

Abstract
Currently 5‐fluorouracil (5‐FU) plays a central role in the chemotherapeutic regimens for colorectal cancers and thus it is important to understand the mechanisms that determine 5‐FU sensitivity. The expression profiles of human colon cancer cell line DLD‐1, its 5‐FU‐resistant subclone DLD‐1/FU and a futher 21 types of colon cancer cell lines were compared to identify the novel genes defining the sensitivity to 5‐FU and to estimate which population of genes is responsible for 5‐FU sensitivity. In the hierarchical clustering, DLD‐1 and DLD‐1/FU were most closely clustered despite over 100 times difference in their 50% inhibitory concentration of 5‐FU. In DLD‐1/FU, the population of genes differentially expressed compared to DLD‐1 was limited to 3.3%, although it ranged from 4.8% to 24.0% in the other 21 cell lines, thus indicating that the difference of 5‐FU sensitivity was defined by a limited number of genes. Next, the role of the cellular inhibitor of apoptosis 2 (cIAP2) gene, which was up‐regulated in DLD‐1/FU, was investigated for 5‐FU resistance using RNA interference. The down‐regulation of cIAP2 efficiently enhanced 5‐FU sensitivity, the activation of caspase 3/7 and apoptosis under exposure to 5‐FU. The immunohistochemistry of cIAP2 in cancer and corresponding normal tissues from colorectal cancer patients in stage III revealed that cIAP2 was more frequently expressed in cancer tissues than in normal tissues, and cIAP2‐positive patients had a trend toward early recurrence after fluorouracil‐based chemotherapy. Although the association between drug sensitivity and the IAP family in colorectal cancer has not yet been discussed, cIAP2 may therefore play an important role as a target therapy in colorectal cancer. (Cancer Sci 2009; 100: 903–913)
Abbreviations:
- 5‐FU
5‐fluorouracil
- TS
thymidylate synthetase
- DPD
dihydropyrimidine dehydrogenase
- TP
thymidine phosphorylase
- IAP
inhibitor of apoptosis protein
- RNAi
RNA interference
- FBS
fetal bovine serum
- MTS
3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfenyl)‐2H‐tetrazolium, inner salt
- IC50
inhibitory concentration 50%
- SD
standard deviation
- RT‐PCR
reverse transcription polymerase chain reaction
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- siRNA
small interfering RNA
- DW
distilled water
- PI
propidium iodide
- FITC
fluorescein isothiocyanate
- CTNNA1
catenin alpha 1
- HBEGF
heparin‐binding EGF‐like growth factor
- PLA2G2A
phospholipase A2 group IIA
- OPRT
orotate phosphoribosyl transferase
- EGFR
epidermal growth factor receptor
- NCBI
National Center for Biotechnology Information
5‐fluorouracil (5‐FU) is an anticancer drug that has been mainly used in the treatment of colorectal cancers. Recently, 5‐FU has been combined with oxaliplatin or irinotecan as the first‐line treatment for advanced colorectal cancers and these have significantly improved the response rates to 40–50% and prolonged overall survival.( 1 , 2 ) Furthermore, novel biological agents including monoclonal antibodies such as cetuximab, which is an antibody against epidermal growth factor receptor (EGFR), and bevacizumab, which is an antibody against vascular endothelial growth factor, have been shown to provide additional clinical benefit for patients with metastatic colorectal cancers.( 3 , 4 , 5 ) However, there are still a large number of patients who do not benefit from the present treatments because of anticancer drug resistance. Elucidating the mechanisms by which 5‐FU resistance arises in colorectal cancer therefore remains an important issue for either overcoming or predicting such resistance.
5‐FU is an analog of uracil and is rapidly incorporated into the cells using the same transport system as uracil.( 6 ) Subsequently, 5‐FU is converted into active metabolites which disrupt the action of thymidylate synthetase (TS) and RNA synthesis. TS and 5‐FU‐metabolizing enzymes such as dihydropyrimidine dehydrogenase (DPD) and thymidine phosphorylase (TP) have been analyzed to elucidate 5‐FU resistance.( 7 ) However the resistance to 5‐FU has not been sufficiently explained by the metabolic pathway of 5‐FU alone, because multiple factors participate in chemoresistance.( 8 ) Recently, complementary DNA (cDNA) microarray technology has been used to identify novel genes regulating 5‐FU resistance, and the potential biomarkers of 5‐FU resistance other than pyrimidine metabolism‐related enzymes have been proposed.( 9 , 10 )
Apoptosis is found to be one of the primary mechanisms of the cytotoxic effect of chemotherapeutic agents and inhibition of the apoptotic pathway is one of the factors that may be responsible for drug resistance.( 11 , 12 , 13 ) In the process of apoptosis, the caspase cascade plays a central role,( 14 , 15 ) and the inhibitor of apoptosis protein (IAP) family is thought to prevent apoptosis through direct caspase and pro‐caspase inhibition (primarily caspase 3 and 7). The IAPs have been described to be abnormally regulated in various types of cancers,( 16 , 17 ) and recently they have been regarded as therapeutic targets of cancer.( 18 , 19 ) Although the association between IAPs and drug resistance has been discussed in cancers of some organs such as lung, pancreas and kidney,( 20 , 21 , 22 ) it has not been fully analyzed in human colorectal cancer.
The present study compared the messenger RNA (mRNA) expression profiles between the human colon cancer cell line DLD‐1 and its 5‐FU‐resistant subclone DLD‐1/FU by cDNA microarray to investigate the novel genes regulating 5‐FU resistance. To estimate which population of genes are responsible for regulating 5‐FU sensitivity or resistance, the expression profiles of DLD‐1 and DLD‐1/FU were also compared to another 21 types of colon cancer cell lines. Next, the role of the cellular IAP 2 (cIAP2) gene, which is most highly expressed among genes of the IAP family in DLD‐1/FU, was investigated using RNA interference (RNAi) on the sensitivity to 5‐FU, the activation of caspase 3/7, and apoptosis in human colon cancer cells. Finally, to identify the association between cIAP2 expression and 5‐FU resistance in human primary colorectal cancer, immunohistochemistry for cIAP2 was analyzed on cancer and corresponding normal tissues from colorectal cancer patients with curative operations followed by fluorouracil‐based adjuvant chemotherapies.
Materials and Methods
Cell lines and reagents. The human colon cancer cell lines (Clone A, COLO205, COLO320, CX‐1, DLD‐1, HCT‐8, HCT‐15, HCT116, HT‐29, KM12C, LoVo, LS174T, LS180, MIP101, RKO, SW48, SW480, SW620, SW948, SW1116, T84 and WiDr‐TC) were either provided from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan), or purchased from RIKEN BioResource Center (Tsukuba, Japan) and the American Type Culture Collection (Manassas, VA, USA). The 5‐FU‐resistant subclone DLD‐1/FU was generously provided from Dr M. Fukushima (Taiho Pharmaceutical, Co. Ltd, Tokyo, Japan). The DLD‐1/FU cell line was originally derived from the DLD‐1 cell line by continuous in vitro exposure of DLD‐1 cells to increasing concentrations of 5‐FU through a number of successive passages, as described earlier.( 23 ) The cells were cultured in either the recommended medium: RPMI1640 (Sigma‐Aldrich, St. Louis, MO, USA), L‐15 Medium Leibovitz (Sigma‐Aldrich), 1 × McCoy 5A (MP Biomedicals, Solon, OH, USA), or D‐MEM/F‐12 (Invitrogen, Carlsbad, CA, USA), containing 10% heat‐inactivated fetal bovine serum (FBS; Sigma‐Aldrich, St Louis, MO, USA) and 1% penicillin‐streptomycin (Invitrogen). 5‐FU was kindly provided by Kyowa Hakko Kogyo (Tokyo, Japan).
Cell proliferation assay and drug cytotoxicity assay. Cells were seeded in 100‐mm culture plate at a density of 5 × 104 cells/plate for the cell proliferation assay. On the indicated days, the viable number of cells was determined using a hemocytometer under a light microscope using the trypan blue exclusion method.
Drug‐induced cytotoxicity was assessed by the 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfenyl)‐2H‐tetrazolium, inner salt (MTS) assay using the CellTiter 96 Aqueous One Solution Proliferation Assay (Promega, Madison, WI, USA). First, 50 µL of cell suspension was seeded in 96‐well plates at a density of 5 × 103 cells/well. All plates were incubated for 24 h at 37°C in a humidified 5% CO2 atmosphere. Subsequently, 10 dilutions of 5‐FU were prepared in growth medium. After incubation, 50 µL of growth medium with diluted 5‐FU or growth medium only (as a control) was distributed in 96‐well plates. The plates were incubated for 72 h at 37°C. Following incubation, the drugs were removed. Then, fresh medium with MTS was added to each well and the cultures were incubated for 2 h at 37°C. The absorbance of formazan at 490 nm, considered to be directly proportional to the number of living cells in the culture,( 24 ) was measured using a plate reader Multiskan JX (Thermo Fisher Scientific, Yokohama, Japan). The cytotoxic effect of 5‐FU was assessed by the 50% inhibitory concentration (IC50: inhibitory drug concentration that results in 50% cell survival) value. With the approximation formula obtained from the straight‐line portion of the graph showing the cell survival rate for each dilution of drug, the IC50 value was calculated for subsequent analysis.
Total RNA isolation and reverse transcription. Total RNA was extracted from cell lines using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). The quality and quantity of the extracted total RNA were confirmed by electrophoresis on 1.2% denaturing agarose gels. cDNA was synthesized using a SuperScript III First‐Strand Synthesis System for Reverse Transcription – Polymerase Chain Reaction (RT‐PCR) Kit (Invitrogen) from 5 µg of total RNA. All the processes were carried out according to the manufacturer's instructions.
cDNA microarray analysis. The CodeLink Uniset Human 20KI Expression Bioarray (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA) was used for the cDNA microarray analysis. cRNA synthesis was carried out following the manufacturer's instructions. All of the following reagents were included in the CodeLink Expression Assay Reagent Kit (GE Healthcare Bio‐Sciences). First‐strand cDNA was generated from 2 µg of total RNA using reverse transcriptase and T7 oligo(dT) primer. Subsequently, second‐strand cDNA was produced using Escherichia coli DNA polymerase mix and RNase H. The resultant double‐stranded cDNA was purified on QIAquick PCR Purification Kit (Qiagen) and cRNA as a probe for microarray was generated by in vitro transcription reaction using T7 RNA polymerase and biotin‐11‐UTP (Perkin Elmer, Boston, MA, USA). cRNA was purified on the RNeasy Mini Kit (Qiagen), quantified by spectrophotometry and 10 µg was then fragmented by heating at 94°C for 20 min in the presence of magnesium ions. The fragmented cRNA was hybridized overnight at 37°C in hybridization buffer to each array in an Innova 4080 Shaking Incubator (New Brunswick Scientific, Edison, NJ, USA) for 18 h. After hybridization, the arrays were washed in 0.75 × TNT buffer (1 × TNT: 0.10 M Tris‐HCl [pH 7.6], 0.15 M NaCl, 0.05% Tween‐20) at 46°C for 1 h followed by incubation with streptavidin‐Cy5 at room temperature for 30 min in the dark. The arrays were then washed in 1 × TNT twice for 5 min each followed by a rinse in 0.05% Tween‐20 in water and then dried. Glass slides were scanned using a GenePix 4000 A Scanner (Axon Instruments, Union City, CA, USA). The grids of the image spots were adjusted and their signals were analyzed using the CodeLink System Software (GE Healthcare Bio‐Sciences). For each cell line, the experiments were performed either two or three times, independently for all of the steps through cell culture to microarray experiment, to confirm the reproducibility of experiments. To compare the gene expression values from the various experiments, the following array‐based normalization was applied. In each array experiment, a set of values was log‐transformed and Z‐normalized by the mean and the standard deviation (SD) calculated from the 5th to 95th percentiles of all non‐marker genes. An unsupervised hierarchical clustering analysis was applied using Cluster 3.0 and Java TreeView programs.( 25 , 26 )
RT‐PCR. Gene‐specific primer sets were designed using the Primer Express Software ver. 2.0 (Applied Biosystems, Foster City, CA, USA). Real‐time RT‐PCR using cDNA of cell lines was carried out using the Power SYBR Green PCR Master Mix (Applied Biosystems) following the manufacturer's instructions. Triplicate cDNA of each cell line was applied to 96‐well reaction plates. Thermal cycling was carried out in the following steps: one cycle of 95°C for 10 min; then 40 cycles of 95°C for 15 s, 60°C for 1 min. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as an endogenous control. The mRNA expression level of cIAP2 was normalized to that of the GAPDH in the corresponding sample.
Western blotting. Human colon cancer cells were harvested with trypsin/EDTA (edetic acid) and phosphated‐buffered saline (PBS)‐washed cell pellets were treated with EBC lysis buffer (1% NP‐40, 40 mM Tris‐HCl pH 8.0, 100 mM NaCl). Electrophoresis was performed using 4–20% Tris‐Glycine Gels (Invitrogen) and proteins were electro‐transferred onto Sequi‐Blot PVDF (polyvinylidene difluoride) Membrane (Bio‐Rad Laboratories, Hercules, CA, USA). The membranes were probed with the following primary antibodies: rabbit polyclonal anticIAP2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or rabbit polyclonal anti‐α actin antibody (AbCam, Cambridge, UK) as a control, at 4°C overnight. The membranes were washed and subsequently incubated with horseradish peroxidase‐coupled donkey antirabbit antibody (Santa Cruz Biotechnology) for 60 min. All proteins were visualized using SuperSignal West Pico Chemiluminescent Substrate (PIERCE, Rockford, IL, USA).
RNA interference (RNAi). Human colon cancer cells were trypsinized and plated into 6‐well plates. After 24 h, the cells were transfected with 50 nM ON‐TARGETplus SMARTpool cIAP2 siRNA (small interfering RNA; Dharmacon, Lafayette, CO, USA) by using DharmaFECT siRNA Transfection Reagent 4 (Dharmacon) according to the manufacturer's instructions. For control experiments, ON‐TARGETplus siCONTROL Non‐targeting siRNA (Dharmacon) was used under the same conditions. Between 48 and 120 h after transfection, gene silencing was examined with real‐time RT‐PCR and Western blotting.
Caspase 3/7 assay and flow cytometry analysis with the annexin V/propidium iodide (PI) staining. Caspase 3 and 7 activation assays were performed using the Caspase‐Glo 3/7 Assay Kit (Promega) according to the manufacturer's instructions. Briefly, the cells were seeded in 96‐well plates at a density of 5 × 103 cells/well. After the cells were treated with 5‐FU or distilled water (DW), Caspase‐Glo 3/7 Reagent (100 µL) was added to each well. The plate was then incubated at room temperature for 1 h and the luminescence of each sample was measured with a Centro LB960 96‐well Luminometer (Berthold Technologies, Natick, MA, USA). Because the luminescence of this assay was proportional to the cell number with the preliminary experiments, caspase 3/7 activity was assessed by the luminescence compensated by cell number.
In addition, apoptosis was measured by the Annexin V‐FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA). Cells were treated with annexin V‐fluorescein isothiocyanate (FITC) and PI according to the manufacturer's protocol and were analyzed by multicolor flow cytometry using FACS Calibur with Cell‐Quest Software (BD Biosciences).
Immunohistochemistry. Primary colorectal cancer and corresponding normal tissue specimens were obtained from 40 colorectal cancer patients with locoregional lymph node metastasis in stage III according to the TNM (tumor–node–metastasis) classification. All patients had undergone curative operations at Tohoku University Hospital, from 1999 to 2004, and were treated with fluorouracil‐based adjuvant chemotherapies (5‐FU/leucovorin, doxifluridine or uracil/tegafur). Written informed consent was obtained from all patients. The median age of the patients was 61.0 years (range 30–76). Nineteen patients were female and 21 were male. The median postoperative follow‐up time was 70.5 months (range 13.6–110.7).
Formalin‐fixed paraffin‐embedded tissue sections (3 µm thick) were deparaffinized in xylene and rehydrated in graded alcohol dilutions. Endogenous peroxidase activity was blocked by 3% hydrogen peroxidase for 10 min at room temperature. Antigen retrieval was performed using an autoclave in 0.01 M citrated buffer (pH 6.0) at 121°C for 5 min. For the reduction of non‐specific staining, the sections were exposed to 10% rabbit serum for 30 min. They were incubated at 4°C overnight with goat polyclonal anticIAP2 antibody (R & D Systems, Minneapolis, MN, USA) at 1:200 dilution. Secondary antibody reaction was performed using biotinylated rabbit antigoat antibody (Dako, Copenhagen, Denmark) at 1:800 dilution for 30 min at room temperature and peroxidase‐conjugated streptavidin (Nichirei Bioscience, Tokyo, Japan) was used according to the manufacturer's instructions. The reacted sections were visualized using 3,3′‐diaminobenzidine solution (1 mM 3,3′‐diaminobenzidine, 50 mM Tris‐HCl [pH 7.6] and 0.006% H2O2) and counterstained with hematoxylin for nuclear staining. Immunoreactivity for cIAP2 expression was graded as positive if >10% of cancer or normal epithelial cells were stained and as negative if <10% of cells were stained.
Statistical analysis. Any statistical significance of the mRNA expression level, caspase activity and time to recurrence was determined using either Student's t‐test or Mann–Whitney U‐test. The Chi‐square test was carried out to test the association between cIAP2 expression and cancer recurrence. The computer program Statcel2 Software (OMS Publishing, Saitama, Japan) and Microsoft Excel 2007 (Microsoft, Redmond, WA, USA) were used for statistical analysis. Values of P < 0.05 were considered to be statistically significant.
Results
The growth rates in DLD‐1 and DLD‐1/FU and cytotoxicity of 5‐FU. The growth rates of parental DLD‐1 cells and its 5‐FU‐resistant subclone DLD‐1/FU were equivalent (Fig. 1A). However, 5‐FU sensitivity of DLD‐1/FU was quite different from that of DLD‐1 and the IC50 of 5‐FU in DLD‐1/FU was higher than that in DLD‐1 by over 100 times: 55.1 ± 10.7 and 0.488 ± 0.013 µg/mL, respectively (1, 2). Among the 21 cell lines other than DLD‐1 and DLD‐1/FU, the IC50 of 5‐FU varied from 0.00998 to 9.96 µg/mL. SW620, HCT‐8 and SW480 showed high IC50 of 5‐FU: 9.96, 4.60 and 4.33 µg/mL, respectively; and WiDr‐TC, HCT116 and SW48 showed low IC50 of 5‐FU: 0.00998, 0.0499 and 0.149 µg/mL, respectively (Fig. 2B).
Figure 1.
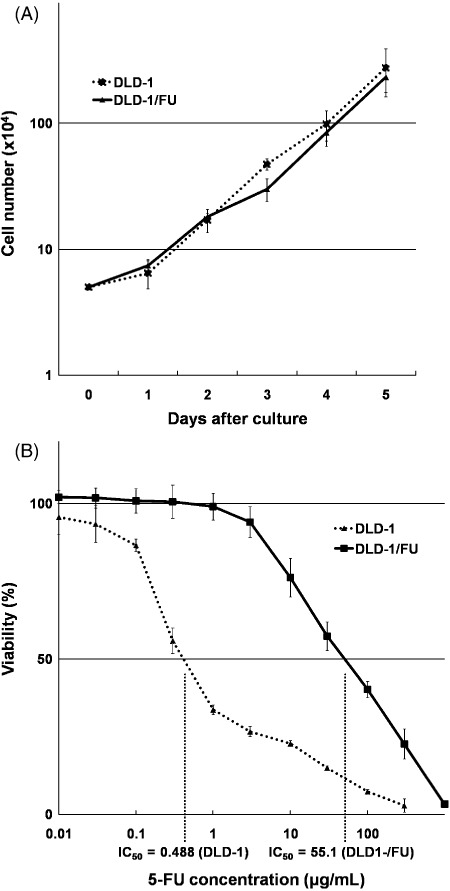
(A) Proliferation assays of DLD‐1 and DLD‐1/FU cells. The cell numbers were determined on the indicated days using trypan blue counting. The data represent the means ± SD of triplicate cultures. (B) Cytotoxity of 5‐FU and 50% inhibitory concentration in DLD‐1 and DLD‐1/FU cells. The MTS assay was performed 72 h after treatment with 5‐FU. The data represent the means ± SD of three independent experiments.
Figure 2.
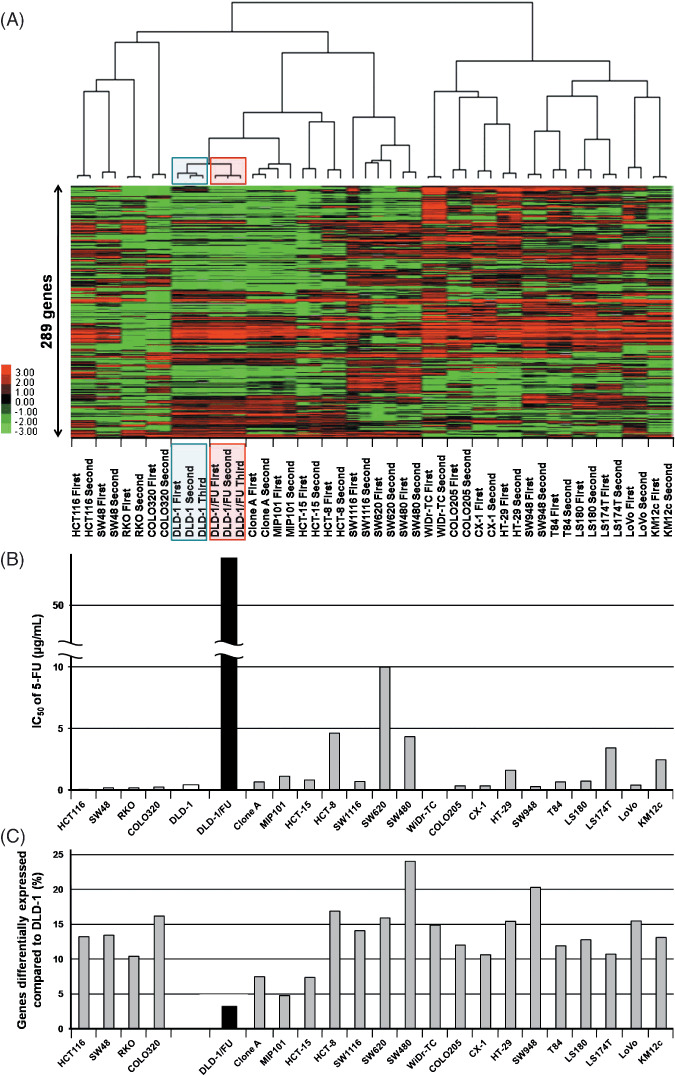
(A) A dendrogram and its image plot of the hierarchical clustering analysis of 23 colon cancer cell lines including DLD‐1 and DLD‐1/FU. A cohort of 289 genes out of 13 472 genes, the expression ratios of which varied by SDs of >1.25, were filtered using program Cluster 3.0. (B) The 50% inhibitory concentration of 5‐FU in 23 colon cancer cell lines ranged from 0.00998 to 55.1 µg/mL. The data represent the means of two or three independent experiments. (C) Percentages of differentially expressed genes compared to DLD‐1. The population of genes with more than a two‐fold change in comparison to DLD‐1 ranged from 3.3% to 24.0%.
cDNA microarray analysis and isolation of genes regulating 5‐FU resistance. On average 13 472 (67.4%) out of the 19 982 probes in the microarray experiments with the 23 colon cancer cell lines were assessed as ‘expressed genes’ that gave larger expression values than the ‘raw threshold’ which was calculated from a set of bacterial marker genes included in the CodeLink microarray as experimental controls.
To evaluate the reproducibility of the microarray experiments and to assess the similarities or differences of gene expression among the colon cancer cells, an unsupervised clustering analysis was performed using 289 genes out of 13 472 genes, the expression ratios of which varied most diversely with SDs of >1.25 (Fig. 2A). In the hierarchical clustering of microarray data of DLD‐1 and DLD‐1/FU combined with those of another 21 colon cancer cell lines, DLD‐1 and DLD‐1/FU were most closely clustered. Clone A, which is a subclone isolated from the DLD‐1 cell line with an assay using a Boyden chamber,( 27 ) was also clustered together with DLD‐1 and DLD‐1/FU. The population of genes differentially expressed between DLD‐1 and DLD‐1/FU cell lines with more than two‐fold change was limited to 3.3% (448/13 472 genes; Fig. 2C). On the other hand, in the other 21 colon cancer cell lines, the population of genes differentially expressed with more than a two‐fold change in comparison to DLD‐1 which was higher than that between DLD‐1 and DLD‐1/FU, ranging from 4.8% to 24.0% (Fig. 2C). In Clone A and MIP101, which were most closely clustered to DLD‐1 and DLD‐1/FU with the hierarchical clustering (Fig. 2A), the population of genes differentially expressed with more than a two‐fold change in comparison to DLD‐1, which was 7.5% and 4.8%, respectively (Fig. 2c). On the other hand, in SW480, SW948, HCT‐8, COLO320, SW620, LoVo and HT‐29, the population of genes differentially expressed with more than a two‐fold change in comparison to DLD‐1 which was higher than 15% (Fig. 2C).
To list genes differentially expressed between DLD‐1 and DLD‐1/FU, two‐sample t‐test was employed. The expression levels of genes in DLD‐1/FU were compared to those of DLD‐1 with fold change and the genes with P‐values < 5.0E‐05 and with fold change >2 or <0.5 are listed in Table 1. Genes highly expressed in DLD‐1/FU which had P‐values <5.0E‐05 and fold change >2 were 16 genes, including catenin alpha 1 (CTNNA1), heparin‐binding EGF‐like growth factor (HBEGF), and cIAP2 (also known as BIRC3). There were 16 genes with a low expression in DLD‐1/FU which had P‐values <5.0E‐05 and a fold change <0.5, including FBP1, ICAM2 and phospholipase A2 group IIA (PLA2G2A). Subsequently, two categories were specifically focused upon: pyrimidine metabolism‐related enzymes and the IAP family (Table 2). In the pyrimidine metabolism‐related enzymes, orotate phosphoribosyl transferase (OPRT) was down‐regulated as had been described in previous reports.( 23 ) In seven genes of IAP family, cIAP2 was most up‐regulated in DLD‐1/FU (2.31 times). Using real‐time RT‐PCR and Western blotting, we reconfirmed the results of cDNA microarray on cIAP2 expression in DLD‐1 and DLD‐1/FU cells (data not shown).
Table 1.
Genes differentially expressed between DLD‐1 and DLD‐1/FU cells
| NCBI Acc. No. | Gene symbol | Gene name | Cytoband | P‐value | Fold change |
|---|---|---|---|---|---|
| Genes highly expressed in DLD1/FU | |||||
| NM_006283 | TACC1 | Transforming, acidic coiled‐coil containing protein 1 | 8p11 | 1.00E‐05 | 4.41 |
| NM_001903 | CTNNA1 | Catenin, alpha 1, 102 kDa | 5q31 | 1.46E‐06 | 3.60 |
| NM_017855 | ODAM | Odontogenic, ameloblast associated | 4q13.3 | 6.62E‐06 | 3.48 |
| NM_001945 | HBEGF | Heparin‐binding EGF‐like growth factor | 5q23 | 3.54E‐06 | 3.18 |
| NM_015570 | AUTS2 | Autism susceptibility candidate 2 | 7q11.22 | 3.28E‐05 | 2.81 |
| NM_005406 | ROCK1 | Rho‐associated, coiled‐coil containing protein kinase 1 | 18q11.1 | 9.22E‐06 | 2.80 |
| NM_004815 | ARHGAP29 | Rho GTPase activationg protein 29 | 1q22.1 | 2.03E‐05 | 2.78 |
| NM_012482 | ZNF281 | Zinc finger protein 281 | 1q32.1 | 1.82E‐06 | 2.72 |
| NM_004848 | C1orf38 | Chromosome 1 open reading frame 38 | 1p35.3 | 2.61E‐05 | 2.68 |
| NM_033439 | IL33 | Interleukin 33 | 9p24.1 | 2.45E‐05 | 2.51 |
| NM_001165 | BIRC3, cIAP2 | Baculoviral IAP repeat‐containing 3 | 11q22 | 1.99E‐05 | 2.31 |
| NM_024796 | FLJ22639 | Hypothetical protein FLJ22639 | 1p36.33 | 3.94E‐05 | 2.23 |
| NM_012328 | DNAJB9 | DnaJ homolog, subfamily B, member 9 | 14q24.2‐q24.3 | 2.51E‐05 | 2.22 |
| NM_006281 | STK3 | Serin/threonine kinase 3 | 8q22.2 | 4.91E‐05 | 2.12 |
| NM_005433 | YES1 | V‐yes‐1 Yamaguchi sarcoma viral oncogene homolog 1 | 18p11.31‐p11.21 | 3.29E‐05 | 2.07 |
| NM_007283 | MGLL | Monoglyceride lipase | 3q21.3 | 5.29E‐06 | 2.00 |
| Genes with low expression in DLD1/FU | |||||
| NM_000507 | FBP1 | Fructose‐1,6‐bisphosphatase 1 | 9q22.3 | 9.33E‐07 | 0.16 |
| NM_000873 | ICAM2 | Intercellular adhesion molecule 2 | 17q23‐q25 | 8.38E‐07 | 0.18 |
| NM_000295 | SERPINA1 | Serpin peptidase inhibitor, clade A, member 1 | 14q32.1 | 1.99E‐05 | 0.19 |
| NM_000802 | FOLR1 | Folate receptor 1 | 11q13.3‐q14.1 | 1.79E‐05 | 0.22 |
| NM_000300 | PLA2G2A | Phospholipase A2, group IIA | 1p35 | 4.37E‐05 | 0.23 |
| NM_030763 | NSBP1 | Nucleosomal binding protein 1 | Xq13.3 | 2.86E‐05 | 0.24 |
| NM_000149 | FUT3 | Fucosyltransferase 3 | 19p13.3 | 1.27E‐05 | 0.34 |
| NM_018725 | IL17RB | Interleukin 17 receptor B | 3p21.1 | 5.57E‐06 | 0.34 |
| NM_002889 | RARRES2 | Retinoic acid receptor responder 2 | 7q36.1 | 2.51E‐05 | 0.38 |
| NM_005739 | RASGRP1 | RAS guanyl releasing protein 1 | 15q14 | 2.89E‐05 | 0.45 |
| NM_032355 | MON1A | MON1 homolog A | 3p21.31 | 2.87E‐05 | 0.47 |
| NM_000407 | GP1BB | Glycoprotein Ib, beta polypeptide | 22q11.21 | 8.30E‐06 | 0.48 |
| NM_003415 | ZNF268 | Zinc finger protein 268 | 12q24.33 | 1.20E‐06 | 0.49 |
| NM_002816 | PSMD12 | Proteasome 26S subunit, non‐ATPase, 12 | 17q24.2 | 4.10E‐05 | 0.49 |
| NM_003360 | UGT8 | UDP glycosyltransferase 8 | 4q26 | 3.93E‐05 | 0.50 |
| NM_005116 | SCL23A2 | Solute carrier family 23, member 2 | 20p13 | 9.12E‐06 | 0.50 |
The expression level of DLD‐1/FU was compared to that of DLD‐1 with fold change. Listed are genes with P‐value < 5.0E‐05 and fold change >2 or <0.5. NCBI Acc. No., National Center for Biotechnology Information accession number.
Table 2.
Genes differentially expressed between DLD‐1 and DLD‐1/FU cells involoved in pyrimidine metabolism‐related enzymes and IAP family
| NCBI Acc. No. | Gene symbol | Gene name | Cytoband | P‐value | Fold change |
|---|---|---|---|---|---|
| Pyrimidine metabolism‐related enzymes | |||||
| NM_001071 | TYMS, TS | Thymidylate synthetase | 18p11.32 | 7.34E‐03 | 0.80 |
| NM_000373 | UMPS, OPRT | Uridine monophosphate synthetase | 3q13 | 3.41E‐04 | 0.57 |
| NM_000110 | DPYD, DPD | Dihydropyrimidine dehydrogenase | 1p22 | – | – |
| NM_001953 | TYMP, TP | Thymidine phosphorylase | 22q13.33 | – | – |
| NM_003258 | TK1 | Thymidine kinase 1, soluble | 17q23.2‐q25.3 | 7.51E‐03 | 0.78 |
| NM_004614 | TK2 | Thymidine kinase 2, mitochondrial | 16q22‐q23.1 | – | – |
| NM_001033 | RRM1 | Ribonucleotide reductase M1 | 11p15.5 | N.S. | 1.21 |
| NM_001034 | RRM2 | Ribonucleotide reductase M2 | 2p25‐p24 | N.S. | 0.83 |
| NM_003364 | UPP1 | Uridine phosphorylase 1 | 7p12.3 | N.S. | 1.08 |
| IAP family | |||||
| NM_004536 | NAIP, BIRC1 | NLR family, apoptosis inhibitory protein | 5q13.1 | – | – |
| NM_001166 | BIRC2, cIAP1 | Baculoviral IAP repeat‐containing 2 | 11q22 | N.S. | 1.25 |
| NM_001165 | BIRC3, cIAP2 | Baculoviral IAP repeat‐containing 3 | 11q22 | 1.99E‐05 | 2.31 |
| NM_001167 | XIAP, BIRC4 | X‐linked inhibitor of apoptosis | Xq25 | – | – |
| NM_001168 | BIRC5, SURVIVIN | Baculoviral IAP repeat‐containing 5 | 17q25 | N.S. | 0.97 |
| NM_016252 | BIRC6, APOLLON | Baculoviral IAP repeat‐containing 6 | 2p22‐p21 | 4.82E‐02 | 1.23 |
| NM_022161 | BIRC7, LIVIN | Baculoviral IAP repeat‐containing 7 | 20q13.3 | N.S. | 1.02 |
The expression level of DLD‐1/FU was compared to that of DLD‐1 with fold change. NCBI Acc. No., National Center for Biotechnology Information accession number; IAP, apoptosis inhibitory protein; N.S., not significant; –, not reliable for low expressions.
Down‐regulation of cIAP2 by siRNA. The role of cIAP2 on 5‐FU resistance in colon cancer cells was further investigated using RNAi. With the data from the microarray and RT‐PCR, HCT‐8, HT‐29, SW948 and WiDr‐TC cells showed high expression of the cIAP2 gene (data not shown). Among the four types of cell lines, HCT‐8 and HT‐29 were successfully transfected with siRNA and the cIAP2 expression was down‐regulated (Fig. 3A). For these reasons, as well as DLD‐1/FU cells, HCT‐8 and HT‐29 cells were chosen for the following experiments using RNAi. At 48 and 120 h after transfection, the down‐regulation of cIAP2 was assessed by real‐time RT‐PCR and 72 h after transfection by Western blotting (Fig. 3A,B). The expression of cIAP2 was always down‐regulated with higher than 50% efficiency, although it could vary depending on the cell types or time points (Fig. 3A,B).
Figure 3.
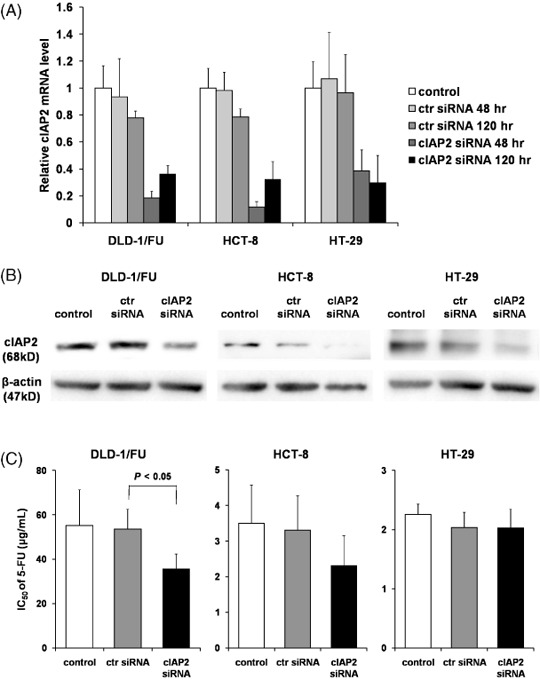
Down‐regulation of cellular inhibitor of apoptosis 2 (cIAP2) by small interfering RNA (siRNA). The messenger RNA (mRNA) and protein levels of cIAP2 were assessed by (A) real‐time reverse transcription – polymerase chain reaction or (B) Western blotting in DLD‐1/FU, HCT‐8 and HT‐29 under transfection of cIAP2 siRNA. As controls, untransfected cells (control) and cells transfected with control siRNA (ctr siRNA) were used. mRNA were isolated 48 or 120 h after transfection (Fig. 3a). Cell lysates were collected 72 h after transfection (Fig. 3b). (C) Effect of the down‐regulation of cIAP2 on 5‐FU sensitivity in human colon cancer cell lines. In vitro cytotoxicity assay was performed using MTS assay. At 48 h after transfection with cIAP2 siRNA or control siRNA, 5‐FU was added at various concentrations. 72 h after treatment with 5‐FU, MTS assay was performed to determine the 50% inhibitory concentration. The results represent the means ± SD of three independent experiments.
Enhanced 5‐FU sensitivity by cIAP2 siRNA. At 48 h after transfection with cIAP2 siRNA or non‐targeting siRNA as control siRNA (‘ctr siRNA’ in 3, 4), DLD‐1/FU, HCT‐8 and HT‐29 cells were cultured in medium with 10 dilutions of 5‐FU and then the IC50 was determined by MTS assay. In all of the cell types, treatment with control siRNA did not have a cytotoxic effect on the IC50 (Fig. 3C). DLD‐1/FU cells transfected with cIAP2 siRNA in the presence of 5‐FU showed a significant decrease of the IC50 in comparison to those transfected with the control siRNA (P < 0.05; Fig. 3C). These results indicate that the down‐regulation of cIAP2 in DLD‐1/FU cells enhanced the cytotoxicity with 5‐FU. The IC50 of 5‐FU in HCT‐8 cells changed in the same manner as in DLD‐1/FU (P = 0.15); but the IC50 of 5‐FU in HT‐29 cells did not change under the down‐regulation of cIAP2 (Fig. 3C).
Figure 4.
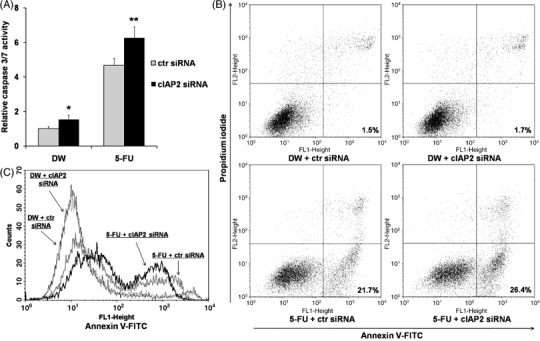
(A) Down‐regulation of cellular inhibitor of apoptosis 2 (cIAP2) induced caspase 3/7 activation. At 48 h after cIAP2 small interfering RNA (siRNA) transfection, cells were exposed to distilled water (DW) or 5‐fluorouracil (5‐FU) for 48 h and measured for caspase 3/7 activity. As controls, the cells transfected with control siRNA (ctr siRNA) were used. The results represent the ratios to caspase 3/7 activity of untransfected cells with common culture. *P < 0.05 versus cells treated with ctr siRNA + DW. **P < 0.05 versus cells treated with each of the other treatments. (B) Incidence of apoptosis in DLD‐1/FU revealed by flow cytometric analysis stained with annexin V‐fluorescein isothiocyanate (V‐FITC) and counterstained with propidium iodide (PI). In cIAP2 siRNA‐transfected cells with exposure to 5‐FU, the percentage of annexin V‐positive/PI‐negative cells (early apoptotic cells) shown in the right lower quadrant was high in comparison to the control siRNA‐transfected cells. (C) The histogram represents the increase of annexin V‐positive fraction in cIAP2 siRNA‐transfected cells with exposure to 5‐FU. A representative assay out of three independent assays was provided.
Activation of caspase 3/7 and induction of apoptosis in DLD‐1/FU by cIAP2 siRNA. At 48 h after transfection with cIAP2 siRNA or control siRNA, 5‐FU (500 µg/mL) or DW was added to DLD‐1/FU cells and the cells were maintained for another 48 h. Then, caspase 3/7 activity was measured and the induction of apoptosis was also analyzed with flow cytometry. As shown in Fig. 4(A), the induction of caspase 3/7 activity was examined in DLD‐1/FU cells under the down‐regulation of cIAP2 with or without exposure to 5‐FU. Without exposure to 5‐FU, the caspase 3/7 activity under the down‐regulation of cIAP2 was enhanced in comparison to that with control siRNA (with arbitrary units 1.53 vs. 0.99, P = 0.03; Fig. 4A). After exposure to 5‐FU, again the caspase 3/7 activity under the down‐regulation of cIAP2 was enhanced in comparison to that with control siRNA (6.26 vs. 4.67, P = 0.02). Next, as shown in Fig. 4(B), the induction of apoptosis with annexin V and PI staining were examined in DLD‐1/FU cells under the down‐regulation of cIAP2 with or without exposure to 5‐FU. Without exposure to 5‐FU, induction of early or late apoptotic cells was not observed under the down‐regulation of cIAP2 (subpopulated in the right lower portion or the right upper portion in each graph of Fig. 4B, respectively); but with exposure to 5‐FU, the induction of early apoptotic cells under the down‐regulation of cIAP2 was seen (26.4%). As shown in Fig. 4(C), with exposure to 5‐FU, the fraction of early and late apoptotic cells in DLD‐1/FU cells was enhanced under the down‐regulation of cIAP2 with two‐peaks curves, although it was not observed without exposure to 5‐FU.
cIAP2 expression in human colorectal cancer and normal tissues with immunohistochemistry and its effect on the prognosis. Immunohistochemistry for cIAP2 was analyzed on colorectal cancer and corresponding normal tissues from 40 patients who underwent curative operations followed by fluorouracil‐based adjuvant chemotherapies. cIAP2 was detected exclusively in the cytoplasm of cancer and normal epithelial cells. Seventy percent of cancer tissues were positive for cIAP2 (28/40 cases; Fig. 5A,D,E), whereas only 20% of the normal tissues were positive (8/40 cases; Fig. 5A,F,G).
Figure 5.
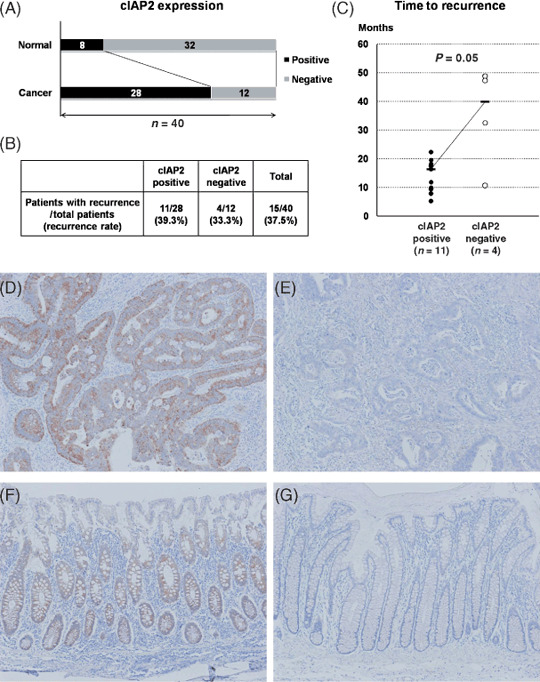
(A) Cellular inhibitor of apoptosis 2 (cIAP2) expression in human colorectal cancer and the corresponding normal tissues by immunohistochemistry. cIAP2 was more frequently expressed in cancer tissues than in normal tissues. (B) The incidence of cancer recurrence after curative operations in cIAP2‐positive and cIAP2‐negative patients. (C) The time to recurrence in cIAP2‐positive and cIAP2‐negative patients. (D–G) Representative image of immunohistochemical staining for cIAP2 in human colorectal cancer and normal tissues (D, cancer tissue/cIAP2‐positive; E, cancer tissue/cIAP2‐negative; F, normal tissue/cIAP2‐positive; G, normal tissue/cIAP2‐negative; original magnification: ×100).
Moreover, we analyzed the 40 colorectal cancer patients on the association between cIAP2 expression in the cancer tissues and cancer recurrence. Of the 40 patients, 15 patients (37.5%) experienced cancer recurrence within 5 years after surgery (Fig. 5B). Although we did not find a difference on incidence of cancer recurrence between cIAP2‐positive and cIAP2‐negative cancer patients (recurrence rate 39.3%vs. 33.3%; Fig. 5B), cIAP2‐positive cancer patients had a trend to recur earlier after operations than cIAP2‐negative cancer patients. Although the number of the cases was limited, we compared the time to recurrence between the two subgroups of the 15 patients: cIAP2‐positive patients (n = 11) and cIAP2‐negative patients (n = 4). The time to recurrence in cIAP2‐positive patients and cIAP2‐negative patients was 16.3 and 39.9 months, respectively (P = 0.05; Fig. 5C).
Discussion
Single‐drug chemotherapy using 5‐FU or its derivatives or combined cytotoxic chemotherapies including 5‐FU have been chosen to treat colon cancer. Although anticancer agents and their regimens have been developed, their efficacies have not yet reached a satisfactory level. As Scheithauer et al. reported in 1993, the randomized comparison in patients with metastatic colorectal cancers revealed that the median survival time of patients without the use of any form of treatment is in the range of 5 months.( 28 ) According to the Cochrane database,( 29 ) when either fluorouracil alone or fluorouracil combined with leucovorin is administered, the median survival time of patients with metastatic colorectal cancers is extended to 10–12 months. As Salts et al.( 30 ) and de Gramont et al.( 31 ) reported, the median survival time is prolonged to 14–16 months when either oxaliplatin or irinotecan is added to 5‐FU‐based chemotherapy. Furthermore, the median survival is prolonged to longer than 20 months when a targeted therapy including bevacizumab or cetuximab is added to the cytotoxic chemotherapies including FOLFOX (folinic acid, FU and oxaliplatin) or FOLFIRI (folinic acid, FU and irinotecan).( 4 ) Currently 5‐FU plays a central role in chemotherapies for colorectal cancers and thus it is important to elucidate the mechanisms defining the sensitivity to 5‐FU for designing successful chemotherapies. Improvement of 5‐FU sensitivity, the selection of agents combined with 5‐FU‐based chemotherapies and the selection of regimens for individual patients are especially important to achieve better prognosis in advanced colorectal cancer cases.
In the current study, focusing specifically on improving the sensitivity to 5‐FU and the selection of types of therapy for each individual patient, to identify novel genes which define the sensitivity or tolerance to 5‐FU in colorectal cancers, the expression profiles of the parental DLD‐1 cell line was compared to its 5‐FU‐resistant subclone DLD‐1/FU. First, a hierarchical clustering was performed with microarray data of DLD‐1 and DLD‐1/FU combined with those of 21 other colon cancer cell lines. As shown in Fig. 2(A), DLD‐1 and DLD‐1/FU were most closely clustered and Clone A and MIP101 were also closely clustered. The expression profiles of genes originating from the entire genome will represent the characteristics of cells and this strategy will be good for prediction of the origin of cell types or derived organs; but this strategy will not be good for more precise analyses, including the prediction of sensitivity to some drugs.( 32 ) For such a purpose, it will be necessary to identify a special set of genes. In Fig. 2(A) 289 genes were selected as representatives from the whole set of genes. The expression profiles of DLD‐1 and DLD‐1/FU cells were quite similar in comparison to the other cell lines, even after the acquisition of 5‐FU resistance in DLD‐1/FU cells as shown in Fig. 2(B). These results suggest that a change of expression profiles in a limited population of genes contributes to the acquisition of 5‐FU resistance in DLD‐1/FU cells. Figure 2(C) demonstrates that 3.3% of the genes (448/13 472 genes) were differentially expressed between DLD‐1 and DLD‐1/FU with more than a two‐fold change. In the other 21 colon cancer cell lines, the population of genes differentially expressed with more than a two‐fold change in comparison to DLD‐1 was higher than that between DLD‐1 and DLD‐1/FU, ranging from 4.8% to 24.0% (Fig. 2C). This revealed that, as expected, the difference of 5‐FU sensitivity was derived from a change of expressions in a limited member of genes. After the acquisition of 5‐FU resistance, the secondary or tertiary effects may have affected the changes in expressions in some of the 448 genes; and therefore the number of genes essential for determining the difference in 5‐FU sensitivity may be much smaller than 448 genes. Schmidt et al. analyzed the change of expression profiles during the establishment of a 5‐FU‐resistant subclone from the human colon cancer cell line SW620 using a cDNA microarray and the number of genes which showed expression changes in their analysis was limited to 4.8% among the genes they used (330/6888 genes).( 33 ) Their result is consistent with the current findings, both of which suggested that the limited number of genes regulate the sensitivity or resistance to 5‐FU in colorectal cancer cells. On the other hand, comparing the hierarchical clustering (Fig. 2A) to the IC50 of 5‐FU (Fig. 2B) or the population of genes differentially expressed among the 23 colon cancer cell lines (Fig. 2C), it is difficult to identify a correlation. These results show that 5‐FU sensitivity is regulated by diverse mechanisms or pathways. As shown in Fig. 3(C), the down‐regulation of cIAP2 induced the HCT‐8 cell line to become more sensitive to 5‐FU, but the 5‐FU sensitivity of HT‐29 did not change. This may suggest the possibility that the sensitivity to anticancer agents including 5‐FU is regulated by a variety of pathways in cancer cells. Harlin et al. reported the compensating pathways of IAP functions using a XIAP knock‐out mouse.( 34 ) In the current experiments using HT‐29, such compensating pathways may therefore have functioned.
In regard to pyrimidine metabolism‐related enzymes (Table 2), a comparison between DLD‐1 and DLD‐1/FU showed that the OPRT gene was down‐regulated in DLD‐1/FU. The correlation between the expression of OPRT and 5‐FU sensitivity was previously described,( 35 , 36 ) and the current result was consistent with those reports. However, our results on TS and DPD did not support the results reported previously, which may suggest the difficulty of such analyses.
To identify novel genes that regulate 5‐FU sensitivity, the expression profiles of DLD‐1 and DLD‐1/FU were compared and the genes differentially expressed between DLD‐1 and DLD‐1/FU were listed (Table 1). Among the genes with low expression in DLD‐1/FU, the PLA2G2A gene, which encodes a group IIA‐secreted phospholipase A2, is of particular interest. The expression of PLA2G2A has been reported to suppress the progression or metastasis of human gastric cancers.( 37 ) Recently, Ganesan et al. also reported that the anti‐invasive effect of PLA2G2A occurs through its ability to inhibit the S100A4 gene, which is a metastasis mediator gene in gastric cancer cells.( 38 ) These results may suggest that the expression of the PLA2G2A gene may affect the character of cancer cells, including their responses to anticancer drugs such as 5‐FU.
In genes highly expressed in DLD‐1/FU, some of these genes are of interest from the standpoint of 5‐FU resistance. HBEGF, which binds to epidermal growth factor receptor (EGFR)/ErbB1 and ErbB4, is a member of the EGF family. The overexpression of HBEGF has been reported in several types of cancers including colon cancer and pancreatic cancer.( 39 , 40 ) Suganuma et al. identified HBEGF as a 5‐FU‐ and cisplatin‐resistance‐related gene in gastric cancer using cDNA microarray,( 41 ) and Wang et al. further investigated the potential role of HBEGF which regulated the chemotherapy‐induced EGFR activation,( 42 ) and our results in Table 1 support their findings. Next, CTNNA1 encodes α‐catenin whose interaction with actin‐based cytoskeleton is required for the strong state of E‐cadherin‐based cell adhesion activity.( 43 ) Matsubara et al. reported α‐catenin expression increases the resistance to apoptosis induced by sphingosine in the colon cancer cell line and α‐catenin mediated transduction of signals from the cadherin‐catenin complex to regulate the apoptotic cascade.( 44 ) CTNNA1 is therefore a gene that can possibly regulate drug resistance through the apoptotic pathway.
Among the genes highly expressed in DLD‐1/FU, the cIAP2 gene has drawn a lot of attention and the role of the cIAP2 gene on 5‐FU sensitivity was further analyzed in colon cancer cell lines. In our analysis using RNAi, the down‐regulation of cIAP2 changed the phenotype of DLD‐1/FU to be more sensitive to 5‐FU (Fig. 3C). Lopes et al. reported that the expression of cIAP2 protein affected the sensitivity to cisplatin and doxorubicin in pancreatic cancer cell lines.( 21 ) In addition, the expression of IAP family genes, such as survivin and XIAP, affect the sensitivity to several types of anticancer drugs in cancers of some organs such as the lung and ovary.( 20 , 21 , 22 , 45 , 46 , 47 ) However, the association between drug sensitivity and the IAP family in colon cancer has not been reported so far. The current result raises the possibility for novel strategies of cancer chemotherapy in colorectal cancer.
After the down‐regulation of cIAP2 by siRNA, the expression level of cIAP2 in DLD‐1/FU was almost equivalent to that in its parental cell DLD‐1 (Table 1, Fig. 3A); however, the IC50 of 5‐FU in DLD‐1/FU (35.5 µg/mL) was still much higher than that in DLD‐1 (0.488 µg/mL). This result suggests that some other genes, such as those listed in Table 1, may also play an important role in regulating 5‐FU resistance as well as cIAP2.
Although the association between IAPs and drug resistance has been previously discussed, there have been few reports discussing overexpression of IAPs. Although we have not shown the association between cIAP2 overexpression and 5‐FU sensitivity in our report, Pratt et al. have analyzed survivin overexpression in breast cancer cell line and showed that unexpectedly the increased survivin did not promote paclitaxel resistance.( 47 ) Their results suggest the difficulty in elucidating the association between cIAP2 induction and drug resistance.
With the down‐regulation of cIAP2 without 5‐FU exposure, the activity of caspase 3/7 was induced, but apoptosis was not induced (Fig. 4). However, with the down‐regulation of cIAP2 with 5‐FU exposure, apoptosis was efficiently induced (Fig. 4). The results indicate that the activation of the caspase 3/7 pathway is essential for the induction of apoptosis in DLD‐1/FU cells; and exposure to 5‐FU was required for the induction of the apoptosis in addition to the activation of the caspase 3/7 pathway. As Johnstone et al. reported previously,( 12 ) the sensitivity or resistance to anticancer drugs is regulated by interactions between drugs and intracellular targets of the drugs (drug–target interaction); however, depending on the severity of the initial insult, drug‐induced damage will also result in catastrophic death or it may initiate a series of secondary effects mediated by various stress signaling pathways leading to cell cycle arrest or cell death.( 12 ) Apoptosis is one of the secondary effects that plays an important role and the caspase cascade is included in the pathways of apoptosis. Recently IAP has been considered as a novel target of cancer therapy and the efficacy of antisense oligonucleotides or small‐molecule IAP antagonists has been reported.( 48 , 49 , 50 ) In colorectal cancer, a target therapy against IAP genes may play an important role in the future.
To further identify the role of cIAP2 and their association with 5‐FU resistance in human colorectal cancer, immunohistochemistry for cIAP2 was performed on colorectal cancer and normal tissues from 40 patients who underwent curative operations followed by fluorouracil‐based adjuvant chemotherapies. As shown in Fig. 5, cIAP2 was more frequently expressed in cancer tissues than in normal tissues. Moreover cIAP2‐positive cancer patients had a trend toward early recurrence after fluorouracil‐based adjuvant chemotherapies and this result sufficiently supported our results in vitro. It suggested that cIAP2 performed a crucial function and could be a potential therapeutic target in human colorectal cancer.
In conclusion, this study indicated that differences of 5‐FU sensitivity could thus be defined by a limited number of genes. Among them, the role of the cIAP2 gene which regulates 5‐FU sensitivity through the apoptotic pathway was elucidated in vitro. Moreover, the expression of cIAP2 was frequently observed in human colorectal cancer tissue, and might contribute to cancer recurrence in stage III colorectal cancer patients after fluorouracil‐based adjuvant chemotherapy. Therefore, cIAP2 may play a key role and may be a good target for the 5‐FU sensitivity in colorectal cancer. On the other hand, in order to develop efficient cancer therapies, it is necessary to consider the diversity of the characteristics of normal cells and hosts, which are regarded as the backgrounds from which cancer cells originate.
Acknowledgments
Grant no. 15659303 and 19659318 for cancer research from Ministry of Education, Culture, Sports, Science and Technology of Japan (K. Miura). We are grateful to Drs S. Ohnuma and K. Masuda (Tohoku University) for helpful discussions and technical advice; Ms. E. Shibuya, H. Fujimura and K. Inabe for technical assistance.
References
- 1. Giacchetti S, Perpoint B, Zidani R et al . Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil‐leucovorin as first‐line treatment of metastatic colorectal cancer. J Clin Oncol 2000; 18: 136–47. [DOI] [PubMed] [Google Scholar]
- 2. Douillard JY, Cunningham D, Roth AD et al . Irinotecan combined with fluorouracil compared with fluorouracil alone as first‐line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000; 355: 1041–7. [DOI] [PubMed] [Google Scholar]
- 3. Cunningham D, Humblet Y, Siena S et al . Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan‐refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337–45. [DOI] [PubMed] [Google Scholar]
- 4. Hurwitz H, Fehrenbacher L, Novotny W et al . Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 5. Hinoda Y, Sasaki S, Ishida T, Imai K. Monoclonal antibodies as effective therapeutic agents for solid tumors. Cancer Sci 2004; 95: 621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wohlhueter RM, McIvor RS, Plagemann PG. Facilitated transport of uracil and 5‐fluorouracil, and permeation of orotic acid into cultured mammalian cells. J Cell Physiol 1980; 104: 309–19. [DOI] [PubMed] [Google Scholar]
- 7. Longley DB, Harkin DP, Johnston PG. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3: 330–8. [DOI] [PubMed] [Google Scholar]
- 8. Fojo T, Bates S. Strategies for reversing drug resistance. Oncogene 2003; 22: 7512–23. [DOI] [PubMed] [Google Scholar]
- 9. Zembutsu H, Ohnishi Y, Tsunoda T et al . Genome‐wide cDNA microarray screening to correlate gene expression profiles with sensitivity of 85 human cancer xenografts to anticancer drugs. Cancer Res 2002; 62: 518–27. [PubMed] [Google Scholar]
- 10. Maxwell PJ, Longley DB, Latif T et al . Identification of 5‐fluorouracil‐inducible target genes using cDNA microarray profiling. Cancer Res 2003; 63: 4602–6. [PubMed] [Google Scholar]
- 11. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis 2000; 21: 485–95. [DOI] [PubMed] [Google Scholar]
- 12. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002; 108: 153–64. [DOI] [PubMed] [Google Scholar]
- 13. Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 1999; 59: 1391–9. [PubMed] [Google Scholar]
- 14. Chang HY, Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev 2000; 64: 821–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuentes‐Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J 2004; 384: 201–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ambrosini G, Adida C, Altieri DC. A novel anti‐apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997; 3: 917–21. [DOI] [PubMed] [Google Scholar]
- 17. Tamm I, Kornblau SM, Segall H et al . Expression and prognostic significance of IAP‐family genes in human cancers and myeloid leukemias. Clin Cancer Res 2000; 6: 1796–803. [PubMed] [Google Scholar]
- 18. Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res 2004; 64: 7183–90. [DOI] [PubMed] [Google Scholar]
- 19. Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007; 12: 1543–68. [DOI] [PubMed] [Google Scholar]
- 20. Yonesaka K, Tamura K, Kurata T et al . Small interfering RNA targeting survivin sensitizes lung cancer cell with mutant p53 to adriamycin. Int J Cancer 2006; 118: 812–20. [DOI] [PubMed] [Google Scholar]
- 21. Lopes RB, Gangeswaran R, McNeish IA, Wang Y, Lemoine NR. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer 2007; 120: 2344–52. [DOI] [PubMed] [Google Scholar]
- 22. Bilim V, Yuuki K, Itoi T et al . Double inhibition of XIAP and Bcl‐2 axis is beneficial for retrieving sensitivity of renal cell cancer to apoptosis. Br J Cancer 2008; 98: 941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol 2000; 17: 277–83. [DOI] [PubMed] [Google Scholar]
- 24. Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 1991; 3: 207–12. [DOI] [PubMed] [Google Scholar]
- 25. de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics 2004; 20: 1453–4. [DOI] [PubMed] [Google Scholar]
- 26. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci USA 1998; 95: 14863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghoda LY, Savarese TM, Dexter DL, Parks RE Jr, Trackman PC, Abeles RH. Characterization of a defect in the pathway for converting 5′‐deoxy‐5′‐methylthioadenosine to methionine in a subline of a cultured heterogeneous human colon carcinoma. J Biol Chem 1984; 259: 6715–19. [PubMed] [Google Scholar]
- 28. Scheithauer W, Rosen H, Kornek GV, Sebesta C, Depisch D. Randomised comparison of combination chemotherapy plus supportive care with supportive care alone in patients with metastatic colorectal cancer. Br Med J 1993; 306: 752–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colorectal Meta‐analysis Collaboration . Palliative chemotherapy for advanced or metastatic colorectal cancer. Cochrane Database Syst Rev 2000: CD001545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saltz LB, Cox JV, Blanke C et al . Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group N Engl J Med 2000; 343: 905–14. [DOI] [PubMed] [Google Scholar]
- 31. de Gramont A, Figer A, Seymour M et al . Leucovorin and fluorouracil with or without oxaliplatin as first‐line treatment in advanced colorectal cancer. J Clin Oncol 2000; 18: 2938–47. [DOI] [PubMed] [Google Scholar]
- 32. Ross DT, Scherf U, Eisen MB et al . Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 2000; 24: 227–35. [DOI] [PubMed] [Google Scholar]
- 33. Schmidt WM, Kalipciyan M, Dornstauder E et al . Dissecting progressive stages of 5‐fluorouracil resistance in vitro using RNA expression profiling. Int J Cancer 2004; 112: 200–12. [DOI] [PubMed] [Google Scholar]
- 34. Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB. Characterization of XIAP‐deficient mice. Mol Cell Biol 2001; 21: 3604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inaba M, Mitsuhashi J, Sawada H et al . Reduced activity of anabolizing enzymes in 5‐fluorouracil‐resistant human stomach cancer cells. Jpn J Cancer Res 1996; 87: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tokunaga Y, Sasaki H, Saito T. Clinical role of orotate phosphoribosyl transferase and dihydropyrimidine dehydrogenase in colorectal cancer treated with postoperative fluoropyrimidine. Surgery 2007; 141: 346–53. [DOI] [PubMed] [Google Scholar]
- 37. Leung SY, Chen X, Chu KM et al . Phospholipase A2 group IIA expression in gastric adenocarcinoma is associated with prolonged survival and less frequent metastasis. Proc Natl Acad Sci USA 2002; 99: 16203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ganesan K, Ivanova T, Wu Y et al . Inhibition of gastric cancer invasion and metastasis by PLA2G2A, a novel beta‐catenin/TCF target gene. Cancer Res 2008; 68: 4277–86. [DOI] [PubMed] [Google Scholar]
- 39. Ito Y, Higashiyama S, Takeda T, Yamamoto Y, Wakasa KI, Matsuura N. Expression of heparin‐binding epidermal growth factor‐like growth factor in pancreatic adenocarcinoma. Int J Pancreatol 2001; 29: 47–52. [DOI] [PubMed] [Google Scholar]
- 40. Itoh Y, Joh T, Tanida S et al . IL‐8 promotes cell proliferation and migration through metalloproteinase‐cleavage proHB‐EGF in human colon carcinoma cells. Cytokine 2005; 29: 275–82. [DOI] [PubMed] [Google Scholar]
- 41. Suganuma K, Kubota T, Saikawa Y et al . Possible chemoresistance‐related genes for gastric cancer detected by cDNA microarray. Cancer Sci 2003; 94: 355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang F, Liu R, Lee SW, Sloss CM, Couget J, Cusack JC. Heparin‐binding EGF‐like growth factor is an early response gene to chemotherapy and contributes to chemotherapy resistance. Oncogene 2007; 26: 2006–16. [DOI] [PubMed] [Google Scholar]
- 43. Imamura Y, Itoh M, Maeno Y, Tsukita S, Nagafuchi A. Functional domains of alpha‐catenin required for the strong state of cadherin‐based cell adhesion. J Cell Biol 1999; 144: 1311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsubara S, Ozawa M. Expression of alpha‐catenin in alpha‐catenin‐deficient cells increases resistance to sphingosine‐induced apoptosis. J Cell Biol 2001; 154: 573–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Down‐regulation of X‐linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res 2000; 60: 5659–66. [PubMed] [Google Scholar]
- 46. Nomura T, Yamasaki M, Nomura Y, Mimata H. Expression of the inhibitors of apoptosis proteins in cisplatin‐resistant prostate cancer cells. Oncol Rep 2005; 14: 993–7. [PubMed] [Google Scholar]
- 47. Pratt MA, Niu MY, Renart LI. Regulation of survivin by retinoic acid and its role in paclitaxel‐mediated cytotoxicity in MCF‐7 breast cancer cells. Apoptosis 2006; 11: 589–605. [DOI] [PubMed] [Google Scholar]
- 48. LaCasse EC, Cherton‐Horvat GG, Hewitt KE et al . Preclinical characterization of AEG35156/GEM 640, a second‐generation antisense oligonucleotide targeting X‐linked inhibitor of apoptosis. Clin Cancer Res 2006; 12: 5231–41. [DOI] [PubMed] [Google Scholar]
- 49. Varfolomeev E, Blankenship JW, Wayson SM et al . IAP antagonists induce autoubiquitination of c‐IAPs, NF‐kappaB activation, and TNFalpha‐dependent apoptosis. Cell 2007; 131: 669–81. [DOI] [PubMed] [Google Scholar]
- 50. Vince JE, Wong WW, Khan N et al . IAP antagonists target cIAP1 to induce TNFalpha‐dependent apoptosis. Cell 2007; 131: 682–93. [DOI] [PubMed] [Google Scholar]