

Abstract
Prostaglandin (PG) E2 promotes gastrointestinal carcinogenesis and tumor progression. We determined the correlations between pattern of expression of 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH), a catabolic enzyme for biological inactivation of PGE2, in gastric adenocarcinoma and various clinicopathological factors and patient outcome in an attempt to elucidate its biological significance. In 35 of 71 cases of gastric adenocarcinoma, expression of 15‐PGDH protein was reduced in tumor tissues. Multivariate analysis revealed reduction of 15‐PGDH expression to be an independent predictor of poor survival. The proportion of Ki67‐positive cells in 15‐PGDH‐negative adenocarcinoma was higher than that in 15‐PGDH‐positive adenocarcinoma. No differences were found in clinicopathological parameters between patients with cyclooxygenase‐2 (COX‐2)‐positive tumors and those with COX‐2 negative tumors. In an in vitro study, use of specific siRNA to silence 15‐PGDH or a specific inhibitor of 15‐PGDH enhanced cell proliferation in the gastric cancer cell line AGS, which expresses 15‐PGDH. These findings suggest that reduction of 15‐PGDH is an independent predictor of poor survival associated with enhancement of cell proliferation in gastric adenocarcinoma. (Cancer Sci 2009)
Prostaglandin (PG) E2 is a bioactive eicosanoid synthesized from arachidonic acid liberated from membrane phospholipids. PGE2 plays important roles in multiple physiological processes, including renal function, vascular homeostasis, bone remodeling, gastrointestinal function, pregnancy, and acute inflammatory responses.( 1 ) Accumulating evidence suggests that PGE2 promotes carcinogenesis and cancer progression by stimulating cell proliferation and angiogenesis and by inhibiting apoptosis.( 2 ) Previous studies of the involvement of PGE2 in gastrointestinal cancer have focused on cyclooxygenase‐2 (COX‐2), an inducible isoform of the late‐limiting enzyme for synthesis of PGs. COX‐2 is overexpressed in gastrointestinal cancer,( 3 ) and epidemiological studies have shown that chronic intake of nonsteroidal anti‐inflammatory drugs and aspirin, which inhibit COX activity, reduces the risk of colon, gastric, and esophageal cancer.( 4 , 5 , 6 , 7 ) However, the total amount of biologically active PGE2 in tumor tissue is regulated by the balance of PGE2 synthesis and degradation.
15‐hydroxyprostaglandin dehydrogenase (15‐PGDH), which catalyzes the oxidation of the 15(S)‐hydroxyl group of PGs, resulting in the production of 15‐keto‐PGs, greatly reduces the biological activity of PGE2.( 8 ) Increasing evidence has recently been obtained for the involvement of reduction of 15‐PGDH in carcinogenesis and cancer progression. Reduced expression of 15‐PGDH has been demonstrated in colorectal,( 9 ) breast,( 10 ) prostate,( 11 ) lung,( 12 ) and medullary thyroid cancer.( 13 ) Experimental studies have revealed that genetic disruption of 15‐PGDH results in increased tumorigenicity in Apc Min/+ mice, an animal model of intestinal neoplasia,( 14 ) while overexpression of 15‐PGDH by transfection with the wild‐type 15‐PGDH gene in non‐small‐cell lung carcinoma cells decreased tumor growth.( 12 ) These findings suggest that 15‐PGDH may have tumor suppressive properties.
A few clinical studies on the expression of 15‐PGDH in gastric cancer have been reported,( 15 , 16 , 17 , 18 ) although the correlations of this expression with prognosis and patient outcome have not been evaluated.
In this study, we determined the correlations between pattern of expression of 15‐PGDH protein and various clinicopathological factors and patient outcome in gastric cancer. We also examined the association of expression of 15‐PGDH with cell proliferation in both clinical samples and gastric cancer cell lines.
Materials and Methods
Patients Tumor specimens were obtained from 71 patients with gastric adenocarcinoma who underwent surgical resection from 2000 to 2003 at the Department of Surgical Oncology in Osaka City University Hospital. Forty‐nine were males and 22 were females, with a median age of 67 (range 31–87) years. None of the patients received chemotherapy or radiation therapy before surgery. Follow‐up time ranged from 1 to 72 months, with a median of 52 months. Patient characteristics are summarized in Table 1. The specimens were subjected to detailed pathologic examination, with determination of depth of invasion, nodal status, involvement or margins, and histological type of tumor. Pathological tumor staging was performed according to the American Joint Committee on Cancer TNM classification. Informed consent was obtained from every patient, and the Ethics Committee of Osaka City University Hospital approved this study.
Table 1.
Relationships between immunohistochemical expression of COX‐2 and 15‐PGDH and clinicopathological parameters
| Clinicopathological parameters | Total cases (n = 71) | Expression of 15‐PGDH | P‐values | Expression of COX‐2 | P‐values | ||
|---|---|---|---|---|---|---|---|
| Negative (n = 35) | Positive (n = 36) | Negative (n = 23) | Positive (n = 48) | ||||
| n (%) | n (%) | n (%) | n (%) | ||||
| Age | |||||||
| < 60 years | 12 | 7 (58.3) | 5 (41.7) | 0.711 | 3 (25.0) | 9 (75.0) | 0.739 |
| ≧60 years | 59 | 28 (47.5) | 31 (52.5) | 20 (33.9) | 39 (66.1) | ||
| Gender | |||||||
| Male | 49 | 24 (49.0) | 25 (51.0) | 0.937 | 17 (34.7) | 32 (65.3) | 0.537 |
| Female | 22 | 11 (50.0) | 11 (50.0) | 6 (27.3) | 16 (72.7) | ||
| Differentiation | |||||||
| Tubular | 39 | 14 (35.9) | 25 (64.1) | 0.013 | 9 (23.1) | 30 (76.9) | 0.204 |
| Poorly | 32 | 21 (65.6) | 11 (34.4) | 14 (43.8) | 18 (56.3) | ||
| Depth of invasion | |||||||
| mp | 13 | 6 (46.2) | 7 (53.8) | 0.269 | 6 (46.2) | 7 (53.8) | 0.775 |
| ss | 16 | 4 (25.0) | 12 (75.0) | 5 (31.3) | 11 (68.8) | ||
| se | 41 | 24 (58.5) | 17 (41.5) | 11 (26.8) | 30 (73.2) | ||
| si | 1 | 1 (100.0) | 0 (0.0) | 1 (100.0) | 0 (0.0) | ||
| Lymph node metastasis | |||||||
| Negative | 69 | 34 (49.3) | 35 (50.7) | 0.984 | 22 (31.9) | 47 (68.1) | 0.546 |
| Positive | 2 | 1 (50.0) | 1 (50.0) | 1 (50.0) | 1 (50.0) | ||
| Disease stage | |||||||
| I | 16 | 3 (18.8) | 13 (81.3) | 0.025 | 6 (37.5) | 10 (62.5) | 0.79 |
| II | 15 | 7 (46.7) | 8 (53.3) | 4 (26.7) | 11 (73.3) | ||
| III | 19 | 13 (68.4) | 6 (31.6) | 5 (26.3) | 14 (73.7) | ||
| IV | 21 | 12 (57.1) | 9 (42.9) | 8 (38.1) | 13 (61.9) | ||
15‐PGDH, 15‐hydroxyprostaglandin dehydrogenase; COX‐2, cyclooxygenase‐2; poorly, poorly differentiated adenocarcinoma; tubular, tubular adenocarcinoma.
Antibodies Antihuman COX‐2 mouse‐monoclonal antibody and antihuman 15‐PGDH rabbit‐polyclonal antibody were purchased from Cayman Chemical (Ann Arbor, MI, USA). Antihuman Ki67 mouse‐monoclonal antibody and 5‐Bromo‐2′‐deoxyuridine (BrdU) antibody were purchased from DakoCytomation (Kyoto, Japan). Antihuman COX‐2 mouse‐monoclonal antibody, antihuman 15‐PGDH rabbit‐polyclonal antibody, and antihuman Ki67 mouse‐monoclonal antibody were diluted to 1:100 with AntibodyDiluent (DakoCytomation) when used for immunohistochemical staining and double‐immunofluorescence staining. Donkey antimouse IgG labeled with AlexaFluor 594 and donkey antirabbit IgG labeled with AlexaFluor 488 were purchased from Invitrogen (Carlsbad, CA, USA). Each antibody was diluted to 1:200 with phosphate‐buffered saline. Antihuman 15‐PGDH goat polyclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) for use in Western blotting.
Immunohistochemistry Immunohistochemical staining for 15‐PGDH, COX‐2, and Ki67 was performed using the EnVision+ system (DakoCytomation). Four‐μm‐thick tissue sections from formalin‐fixed, paraffin‐embedded tissue specimens were incubated in Target Retrieval Solution (pH 6.0; DakoCytomation) heated at 95°C for 40 min. After endogenous peroxidase activity was quenched with 3% hydrogen peroxide, the sections were treated with ProteinBlock (DakoCytomation). They were incubated with primary antibody overnight at 4°C. The sections were then incubated with peroxidase‐labeled polymer conjugated with goat antimouse or antirabbit immunoglobulins for 30 min. The sections were finally treated with 0.03% 3,3′‐diaminobenzedine (Wako Pure Chemical Industries, Osaka, Japan) containing 0.005% hydrogen peroxide. Counterstaining was performed with Mayer’s hematoxylin.
To evaluate co‐localization of 15‐PGDH protein with Ki67 protein, double labeling by immunofluorescence was performed according to our previous procedure.( 19 ) In brief, sections were incubated overnight with anti‐15‐PGDH antibody and anti‐Ki67 antibody. Primary antibody were reacted with donkey antimouse IgG labeled with Alexa Fluor 594 (Invitrogen) for detection of Ki67 and donkey antirabbit IgG labeled with Alexa Fluor 488 (Invitrogen) for detection of 15‐PGDH. The sections were examined with a confocal microscope.
Scoring system For assessment of the patterns of expression of 15‐PGDH and COX‐2, staining intensity and percentage stained tumor area were determined. Staining intensity was scored as 0 (absent), 1 (weak), 2 (moderate), or 3 (strong). Percentage stained tumor area was estimated (0, none; 1, <10%; 2, 10–50%; 3, >50% of total tumor area). The scores for intensity of staining and scores for percentage stained area were multiplied, and results were classified into four groups: absent (0), weak (1–3), moderate (4–6), and strong (7–9) expression. Tumors exhibiting moderate or strong expression of 15‐PGDH or COX‐2 were considered positive, while those exhibiting absent or weak expression of 15‐PGDH or COX‐2 were considered negative.
For assessment of cell proliferation in tumor tissue, numbers of Ki67‐positive tumor cells and of all tumor cells in five randomly selected representative tumor fields containing at least 250 tumor cells were counted in each section. Ki67 labeling index was expressed as the number of Ki67‐positive tumor cells in a total of 1000 tumor cells.
For analysis of the association of cell proliferation in tumors and cumulative survival rates, the patients were divided into two groups according to the Ki67 labeling index: ≥500 and <500; this cut‐off point corresponded to the 25% percentile, which is optimal for assessment of cell proliferation for highly proliferative tumors( 20 ) The former was considered to have high Ki67 expression, while the latter was considered to have low Ki67 expression, respectively.
Cell lines and cell culture Human gastric carcinoma cell lines AGS and NCI‐N87 (American Type Culture Collection, Manassas, MD, USA) and MKN7, MKN45, and NUGC3 (RIKEN Bio Resource Center, Tsukuba, Japan) were routinely cultured in RPMI‐1640 (Wako Pure Chemical Industries) containing 10% heat‐inactivated fetal bovine serum (FBS).
Western blotting Gastric cancer cells were harvested and lysed on ice with lysis buffer containing 0.5% NP‐40, 40 mm Tris HCl (pH 8.0), 120 mm NaCl, and a protease cocktail inhibitor (Complete Mini, Pierce, Rockford, IL, USA). After centrifugation, the supernatant was collected and subjected to SDS‐PAGE followed by Western blotting according to our previous procedure.( 21 ) In brief, proteins were denatured with SDS sample buffer and subjected to SDS–polyacrylamide gel electrophoresis, and then transferred to a PVDF membrane. Membranes were then blocked with blocking buffer containing 5% skim‐milk and incubated with antihuman 15‐PGDH antibody diluted to 1:250 overnight at 4°C. The bound antigen–antibody complexes were detected with antigoat IgG‐HRP using enhanced chemiluminescence in accordance with the manufacturer’s instructions (Amersham, Arlington Heights, IL, USA).
Determination of expression of 15‐PGDH mRNA in gastric cancer cell lines by real‐time quantitative RT‐PCR Total RNA was isolated from gastric cancer cell lines using an ISOGEN kit (Nippon Gene, Tokyo, Japan) according to the manufacturer’s protocol. After precipitation, the RNA was resuspended in RNase‐free Tris‐HCl EDTA buffer. PCR primers and TaqMan probes for human 15‐PGDH were designed using Primer Express, a software program (PE Applied Biosystems, Foster City, CA, USA). For human 15‐PGDH, the sense primer was 5′‐AAGCAAAATGGAGGTGAAGGC‐3′ and the antisense primer was 5′‐TGGCATTCAGTCTCACACCAC‐3′. Real‐time quantitative RT‐PCR analyses were performed using an ABI Prism 7700 Sequence Detection System instrument and software (PE Applied Biosystems).
The reaction mixture was prepared according to the manufacturer’s protocol using the Platinum qRT‐PCR ThermoScript One‐Step System (Invitrogen). Thermal cycling conditions were 50°C for 15 min and 95°C for 2 min, followed by 40 cycles of amplification at 95°C for 15 s and 60°C for 30 s. Total RNA was subjected to real‐time quantitative RT‐PCR for measurement of target genes and GAPDH as an internal standard using TaqMan GAPDH control reagents (PE Applied Biosystems). Expression of mRNA for 15‐PGDH in gastric cancer cell lines was standardized to GAPDH mRNA.
Silencing of 15‐PGDH gene expression AGS and MKN7 cells were seeded at a density of 1.0 × 105 cells/mL in RPMI‐1640 containing 10% FBS and cultured overnight. Medium was then removed and replaced with fresh RPMI‐1640 without FBS. After 24‐h incubation, 15‐PGDH‐specific siRNA or negative control siRNA was transfected according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA).
Assessment of cell proliferation by Water‐soluble Tetrazolium Salt (WST‐1) assay AGS and MKN7 cells were seeded in 96‐well plates at a density of 1.0 × 105 cells /mL in 150 μL of RPMI‐1640 containing 10% FBS and cultured for 24 h. Medium was then removed and replaced with 150 μL of fresh RPMI‐1640 without FBS. After 24‐h incubation, addition or not of PGE2 at 1 or 10 μm (Sigma, St. Louis, MO, USA), 16,16‐dimethyl PGE2 at 1 or 10 nm (Sigma), or 10 μm of CAY10397, a specific 15‐PGDH inhibitor (Cayman Chemical, Ann Arbor, MI, USA), was performed and incubation continued for 24 h. Finally, 10 μL of WST‐1 (Roche, Mannheim, Germany) was added to the medium for the last 2 h of incubation. The absorbance of the formazan product formed was detected at 450 nm using a 96‐well spectrophotometric plate reader. In another series of experiments, AGS and MKN7 cells were transfected with 15‐PGDH‐specific siRNA, and at 24 h after transfection cell proliferation was determined by WST‐1 assay. Absorbance in the control cells was considered 100%. Eight experiments were performed for each study group.
Assessment of cell proliferation assay by BrdU staining AGS cells were cultured in LAB‐TEK chamber slide glasses (Nalge Nunc International, Rochester, NY, USA) for 24 h, and medium was then removed and replaced with fresh RPMI‐1640 without FBS. After 24 h incubation, addition or not of 10 μm of CAY10397 (Cayman Chemical) was performed. To label the cells in the DNA synthetic phase, 1 mm of BrdU (Sigma) was added to the medium for the last 1 h of incubation. BrdU staining was performed according to our previous report.( 22 ) In brief, cells were fixed in 4% paraformaldehyde followed by cold acetone. Endogenous peroxidase was quenched using 3% H2O2 for 20 min, and slides were then incubated in 4N HCl for 20 min and rinsed with 0.1M sodium tetraborate buffer (pH 8.5) for 5 min to neutralize acid. After washing, slides were incubated with monoclonal anti‐BrdU antibody (1:100 dilution) overnight at 4°C. Immunohistochemical staining for BrdU was performed using the EnVision+ system (DakoCytomation). Finally, counterstaining of nuclei was performed using Mayer’s hematoxylin. The total number of BrdU‐labeled cells and total number of cells were counted in six fields in each experiment, with four experiments performed for each study group.
Statistical analysis Statistical analysis of the relationships between expression of 15‐PGDH and COX‐2 and clinicopathological parameters was performed using the χ2‐test for independence. Cumulative survival rates stratified by disease stage, Ki67 labeling index, and COX‐2 or 15‐PGDH immunoreactivity were calculated by the Kaplan–Meier method, and the significance of differences in them between groups was determined using the log‐rank test. Univariate and multivariate survival analyses were performed with Cox’s proportional hazards model. In the in vitro study, between‐group differences were examined using the nonparametric Mann‐Whitney U‐test. Values are expressed as means ± SD, with findings of P < 0.05 considered significant.
Results
Expression of 15‐PGDH and COX‐2 in gastric cancer tissue. Immunoreactivity for 15‐PGDH protein was observed mainly in the cytoplasm of epithelial cells and inflammatory cells in the lamina propria in noncancerous epithelium (Fig. 1a,b). Of the 71 cases, 36 (50.7%) had 15‐PGDH‐positive cancerous tissue. In 15‐PGDH‐positive cases, immunoreactivity for 15‐PGDH protein was observed in the cytoplasm of cancer cells as well as noncancerous epithelial cells (Fig. 1c–f).
Figure 1.
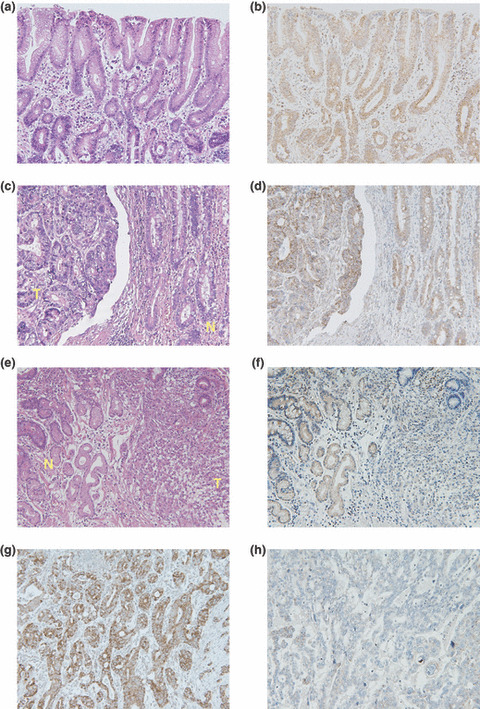
Expression of 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH) and cyclooxygenase‐2 (COX‐2) protein in gastric adenocarcinoma. (a,b) In noncancerous gastric epithelial cells, immunoreactivity for 15‐PGDH protein was observed mainly in the cytoplasm of epithelial cells and inflammatory cells in the lamina propria. (c,d) In case immunoreactivity for 15‐PGDH was observed in tumor tissue, 15‐PGDH protein was also observed in cytoplasm in cancer cells. (e,f) Immunoreactivity for 15‐PGDH protein was very weak in some cancer tissues. (a, c, e) H&E staining; (b, d, f) immunostaining for 15‐PGDH. N, noncancerous epithelium; T, tumor tissue. (g) COX‐2 protein was observed in cancer cells. (h) Immunoreactivity for COX‐2 protein was very weak in some cancer tissues (original magnification, ×200).
Of the 71 cases, 48 (67.6%) had COX‐2‐positive cancerous tissue. In adenocarcinoma cells, COX‐2 protein was located in the cytoplasm (Fig. 1g,h).
Relationships between immunohistochemical expression of 15‐PGDH and COX‐2 and clinicopathological parameters in gastric cancer The percentage of 15‐PGDH‐positive tumors was significantly higher in tubular adenocarcinoma than in poorly differentiated adenocarcinoma (35.9%vs 65.6%; P = 0.013). There was significant difference in pattern of expression of 15‐PGDH between the pathological tumor stages (Table 1).
In contrast to 15‐PGDH expression, there were no significant relationships between expression of COX‐2 and any clinicopathological parameters examined (Table 1).
Univariate and multivariate analyses of prognosis of patients with advanced gastric cancer On univariate analysis, disease stage, and 15‐PGDH immunoreactivity were prognostic factors significantly influencing survival (Table 2).
Table 2.
Determination of predictive factors for long‐term survival of gastric adenocarcinoma patients with curative surgical resection by univariate analysis
| Valuables | n | Relative risk (95% CI) | P‐values |
|---|---|---|---|
| Age | |||
| < 60 years | 12 | 1 | |
| ≧60 years | 59 | 1.00 (0.97–1.04) | NS |
| Gender | |||
| Female | 22 | 1 | |
| Male | 49 | 1.18 (0.54–2.58) | NS |
| Histological type | |||
| Tubular | 39 | 1 | |
| Poorly | 32 | 1.38 (0.68–2.79) | NS |
| Gastric cancer stage | |||
| Stage I | 16 | 1 | |
| Stage II | 15 | 1.16 (0.07–18.46) | NS |
| Stage III | 19 | 13.10 (1.67–102.92) | <0.01 |
| Stage IV | 21 | 49.26 (6.40–379.41) | <0.01 |
| Expression of 15‐PGDH in tumor tissue | |||
| Positive | 36 | 1 | |
| Negative | 35 | 3.50 (1.59–6.63) | 0.02 |
| Expression of COX‐2 in tumor tissue | |||
| Positive | 48 | 1 | |
| Negative | 23 | 1.69 (0.81–3.52) | NS |
15‐PGDH, 15‐hydroxyprostaglandin dehydrogenase; CI, confidence interval; COX‐2, cyclooxygenase‐2; NS, not significant; poorly, poorly differentiated adenocarcinoma; tubular, tubular adenocarcinoma.
On multivariate analysis using Cox’s proportional hazards model, disease stage and expression of 15‐PGDH were independent predictive factors for long‐term survival (Table 3).
Table 3.
Determination of predictive factors for long‐term survival of gastric adenocarcinoma patients with curative surgical resection by multivariate analysis
| Valuables | Relative risk (95% CI) | P‐values |
|---|---|---|
| Gastric cancer stage | ||
| Stage I | 1 | |
| Stage II | 0.94 (0.58–16.10) | NS |
| Stage III | 8.75 (1.09–70.60) | 0.04 |
| Stage IV | 42.60 (5.42–334.35) | <0.01 |
| Histological staining of 15‐PGDH | ||
| Positive | 1 | |
| Negative | 2.81 (1.21–6.56) | 0.02 |
15‐PGDH, 15‐hydroxyprostaglandin dehydrogenase; CI, confidence interval; COX‐2, cyclooxygenase‐2; NS, not significant.
On univariate analyses, no significant differences in any clinicopathological parameters were found between patients with COX‐2‐positive tumors and those with COX‐2‐negative tumors (Table 2).
Cumulative survival rate of patients with advanced gastric cancer by expression of 15‐PGDH, expression of COX‐2, Ki67 labeling index, and disease stage There was significant difference in survival among patients with pathological tumor stages (Fig. 2a). Cumulative survival was significantly shorter for patients with 15‐PGDH‐negative tumors than for those with 15‐PGDH‐positive tumors (Fig. 2b). There was no significant difference in survival rate between patients with COX‐2‐positive tumors and those with COX‐2‐negative tumors (Fig. 2c). By subgroup analysis according to the pattern of expression of COX‐2 in tumor tissue, in patients with COX‐2‐positive tumors cumulative survival tended to be shorter for patients with 15‐PGDH‐negative tumors than for those with 15‐PGDH‐positive tumors, although this was statistically not significant (P = 0.062) (Fig. 2d,e).
Figure 2.
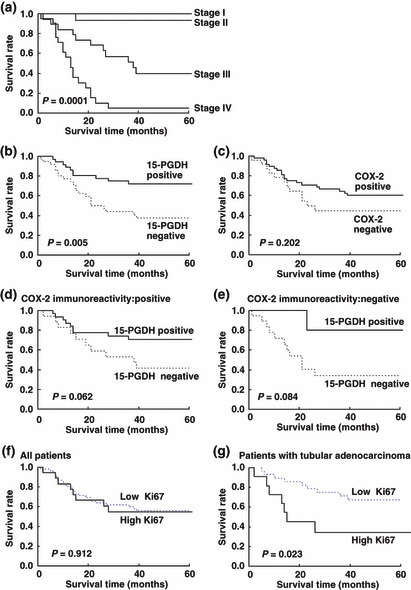
Kaplan–Meier analysis of survival according to (a) disease stage, (b) 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH) expression, (c) cyclooxygenase‐2 (COX‐2) expression, (d) 15‐PGDH expression in a subgroup of patients with COX‐2‐positive tumors, and (e) in a subgroup of patients with COX‐2‐negative tumors. (f) Ki67 labeling index in all patients and (g) Ki67 labeling index in a subgroup of patients with tubular adenocarcinoma.
Kaplan–Meier analyses of all patients revealed that high Ki67 expression was not associated with an increased probability of gastric cancer–specific mortality (Fig 2f). Regarding histological subtypes, in the subgroup of patients with tubular adenocarcinoma, Ki67 was significantly correlated with cancer‐specific mortality (Fig 2g), with risk ratio of 3.07 (95% CI, 1.13–8.37) by univariate analysis. In multivariable Cox proportional hazard regression analyses of patients with tubular adenocarcinoma, Ki67 was not an independent prognostic factor associated with cancer‐specific mortality (data not shown).
Distribution of 15‐PGDH protein and Ki67‐positive cells in gastric cancer Double‐immunofluorescence staining for 15‐PGDH and Ki67 revealed that expression of 15‐PGDH protein was decreased in cancer tissue compared with noncancerous mucosa, while the proportion of Ki67‐positive cells was much higher in cancer tissue than in noncancerous mucosa (Fig. 3a–d). In tumors, the number of Ki67‐positive cells was larger in areas exhibiting negative immunoreactivity for 15‐PGDH than in areas exhibiting positive immunoreactivity for 15‐PGDH (Fig. 3e–h).
Figure 3.
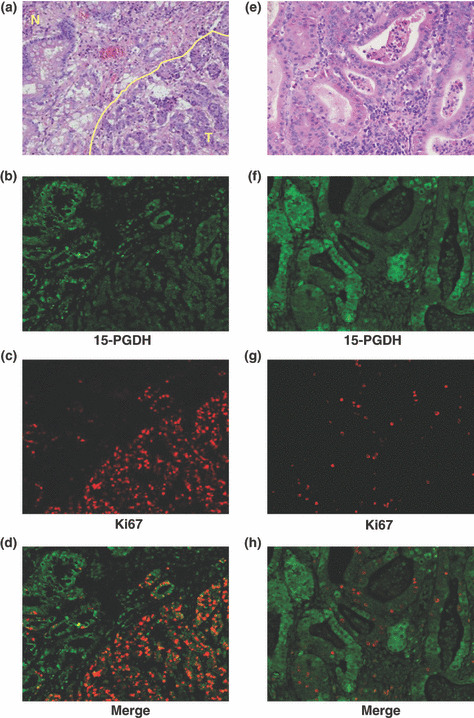
Distributions of 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH) protein‐ and Ki67‐positive cells in cancer tissue. (a) H&E staining of noncancerous mucosa (N, noncancerous epithelium, upper left) and gastric cancer tissue (T, tumor tissue, lower right). (b) 15‐PGDH protein (green area). (c) Ki67‐positive cells (red dots). (d) Merged image of Fig. 2(b,c). (e) H&E staining of tumor tissue. (f) Immunoreactivity for 15‐PGDH protein was observed in tumor tissue (green area). (g) Ki67‐positive cells (red dots) in tumor tissue. (h) Merged image of Fig. 2(e,f). Ki67‐positive cells show negative or very weak immunoreactivity for 15‐PGDH.
Relationships between Ki67 labeling index and expression of 15‐PGDH and COX‐2 in gastric cancer The Ki67 labeling index in the group with 15‐PGDH‐negative tumors was significantly higher than that in the group with 15‐PGDH‐positive tumors (Fig. 4a). In contrast, there was no difference in Ki67 labeling index between the group with COX‐2‐positive tumors and that with COX‐2‐negative tumors (Fig. 4b). Combined analysis of the pattern of expression of 15‐PGDH and COX‐2 showed that either in the group with COX‐2‐positive tumors or in the group with COX‐2‐negative tumors, the Ki67 labeling index in the subgroup with 15‐PGDH‐negative tumors was significantly higher than that in the subgroup with 15‐PGDH‐positive tumors (Fig. 4c).
Figure 4.
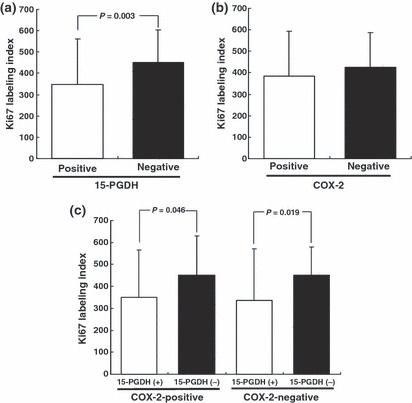
Ki67 labeling index in gastric adenocarcinoma according to expression of (a) 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH), (b) cyclooxygenase‐2 (COX‐2), and (c) expression of 15‐PGDH in subgroups of patients with COX‐2‐positive and ‐negative tumors. Values are the mean ± SD.
Expression of 15‐PGDH mRNA and protein in gastric cancer cell lines As shown in Figure 5, 15‐PGDH mRNA was expressed in AGS and MKN45, but was undetectable in MKN7, NUGC3, and NCI‐N87 cells by real‐time RT‐PCR (Fig. 5a). Consistent with this, 15‐PGDH protein was also expressed in AGS and MKN45 cells, but was not detected in MKN7, NUGC3, or NCI‐N87 cells by Western blotting (Fig. 5b).
Figure 5.
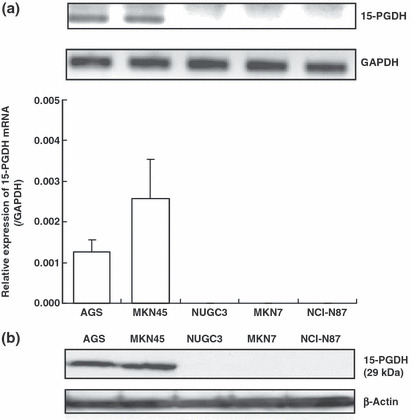
Expression of 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH) mRNA and protein in gastric adenocarcinoma cell lines detected by (a) real‐time RT‐PCR and (b) Western blotting.
Effects of PGE2, 16,16‐dimethyl PGE2, CAY10397, and 15‐PGDH‐specific siRNA on proliferation of gastric cancer cells Because AGS cells constitutively expressed 15‐PGDH mRNA and protein, we examined the effects of inhibition and reduction of 15‐PGDH on proliferation of AGS cells. Treatment with PGE2 did not enhance cell proliferation (Fig. 6a). In contrast, treatment with 16,16‐dimethyl PGE2, which is not catabolized by 15‐PGDH, enhanced cell proliferation (Fig. 6b).
Figure 6.
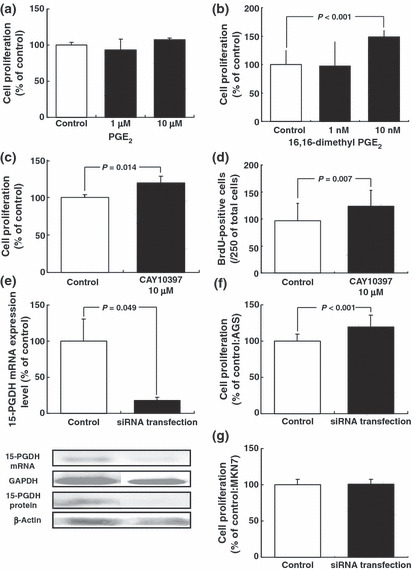
Involvement of 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH) in proliferation in gastric adenocarcinoma cells. (a) Effects of prostaglandin (PG) E2 on proliferation of AGS cells. (b) Effects of 16, 16‐dimethyl PGE2 on proliferation of AGS cells. Cell proliferation was estimated by WST‐1 cell proliferation assay. (c) Effect of 15‐PGDH inhibitor (CAY 10397) on proliferation of AGS cells. Cell proliferation was estimated by WST‐1 cell proliferation assay. (d) Effect of 15‐PGDH inhibitor (CAY 10397) on proliferation of AGS cells. Cell proliferation was estimated by BrdU staining. (e) AGS cells were subjected to Western blotting and real‐time RT‐PCR 24 h after transfection with 15‐PGDH‐specific siRNA. (f) Effect of transfection with 15‐PGDH‐specific siRNA on proliferation of AGS cells. (g) Effect of transfection with 15‐PGDH‐specific siRNA on proliferation of MKN7 cells. Cell proliferation was estimated by WST‐1 cell proliferation assay. Values are the mean ± SD.
Inhibition of 15‐PGDH by treatment with CAY10397 enhanced cell proliferation (Fig. 6c). Because the results of the WST‐1 assay reflect rates of metabolism in the cytoplasm as well as cell proliferation, BrdU incorporation in the nucleus was also determined to assess DNA synthesis. Consistent with the results of WST‐1 assay, treatment of AGS cells with CAY10397 increased the number of cells with BrdU incorporation (Fig. 6d).
Transfection with 15‐PGDH‐specific siRNA markedly reduced the expression of 15‐PGDH mRNA and protein (Fig. 6e) and enhanced the proliferation of AGS cells (Fig. 6f). In contrast, transfection with 15‐PGDH‐specific siRNA did not affect the cell proliferation of MKN7 cells, which exhibited no expression of 15‐PGDH (Fig 6g).
Discussion
In the present study, we found that 15‐PGDH expression was reduced in half of the gastric adenocarcinomas examined, and that reduction of 15‐PGDH protein expression was correlated with differentiation, disease stage, and prognosis of gastric cancer. We also showed that reduced expression of 15‐PGDH is associated with enhanced cancer cell proliferation in tumor tissue, as also demonstrated in an experimental study on a gastric cancer cell line by silencing of gene expression or enzymatic inhibition of 15‐PGDH. Together, our findings suggest that reduction of 15‐PGDH is a critical step in the acquisition of aggressive phenotype and poor prognosis in gastric cancer via enhancement of tumor cell proliferation.
PGE2 is an important chemical mediator in gastrointestinal carcinogenesis and tumor progression. PGE2 levels are elevated in intestinal adenoma and colon cancer tissues.( 23 , 24 ) In an experimental study of an animal model of intestinal neoplasia, the antitumorigenic effect of NSAIDs was reversed by exogenous administration of PGE2.( 25 ) These findings indicate that the amount of PGE2 in tumor tissue is the principal determinant of promotion of carcinogenesis and tumor progression by PGE2.
Although evidence is accumulating that PGE2 is involved in gastrointestinal carcinogenesis and tumor progression, the mechanisms responsible for modulation of the production and degradation of PGE2 in tumor tissue are not completely understood. The roles played by COX in the involvement of PGs in tumor biology have been extensively investigated. In addition to COX, the concentration of biologically active PGE2 is determined not only by its synthetic enzyme but its catabolic enzyme, 15‐PGDH.( 8 ) Recent studies have shown that reduction of 15‐PGDH occurs in association with aggressive phenotype and unfavorable prognostic factors in breast cancer,( 10 ) prostate cancer,( 11 ) lung cancer,( 12 ) colorectal cancer( 9 , 14 ) and medullary thyroid cancer.( 13 ) Genetic silencing by methylation or histone deacetylation of 15‐PGDH promoter( 8 , 10 , 26 )and activation of the epidermal growth factor receptor signaling pathway ( 27 , 28 )have been proposed as mechanisms by which expression of 15‐PGDH might be reduced in cancer. Experimental studies have demonstrated the involvement of 15‐PGDH in carcinogenesis and cancer progression. In breast, colon, and lung cancer cells, overexpression of 15‐PGDH by transfection with plasmid encoding 15‐PGDH reduces tumorigenicity, while silencing of 15‐PGDH using siRNA enhanced the growth of breast cancer.( 10 ) In addition, 15‐PGDH gene knockout markedly increases the number of colon tumors arising in the Apc Min/+ mouse model.( 14 ) These findings thus suggest that 15‐PGDH may function as a tumor suppressor.
Previously, it has been shown that 65 to 80% of gastric cancers exhibit down‐regulation of 15‐PGDH protein expression as detected by immunohistochemistry. ( 15 , 17 , 18 ) Although in one report 15‐PGDH protein expression was lost only in 10% of the gastric cancer samples,( 16 ) it may be possible that immunoreactivity for 15‐PGDH in gastric cancer is overestimated, because in the study tumors were interpreted as positive when at least weak to moderate cytoplasmic staining was seen by immunohistochemistry.
In regard to prognostic significance of expression of 15‐PGDH in gastric cancer, previous studies have yielded conflicting results. Liu et al. ( 15 ) and Jang et al. ( 17 ) reported that expression of 15‐PGDH is reduced and correlated with tumor differentiation, lymph node metastasis, and clinical stage in gastric cancer. Similar findings were obtained in a clinicopathological analysis of breast cancer by status of expression of 15‐PGDH( 10 ) and the present study. However, Thiel et al. reported that no association with 15‐PGDH immunostaining in gastric cancer and clinicopathologic parameter or prognosis was found.( 18 ) The reason why such a discrepancy exists is possibly because of genetic background difference and/or influence of different environmental factors and status of Helicobacter pylori infection. The present study clearly demonstrated that reduction of 15‐PGDH expression is positively correlated with disease stage and differentiation. Univariate and multivariate analyses revealed that reduction of 15‐PGDH expression in gastric cancer is associated with poorer prognosis than presence of such expression, and is an independent prognostic factor. These findings suggest that status of expression of 15‐PGDH may be useful as a prognostic biomarker in gastric cancer.
Several previous studies of the expression of COX‐2 in gastric cancer have shown a high frequency (43–100%) of COX‐2 immunoreactivity in gastric cancer.( 29 ) The correlations between COX‐2 expression and various clinicopathological characteristics and patient outcome in gastric cancer have been controversial. ( 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 ) In the present study, overexpression of COX‐2 was observed in 68% of the cases examined, and correlated with none of a series of clinicopathological features and patient outcome. Interestingly, in patients with COX‐2‐positive tumors, cumulative survival tended to be shorter for patients with 15‐PGDH‐negative tumors than for those with 15‐PGDH‐positive tumors, and the Ki67 labeling index in COX‐2‐overexpressing gastric adenocarcinomas negative for 15‐PGDH expression was higher than that in COX‐2‐overexpressing gastric adenocarcinomas positive for expression of 15‐PGDH. These findings suggest that reduction of 15‐PGDH enhances tumor progression and aggressive phenotype in cooperation with overexpression of COX‐2 via increase in amount of bioactive PGE2 in gastric adenocarcinoma.
Enhancement of cell proliferation is essential for the growth of solid tumors and affects the clinical course. One possible mechanism by which PGE2 promotes carcinogenesis is enhancement of cancer cell proliferation.( 2 ) Previous studies have shown that Ki67 index is correlated with poor prognosis in some types of cancer,( 38 , 39 , 40 , 41 , 42 , 43 ) although in gastric cancer immunostaining with Ki67 has limited independent value for predicting prognosis, possibly because the prognostic value of Ki67 labeling index in gastric cancer may vary depending on histological type.( 44 ) Our present study showed that high value of Ki67 labeling index was not associated with an increased probability of gastric cancer–specific mortality in all patients, and in the subgroup of patients with tubular adenocarcinoma Ki67 was associated with cancer‐specific mortality, although it was not independent prognostic factor for poor survival by multivariate analysis. In regard to 15‐PGDH, reduction of 15‐PGDH is associated with enhanced tumor cell proliferation in gastric adenocarcinoma. Consistent with the findings of the present immunohistochemical examination of human gastric carcinoma tissue, an in vitro study clearly demonstrated that inhibition of 15‐PGDH and silencing of gene expression of 15‐PGDH resulted in enhanced cell proliferation. Moreover, it is reported that overexpression of 15‐PGDH by transfection with plasmid encoding 15‐PGDH suppresses cell growth in glioblastoma( 45 ) and a breast cancer cell line.( 10 ) Taken together, these findings suggest that increase in biologically active PGE2 in tumor tissue via reduction of 15‐PGDH results in enhancement of tumor cell proliferation.
In conclusion, our findings showed that reduction of 15‐PGDH is an independent prognostic factor in gastric cancer. In addition, reduction of 15‐PGDH was found to enhance cell proliferation in gastric adenocarcinoma. Evaluation of the pattern of expression of 15‐PGDH in tumor tissues may be useful as a diagnostic or prognostic marker for gastric adenocarcinoma. Our findings also suggest that up‐regulation or recovery of expression of 15‐PGDH could be targeted for treatment and chemoprevention of gastric adenocarcinoma.
Acknowledgments
This study was supported by a Grant‐in‐Aid from the Ministry of Education, Science, and Culture of Japan.
References
- 1. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev 1999; 79: 1193–226. [DOI] [PubMed] [Google Scholar]
- 2. Wang D, Dubois RN. Prostaglandins and cancer. Gut 2006; 55: 115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams C, Shattuck‐Brandt RL, DuBois RN. The role of COX‐2 in intestinal cancer. Ann N Y Acad Sci 1999; 889: 72–83. [DOI] [PubMed] [Google Scholar]
- 4. Farrow DC, Vaughan TL, Hansten PD et al. Use of aspirin and other nonsteroidal anti‐inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev 1998; 7: 97–102. [PubMed] [Google Scholar]
- 5. Giovannucci E, Egan KM, Hunter DJ et al. Aspirin and the risk of colorectal cancer in women. N Engl J Med 1995; 333: 609–14. [DOI] [PubMed] [Google Scholar]
- 6. Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti‐inflammatory drugs. Adv Pharmacol 1997; 39: 1–20. [DOI] [PubMed] [Google Scholar]
- 7. Thun MJ. Aspirin, NSAIDs, and digestive tract cancers. Cancer Metastasis Rev 1994; 13: 269–77. [DOI] [PubMed] [Google Scholar]
- 8. Ensor CM, Tai HH. 15‐Hydroxyprostaglandin dehydrogenase. J Lipid Mediat Cell Signal 1995; 12: 313–9. [DOI] [PubMed] [Google Scholar]
- 9. Backlund MG, Mann JR, Holla VR et al. 15‐Hydroxyprostaglandin dehydrogenase is down‐regulated in colorectal cancer. J Biol Chem 2005; 280: 3217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolf I, O’Kelly J, Rubinek T et al. 15‐hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res 2006; 66: 7818–23. [DOI] [PubMed] [Google Scholar]
- 11. Swami S, Krishnan AV, Moreno J, Bhattacharyya RB, Peehl DM, Feldman D. Calcitriol and genistein actions to inhibit the prostaglandin pathway: potential combination therapy to treat prostate cancer. J Nutr 2007; 137: 205S–10S. [DOI] [PubMed] [Google Scholar]
- 12. Ding Y, Tong M, Liu S, Moscow JA, Tai HH. NAD+‐linked 15‐hydroxyprostaglandin dehydrogenase (15‐PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis 2005; 26: 65–72. [DOI] [PubMed] [Google Scholar]
- 13. Quidville V, Segond N, Lausson S, Frenkian M, Cohen R, Jullienne A. 15‐Hydroxyprostaglandin‐dehydrogenase is involved in anti‐proliferative effect of non‐steroidal anti‐inflammatory drugs COX‐1 inhibitors on a human medullary thyroid carcinoma cell line. Prostaglandins Other Lipid Mediat 2006; 81: 14–30. [DOI] [PubMed] [Google Scholar]
- 14. Myung SJ, Rerko RM, Yan M et al. 15‐Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A 2006; 103: 12098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Z, Wang X, Lu Y et al. Expression of 15‐PGDH is downregulated by COX‐2 in gastric cancer. Carcinogenesis 2008; 29: 1219–27. [DOI] [PubMed] [Google Scholar]
- 16. Yoo NJ, Jeong EG, Lee SH. Expression of 15‐hydroxyprostaglandin dehydrogenase, a COX‐2 antagonist and tumour suppressor, is not altered in gastric carcinomas. Pathology 2007; 39: 174–5. [DOI] [PubMed] [Google Scholar]
- 17. Jang TJ, Ji YS, Jung KH. Decreased expression of 15‐hydroxyprostaglandin dehydrogenase in gastric carcinomas. Yonsei Med J 2008; 49: 917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thiel A, Ganesan A, Mrena J et al. 15‐hydroxyprostaglandin dehydrogenase is down‐regulated in gastric cancer. Clin Cancer Res 2009; 15: 4572–80. [DOI] [PubMed] [Google Scholar]
- 19. Watanabe T, Higuchi K, Kobata A et al. Non‐steroidal anti‐inflammatory drug‐induced small intestinal damage is Toll‐like receptor 4 dependent. Gut 2008; 57: 181–7. [DOI] [PubMed] [Google Scholar]
- 20. Spyratos F, Ferrero‐Pous M, Trassard M et al. Correlation between MIB‐1 and other proliferation markers: clinical implications of the MIB‐1 cutoff value. Cancer 2002; 94: 2151–9. [DOI] [PubMed] [Google Scholar]
- 21. Tanigawa T, Watanabe T, Hamaguchi M et al. Anti‐inflammatory effect of two isoforms of COX in H. pylori‐induced gastritis in mice: possible involvement of PGE2. Am J Physiol Gastrointest Liver Physiol 2004; 286: G148–56. [DOI] [PubMed] [Google Scholar]
- 22. Tanigawa T, Pai R, Arakawa T, Tarnawski AS. Rebamipide inhibits gastric cancer cell growth. Dig Dis Sci 2007; 52: 240–7. [DOI] [PubMed] [Google Scholar]
- 23. Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut 1994; 35: 675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med 1993; 122: 518–23. [PubMed] [Google Scholar]
- 25. Hansen‐Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti‐inflammatory drug‐induced regression in Apc(Min/+) mice. Cancer Res 2002; 62: 403–8. [PubMed] [Google Scholar]
- 26. Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res 2005; 65: 4218–27. [DOI] [PubMed] [Google Scholar]
- 27. Mann JR, Backlund MG, Buchanan FG et al. Repression of prostaglandin dehydrogenase by epidermal growth factor and snail increases prostaglandin E2 and promotes cancer progression. Cancer Res 2006; 66: 6649–56. [DOI] [PubMed] [Google Scholar]
- 28. Yang L, Amann JM, Kikuchi T et al. Inhibition of epidermal growth factor receptor signaling elevates 15‐hydroxyprostaglandin dehydrogenase in non‐small‐cell lung cancer. Cancer Res 2007; 67: 5587–93. [DOI] [PubMed] [Google Scholar]
- 29. Saukkonen K, Rintahaka J, Sivula A et al. Cyclooxygenase‐2 and gastric carcinogenesis. APMIS 2003; 111: 915–25. [DOI] [PubMed] [Google Scholar]
- 30. Chen CN, Sung CT, Lin MT, Lee PH, Chang KJ. Clinicopathologic association of cyclooxygenase 1 and cyclooxygenase 2 expression in gastric adenocarcinoma. Ann Surg 2001; 233: 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joo YE, Oh WT, Rew JS, Park CS, Choi SK, Kim SJ. Cyclooxygenase‐2 expression is associated with well‐differentiated and intestinal‐type pathways in gastric carcinogenesis. Digestion 2002; 66: 222–9. [DOI] [PubMed] [Google Scholar]
- 32. Lee TL, Leung WK, Lau JY et al. Inverse association between cyclooxygenase‐2 overexpression and microsatellite instability in gastric cancer. Cancer Lett 2001; 168: 133–40. [DOI] [PubMed] [Google Scholar]
- 33. Leung WK, To KF, Ng YP et al. Association between cyclo‐oxygenase‐2 overexpression and missense p53 mutations in gastric cancer. Br J Cancer 2001; 84: 335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lim HY, Joo HJ, Choi JH et al. Increased expression of cyclooxygenase‐2 protein in human gastric carcinoma. Clin Cancer Res 2000; 6: 519–25. [PubMed] [Google Scholar]
- 35. Murata H, Kawano S, Tsuji S et al. Cyclooxygenase‐2 overexpression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol 1999; 94: 451–5. [DOI] [PubMed] [Google Scholar]
- 36. Okano H, Shinohara H, Miyamoto A, Takaori K, Tanigawa N. Concomitant overexpression of cyclooxygenase‐2 in HER‐2‐positive on Smad4‐reduced human gastric carcinomas is associated with a poor patient outcome. Clin Cancer Res 2004; 10: 6938–45. [DOI] [PubMed] [Google Scholar]
- 37. Sung JJ, Leung WK, Go MY et al. Cyclooxygenase‐2 expression in Helicobacter pylori‐associated premalignant and malignant gastric lesions. Am J Pathol 2000; 157: 729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen L, Li X, Wang GL, Wang Y, Zhu YY, Zhu J. Clinicopathological significance of overexpression of TSPAN1, Ki67 and CD34 in gastric carcinoma. Tumori 2008; 94: 531–8. [DOI] [PubMed] [Google Scholar]
- 39. Fernandez‐Cebrian JM, Nevado Santos M, Vorwald Kuborn P et al. Can the clinical outcome in stage II colon carcinomas be predicted by determination of molecular marker expression? Clin Transl Oncol 2007; 9: 663–70. [DOI] [PubMed] [Google Scholar]
- 40. Fujimoto Y, Nakanishi Y, Yoshimura K, Shimoda T. Clinicopathologic study of primary malignant gastrointestinal stromal tumor of the stomach, with special reference to prognostic factors: analysis of results in 140 surgically resected patients. Gastric Cancer 2003; 6: 39–48. [DOI] [PubMed] [Google Scholar]
- 41. Ishida H, Miwa H, Tatsuta M et al. Ki67 and CEA expression as prognostic markers in Dukes’ C colorectal cancer. Cancer Lett 2004; 207: 109–15. [DOI] [PubMed] [Google Scholar]
- 42. Ito Y, Matsuura N, Sakon M et al. Both cell proliferation and apoptosis significantly predict shortened disease‐free survival in hepatocellular carcinoma. Br J Cancer 1999; 81: 747–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skalova A, Lehtonen H, Von Boguslawsky K, Leivo I. Prognostic significance of cell proliferation in mucoepidermoid carcinomas of the salivary gland: clinicopathological study using MIB 1 antibody in paraffin sections. Hum Pathol 1994; 25: 929–35. [DOI] [PubMed] [Google Scholar]
- 44. Muller W, Schneiders A, Meier S, Hommel G, Gabbert HE. Immunohistochemical study on the prognostic value of MIB‐1 in gastric carcinoma. Br J Cancer 1996; 74: 759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wakimoto N, Wolf I, Yin D et al. Nonsteroidal anti‐inflammatory drugs suppress glioma via 15‐hydroxyprostaglandin dehydrogenase. Cancer Res 2008; 68: 6978–86. [DOI] [PubMed] [Google Scholar]