOI] [PubMed] [Google Scholar]
- 32. Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther 2008; 30: 1426–47. [DOI] [PubMed] [Google Scholar]
- 33. Moasser MM. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene 2007; 26: 6577–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sergina NV, Rausch M, Wang D et al. Escape from HER‐family tyrosine kinase inhibitor therapy by the kinase‐inactive HER3. Nature 2007; 445: 437–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ritter CA, Perez‐Torres M, Rinehart C et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res 2007; 13: 4909–19. [DOI] [PubMed] [Google Scholar]
- 36. Nahta R, Yuan LXH, Zhang B et al. Insulin‐like growth factor‐I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 2005; 65: 11118–28. [DOI] [PubMed] [Google Scholar]
- 37. Nahta R, Yuan LX, Du Y et al. Lapatinib induces apoptosis in trastuzumab‐resistant breast cancer cells: effects on insulin‐like growth factor I signaling. Mol Cancer Ther 2007; 6: 667–74. [DOI] [PubMed] [Google Scholar]
- 38. Biswas DK, Dai SC, Cruz A et al. The nuclear factor kappa B (NF‐κB): a potential therapeutic target for estrogen receptor negative breast cancers. Proc Natl Acad Sci USA 2001; 98: 10386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]