

Abstract
Transforming growth factor (TGF)‐β signaling has been shown to promote tumor growth and metastasis in advanced cancer. Use of inhibitors of TGF‐β signaling may thus be a novel strategy for treatment of patients with such cancers. In this study, we investigated the effects of a novel TGF‐β type I receptor (TβR‐I) kinase inhibitor, Ki26894, on bone metastasis of a highly bone‐metastatic variant of human breast cancer MDA‐MB‐231 cells, termed MDA‐MB‐231–5a‐D (MDA‐231‐D). Ki26894 blocked TGF‐β signaling in MDA‐231‐D cells, as detected by suppression of phosphorylation of Smad2 and inhibition of TGF‐β‐responsive reporter activity. Moreover, Ki26894 decreased the motility and the invasion of MDA‐231‐D cells induced by TGF‐βin vitro. Ki26894 also suppressed transcription of plasminogen activator inhibitor‐1 (PAI‐1), parathyroid hormone‐related protein (PTHrP), and interleukin‐11 (IL‐11) mRNA of MDA‐231‐D cells, which were stimulated by TGF‐β. X‐ray radiography revealed that systemic Ki26894 treatment initiated 1 day before the inoculation of MDA‐231‐D cells into the left ventricle of BALB/c nu/nu female mice resulted in decreased bone metastasis of breast cancer cells. Moreover, Ki26894 prolonged the survival of mice inoculated with MDA‐231‐D cells compared to vehicle‐treated mice. These findings suggest that TβR‐I kinase inhibitors such as Ki26894 may be useful for blocking the progression of advanced cancers. (Cancer Sci 2007; 98: 127–133)
Transforming growth factor (TGF)‐β is a multifunctional cytokine that regulates a wide range of cellular responses, including cell proliferation, differentiation, adhesion, migration, and apoptosis.( 1 , 2 ) It is a potent inhibitor of various types of cells, including most epithelial cells, whereas it stimulates deposition of extracellular matrix proteins and induction of epithelial‐to‐mesenchymal transition (EMT). TGF‐β thus plays two distinct and opposing roles in cancer progression. In early stages of carcinogenesis, it acts as a tumor suppressor by preventing cell proliferation, although in advanced stages of cancer, tumor cells often become refractory to TGF‐β‐mediated growth inhibition. TGF‐β is often overexpressed in tumor cells, and induces migration, invasion, and EMT of tumor cells and facilitates immunosuppression, deposition of extracellular matrix proteins and angiogenesis.( 2 , 3 , 4 ) Blocking of TGF‐β signaling in advanced stages of cancer may thus yield beneficial effects through inhibition of invasion and metastasis of cancer.( 5 )
TGF‐β transduces signals through two distinct serine‐threonine kinase receptors, termed type I (TβR‐I) and type II (TβR‐II).( 6 , 7 ) TβR‐II is the primary ligand‐binding receptor at the cell surface and contains constitutively active kinase. Upon ligand binding, the TβR‐II kinase transphosphorylates TβR‐I, and TβR‐I then transduces intracellular signals through various proteins, of which Smad proteins are the major signaling transducers for TGF‐β.( 6 , 7 ) Activated TβR‐I phosphorylates receptor‐regulated Smads (R‐Smads), i.e. Smad2 and Smad3, which interact with common‐mediator Smad (Smad4) and translocate to the nucleus where they regulate transcription of various target genes. Smad7 is an inhibitory Smad that inhibits TGF‐β signaling through interaction with activated TβR‐I and other mechanisms.
TGF‐β signaling can be regulated by various molecules, including antisense oligonucleotides to certain TGF‐β isoforms, monoclonal antibodies to TGF‐βs, soluble forms of TβR‐II, and small‐molecule compounds that act on TGF‐β receptor kinases.( 8 ) Of these, TβR‐I kinase is one potential target for blockade of TGF‐β signaling pathway. Recently, several small‐molecule compounds that bind to and inhibit TβR‐I kinase activity have been generated and shown to potently inhibit TGF‐β activity in vitro and in vivo.( 8 ) Small‐molecule inhibitors of TβR‐I kinase are highly specific for TβR‐I, although they also inhibit kinase activities of closely related molecules, that is, type I receptors for activin and nodal (activin‐receptor like kinase‐4; ALK‐4 and ALK‐7).( 9 )
An in vivo experimental bone metastasis model has been established using an intracardiac injection of cancer cells, including MDA‐MB‐231 cells.( 10 , 11 ) In this model, growth of cancer cells arrested in bone is easily observed as the progression of osteolysis on radiographs. This model is thus frequently used to explore the molecular interactions between the cancer cells and bone microenvironment. Several studies using this in vivo model suggested that TGF‐β and its transcriptional targets, including parathyroid hormone‐related protein (PTHrP) and interleukin‐11 (IL‐11), are the essential mediators of bone metastasis.( 12 , 13 ) More recent studies have revealed that Smad signaling is essential for the development of bone metastasis of breast cancers.( 14 , 15 )
Since TGF‐β signaling plays roles in breast cancer metastasis, inhibition of it may be a novel strategy for treatment of breast cancer metastasis.( 16 , 17 ) To examine the effects of TβR‐I inhibitors on bone metastasis in vivo, we first established a breast cancer cell line that can metastasize to bone in highly efficient fashion. We then showed that a novel small‐molecule inhibitor, Ki26894, decreased bone metastasis of breast cancer cells in vivo, suggesting that it and related molecules may be useful for treatment of advanced‐stage cancers.
Materials and methods
Reagents and cell lines. Ki26894 is a TβR‐I kinase inhibitor developed by Kirin Brewery Company (Gunma, Japan). A human breast cancer cell line, MDA‐MB‐231–5a,( 18 ) was kindly provided by Dr H. Yamada‐Okabe (Chugai Pharmaceutical Company, Tokyo, Japan). A human keratinocyte cell line, HaCaT, was provided by Dr Norbert E. Fusenig (DKFZ, Heidelberg, Germany). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin G, and 100 µg/mL streptomycin. Cells were grown in a 5% CO2 atmosphere at 37°C.
Establishment of a highly metastatic clone of MDA‐MB‐231–5a‐D. A breast cancer cell line that can metastasize to bone in highly efficient fashion was established from MDA‐MB‐231–5a, a subline with intermediate metastatic capacity derived from a human breast cancer, MDA‐MB‐231.( 18 ) Several clones from MDA‐MB‐231–5a were isolated by limiting dilution. Isolated cells were tested for bone metastatic capacity, and one of the clones, MDA‐MB‐231–5a‐D (MDA‐231‐D), was found to cause severe and highly efficient bone metastasis.
Immunoblotting. Immunoblotting was performed as described.( 9 ) Cell extracts were prepared using Nonidet P‐40 lysis buffer containing 20 mM Tris‐HCl pH 7.5, 150 mM NaCl, 1% Nonidet P‐40, 1% aprotinin, and 1 mM phenylmethylsulfonyl fluoride. Anti‐phospho‐Smad2 antibody (Upstate Biotechnology, Lake Placid, NY, USA) and anti‐Smad2/3 antibody (BD Transduction Laboratories, Lexington, KY, USA) were used for immunoblotting.
Lentiviral production and infection. To establish MDA‐231‐D cells that express the TGF‐β‐responsive reporter firefly luciferase and a control renilla luciferase (MDA‐231‐D‐TβFluc/Rluc), we used a lentiviral expression system.( 19 ) Briefly, cDNAs encoding 9xCAGA‐firefly luciferase and CMV‐renilla luciferase were inserted into the multicloning site (MCS) of the lentiviral vector construct CS‐CDF‐CG‐PRE.
For production of defective lentiviral vectors, 293FT cells (Invitrogen, Carlsbad, CA, USA) were transfected using Lipofectamine2000 (Invitrogen) with three plasmids: vector construct, VSV‐G and Rev expressing construct (pCMV‐VSV‐G‐RSV‐Rev), and packaging construct (pCAG‐HIVgp). The culture supernatants were collected 48 h after transfection, and viral particles were concentrated by centrifugation. For lentiviral infection, 7 × 105 MDA‐231‐D cells/dish in 10 cm culture dishes were infected with lentivirus vectors at 50 plaque‐forming units/cell.
Luciferase assay. MDA‐231‐D‐TβFluc/Rluc cells were seeded in duplicate in 12‐well plates. Luciferase activity was measured using the Dual‐Luciferase Reporter System (Promega, Madison, WI, USA), and normalized to renilla luciferase activity as described.( 9 )
Cell proliferation assay. HaCaT cells were seeded in duplicate at a density of 2.5 × 104 cells/well in 24‐well plates. On the following day, cells were pretreated for 1 h with 10 µM Ki26894 and then cultured with TGF‐β3 (1 ng/mL) for 4 days. Cells were trypsinized and counted with a Coulter Counter (Beckman Coulter, Fullerton, CA, USA).
Wound closure assay. MDA‐231‐D cells were seeded at 1 × 105 cells/well in 6‐well plates. After cells reached 80% confluency, a wound was incised with a pipette tip in the central area of culture, and TGF‐β3 (1 ng/mL) and Ki26894 (10 µM) were added. Photographs were taken under phase‐contrast microscopy immediately after the incision, and after 24 h of TGF‐β3 and Ki26894 treatment.
Migration and invasion assays. To assess cell motility, chamber assays were performed using a Cell Culture Insert (8‐µm pore size, 12‐well format; Becton Dickinson Labware, Franklin Lakes, NJ, USA), according to the manufacturer's instructions. To assess cell invasion, type I collagen (Nitta Gelatin, Osaka, Japan) was coated on the Cell Culture Insert. MDA‐231‐D cells were seeded at a concentration of 3 × 105 cells/chamber, and TGF‐β3 (1 ng/mL) was added to the lower wells as a chemo‐attractant. After 24‐h incubation with or without Ki26894 (10 µM), cells that had not moved to the lower wells were removed from the upper face of the filters using cotton swabs, and the cells that had moved to the lower surface of the filter were stained with a Diff‐Quik kit (Sysmex, Hyogo, Japan). Motility and invasion were quantified by visual counting after being photographed. Experiments were performed in duplicate. Mean values for three random fields were obtained for each well.
Quantitative real‐time reverse transcription polymerase chain reaction (RT‐PCR). Quantitative real‐time RT‐PCR was performed as described.( 20 ) Briefly, MDA‐231‐D cells were seeded at 1 × 105 cells/well in 6‐well plates. On the following day, cells were pretreated for 1 h with 10 µM Ki26894 and then cultured with TGF‐β3 (1 ng/mL). Twenty‐four hours after TGF‐β3 stimulation, total RNA was extracted from cells using Trizol reagent (Invitrogen). Total RNA was reverse transcribed by oligo‐dT priming using Superscript First‐Strand Synthesis System (Invitrogen). Quantitative real‐time PCR was then performed using Platinum SYBR Green qPCR SuperMix and the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primer sequences used were as follows: human hypoxanthine phosphoribosyltransferase 1 (HPRT1): forward 5′‐TTTGCTTTCCTTGGTCAGGC, reverse 5′‐GCTTGCGACCTTGACCATCT; human plasminogen activator inhibitor (PAI)‐1: forward 5′‐GGCTGACTTCACGAGTCTTTCA, reverse 5′‐ATGCGGGCTGAGACTATGACA; human PTHrP exon IV: forward 5′‐ACCTCGGAGGTGTCCCCTAAC, reverse 5′‐TCAGACCCAAATCGGACG; human IL‐11: forward 5′‐AGCGAGTGGATCACTGAAGTCC, reverse 5′‐TTGCCATGTCTCGCAGGC. The specificity of detected signals was confirmed by a dissociation curve consisting of a single peak. All samples were run in duplicate in each experiment. Values were normalized by human HPRT1.
Mouse strain, animal care, drug administration, and ex vivo bioassay of TGF‐β activity. All animal procedures were performed in the animal experiment room of the Japanese Foundation for Cancer Research (JFCR) according to the guideline proposed by the Science Council of Japan. Female BALB/c nu/nu mice (4 weeks old) were purchased from Charles River Japan, Inc. (Kanagawa, Japan). Mice were maintained under specific‐pathogen‐free conditions. Ki26894 was given orally mixed in chow (0.08%). The bioavailability of administered Ki26894 was examined using mouse serum. Blood was drawn by cardiac puncture from control mice or those administrated Ki26894 for 3 days orally, and centrifuged at 8000 rpm for 10 min in blood collection tubes (Microtainer; Becton Dickinson). Separated mouse serum was diluted four‐fold with DMEM containing 10% FBS and added to promoter‐reporter expressing cells (MDA‐231‐D‐TβFluc/Rluc) in duplicate in 24‐well plates, followed by TGF‐β3 stimulation for 24 h. Luciferase activity was measured using the Dual‐Luciferase Reporter System (Promega) as described previously.( 9 )
Intracardiac experimental metastasis model, radiographic analysis of bone metastasis, and survival studies in vivo. Subconfluent MDA‐231‐D cells (1 × 105) were suspended in 200 µL sterile DMEM containing 10% FBS and injected into the left ventricle of the heart with a 26‐gauge needle under anesthesia with diethylether. Development of bone metastasis was monitored by X‐ray radiography once a week for up to 6 week. Mice were anesthetized (50 mg/kg sodium pentobarbital), placed in prone position on wrapped films (Fuji Industrial X‐ray Film FR; Fuji Photo Film, Kanagawa, Japan), and exposed to X‐irradiation at 23 kV for 90 s using a Soft X‐ray SOFRON Apparatus (Sofron, Tokyo, Japan). Films were developed using a Fuji Medical Film Processor SEPROS SV (Fuji Photo Film) and inspected for visible bone lesions. All of the radiographs of bones in nude mice were analyzed carefully by three individuals including one orthopedist. Mouse survival was evaluated by Kaplan‐Meier analysis and the log‐rank test. P‐values less than 0.05 were considered significant. All statistical tests were two‐sided.
Histological examination of bone metastasis. Mouse hindlimbs, which contained osteolytic lesions on radiographs, were harvested and fixed in 10% formalin for 2 days. After demineralization using 10% ethylenediaminetetraacetic acid (EDTA) pH 7.2 solution for 3 days, paraffin sections were made following conventional methods. Sections were stained with hematoxylin and eosin.
Results
Ki26894 is a functional TGF‐β antagonist in vitro. To determine whether the small‐molecule compound, Ki26894, inhibits TGF‐β signaling, we examined the effect of Ki26894 on TGF‐β‐induced Smad phosphorylation. MDA‐231‐D cells were treated with TGF‐β3 and Ki26894, followed by immunoblotting of endogenous phospho‐Smad2. As shown in Fig. 1(a), phosphorylation of Smad2 induced by TGF‐β3 was completely inhibited by 10 µM Ki26894. We next examined the effects of Ki26894 on TGF‐β‐induced transcriptional activation in MDA‐231‐D‐TβFluc/Rluc cells. Ki26894 inhibited the luciferase activity induced by TGF‐β3 in dose‐dependent fashion (Fig. 1b). The concentration of Ki26894 required to reduce the level of TGF‐β‐induced transcription to basal level was 3 µM, and half‐maximal inhibition of the transcriptional activity induced by TGF‐β3 was obtained with 0.49 µM Ki26894. We then examined the effect of Ki26894 on cell proliferation. Since MDA‐231‐D cells are not sensitive to the growth‐inhibitory effects of TGF‐β3 (data not shown), human keratinocyte HaCaT cells were used here to verify the TGF‐β‐antagonistic properties of Ki26894. As shown in Fig. 1(c) and 10 µM Ki26894 did not affect the basal growth rate of HaCaT cells, and rescued the inhibition of growth mediated by 1 ng/mL of TGF‐β3.
Figure 1.
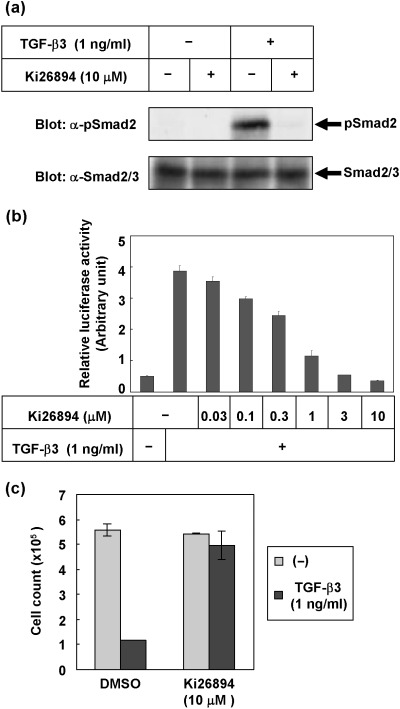
Ki26894 is a potent inhibitor of TGF‐β signaling in vitro. (a) Prevention of TGF‐β‐induced phosphorylation of Smad2 by Ki26894. MDA‐231‐D cells were treated with 1 ng/mL TGF‐β3 in the presence or absence of Ki26894 (10 µM) for 1 h. Cell lysates were subjected to immunoblotting with antiphospho‐Smad2 antibody (top panel) and anti‐Smad2/3 antibody (lower panel). (b) Inhibition of TGF‐β‐induced transcriptional activity by Ki26894. MDA‐231‐D‐TβFluc/Rluc cells were treated with 1 ng/mL TGF‐β3 in the presence or absence of various concentrations of Ki26894 (between 0.03 and 10 µM) for 1 h. Firefly luciferase activity was normalized to renilla luciferase activity, and relative luciferase activity is expressed as the mean ± SD of duplicate measurements. (c) Inhibition of TGF‐β‐induced growth suppression by Ki26894 in a human keratinocyte cell line, HaCaT. HaCaT cells were cultured in DMEM containing 10% fetal bovine serum and treated with 1 ng/mL TGF‐β3 in the presence or absence of Ki26894 (10 µM). Cells were counted 4 days after TGF‐β3 stimulation and cell numbers were plotted. Individual data points are the mean ± SD of triplicate determinations.
Ki26894 decreases motility and invasion of MDA‐231‐D cells induced by TGF‐β. Cell motility and invasion are critical metastatic events that occur in various epithelial cells during cancer progression. Since TGF‐β has been reported to induce motility and invasion of various cancer cell lines, we examined whether Ki26894 decreases the motility and the invasion of MDA‐231‐D cells induced by TGF‐β3. In a wound closure assay, TGF‐β3 caused migration of MDA‐231‐D cells, and this was inhibited by addition of 10 µM Ki26894 (Fig. 2a). Cell motility was also examined using a non‐coated chamber assay. As shown in Fig. 2(b), Ki26894 inhibited the migration of MDA‐231‐D cells in the absence as well as the presence of TGF‐β3, suggesting that Ki26894 inhibited the migration of cells induced by TGF‐β.
Figure 2.
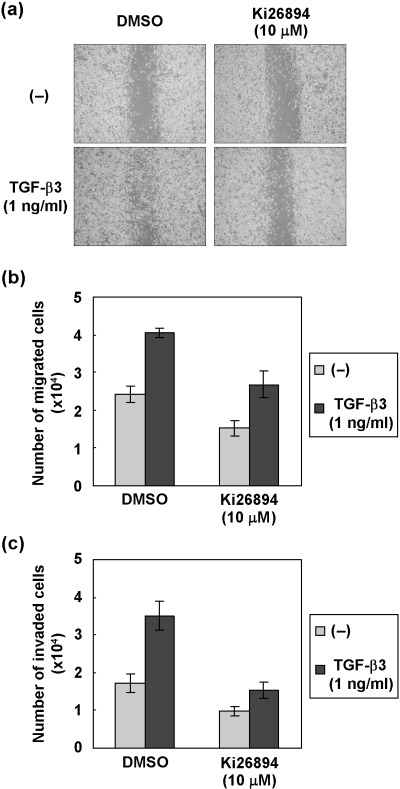
Ki26894 decreases the motility and inhibits invasion of MDA‐231‐D cells. (a) Motility of MDA‐231‐D cells was assessed by wound closure assay. Representative pictures from three experiments are shown. (b) Motility of MDA‐231‐D cells in the absence or presence of TGF‐β3 and/or Ki26894 was examined in an uncoated chamber assay. Results are shown as absolute numbers of counted cells. (c) Invasive behavior of MDA‐231‐D cells in the absence or presence of TGF‐β3 and/or Ki26894 was examined in a chamber assay with type I collagen‐coated membrane. Results are shown as absolute numbers of counted cells.
Bone tissue is composed of fibrillar type I collagen, and degradation of this matrix protein is thought to be one of the critical steps in the process of bone metastasis. We therefore examined the effects of Ki26894 on MDA‐231‐D cells in an in vitro invasion paradigm using a chamber coated with type I collagen, which mimics the native extracellular matrix in bone.( 21 ) As in the migration assays, Ki26894 reduced the invasion of MDA‐231‐D cells in the absence as well as the presence of TGF‐β3 (Fig. 2c). These findings suggest that Ki26894 prevented the TGF‐β‐induced invasive phenotype of MDA‐231‐D cells in vitro.
Ki26894 inhibits TGF‐β‐induced transcription of PAI‐1, PTHrP and IL‐11 in MDA‐231‐D cells. TGF‐β induces transcription of bone metastasis‐related genes, including cytokines and chemokines.( 13 , 14 , 22 ) In order to determine whether Ki26894 affects the expression of bone metastasis‐related genes, we performed quantitative real‐time RT‐PCR using MDA‐231‐D cells (Fig. 3). In MDA‐231‐D cells, TGF‐β induced mRNA of PAI‐1 (Fig. 3a), PTHrP (Fig. 3b) and IL‐11 (Fig. 3c). Ki26894 decreased TGF‐β‐induced as well as basal levels of mRNA of these genes, suggesting that Ki26894 suppresses induction of metastasis‐related genes of breast cancer cells.
Figure 3.
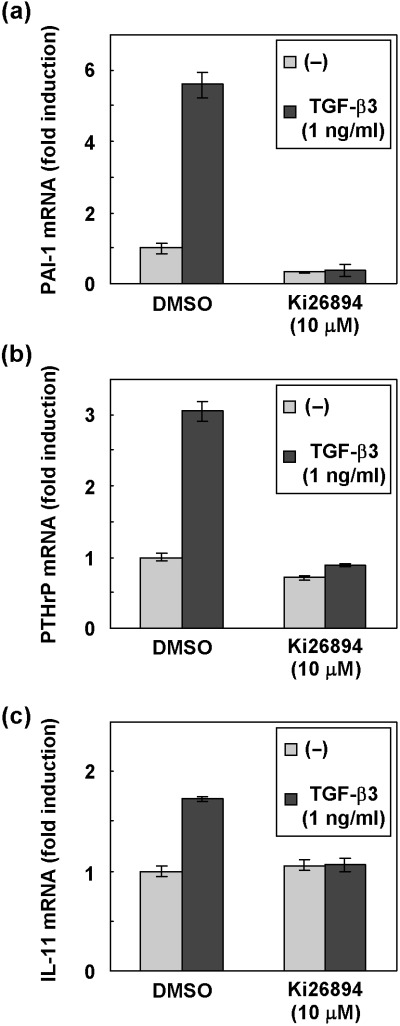
Effects of Ki26894 on the transcription of PAI‐1, PTHrP, and IL‐11. MDA‐231‐D cells were treated with TGF‐β3 (1 ng/mL) for 24 h in the presence or the absence of Ki26894 (10 µM). Total RNA was extracted, and the expression of PAI‐1 (a), PTHrP (b) and IL‐11 (c) was examined by quantitative real‐time PCR. Fold‐induction by TGF‐β3 stimulation is indicated. Each value is normalized to the expression of HPRT1 and represents a mean of duplicate determination.
Monitoring in vivo development of metastasis following intracardiac injection of MDA‐231‐D cells. Bone metastasis in breast cancer‐bearing mice was monitored by radiographic analysis once a week. Around knee joints, demarcated radiolucent areas were detected 5 weeks after inoculation, and the area expanded at 6 weeks (Fig. 4a). The entire animal body radiographs taken at 6 weeks revealed that the breast cancer cells metastasized to various bones, including brachia, scapula, ilium, femur, tibia and fibula (Fig. 4b). Bone metastasis was confirmed by histological examination of the samples from osteolytic lesions (Fig. 4c).
Figure 4.
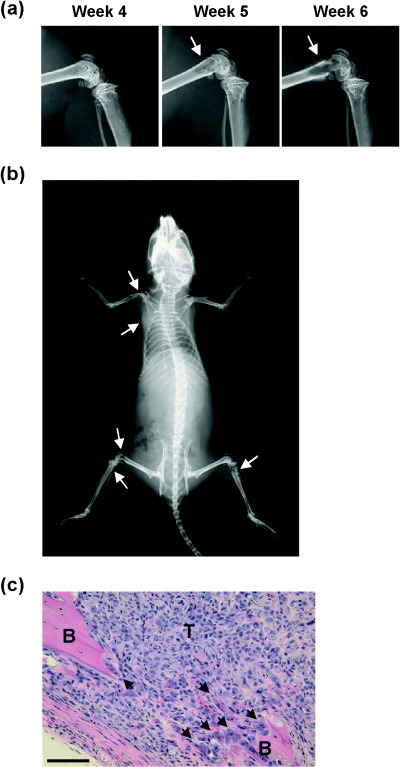
Detection of systemic bone metastasis in vivo following intracardiac injection of breast cancer cells. (a) Progression of bone destruction of femur by breast cancer detected by X‐ray radiography. MDA‐231‐D cells (1 × 105 cells/mouse) were injected into female nude mice, and subsequent metastasis was monitored by radiographic analysis once a week. Radiographs of femur from the same mouse on 4, 5, or 6 weeks are presented. Progressive demarcated radiolucent area (arrows) was seen in the femur. (b) Representative radiograph of a whole mouse. Radiograph was taken 6 weeks after inoculation. Osteolytic lesions were observed in left brachia, left scapula, left femur, left fibula, and right tibia (arrows). (c) Histological analysis. Mice were sacrificed, and their tibiae were subjected to histological examination. Bone marrow cavity was completely replaced by metastatic tumor cells (T). Osteoclasts (arrows) were observed at the endosteal bone surface, and tumor cells developed out of bone tissue (B). Bars, 100 µm. Hematoxylin and eosin staining.
Ki26894 prevents metastasis of MDA‐231‐D cells and prolongs the survival of MDA‐231‐D‐bearing mice. In order to examine the effects of TβR‐I inhibition on bone metastasis of MDA‐231‐D cells, Ki26894 was orally administered via food (0.08%) to MDA‐231‐D‐bearing mice. To verify the bioavailability of orally administered Ki26894, we first examined the effects of serum from Ki26894‐treated mice on TGF‐β‐induced reporter activity of MDA‐231‐D cells in vitro. As shown in Fig. 5(a), serum from Ki26894‐treated mice effectively decreased TGF‐β‐induced reporter activity.
Figure 5.
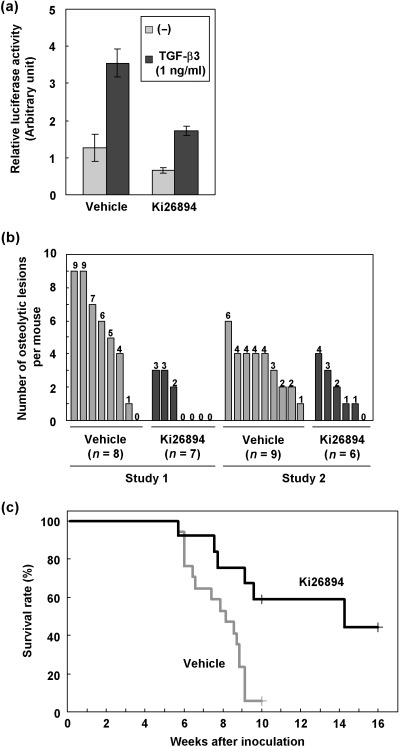
Orally administered Ki26894 decreases experimental metastasis of breast cancer. (a) Effects of Ki26894 in serum of mice treated with Ki26894 on TGF‐β‐induced transcription. MDA‐231‐D‐TβFluc/Rluc cells were pretreated with 100 µL of mouse serum collected from untreated or Ki26894‐treated mice as described in Materials and Methods. The cells were then stimulated or not with 1 ng/mL TGF‐β3, and harvested 24 h after initiation of stimulation. Firefly luciferase activities are normalized to renilla luciferase activities. (b) Effects of orally administered Ki26894 on bone metastasis in mice undergoing intracardiac injection of breast cancer cells. Ki26894 administration was initiated a day before tumor inoculation. Vehicle‐treated and Ki26894‐treated mice were carefully monitored, and subsequent metastasis at 6 weeks was determined on radiographs as described in Materials and Methods. Independent two studies were shown (Studies 1 and 2). (c) Survival rate of tumor‐bearing mice in Fig. 5(b) was evaluated by Kaplan‐Meier analysis. Survival of the Ki26894‐treated group (n = 13) was significantly longer than that of the vehicle‐treated group (n = 17) (P < 0.05, log‐rank test).
The therapeutic effects of Ki26894 were then assessed in the model of metastasis monitored by X‐ray radiography. The frequency of bone metastasis in Ki26894‐treated mice was lower than that in vehicle‐treated mice (Fig. 5b). Moreover, mean survival was prolonged to 86.6 days compared with 56.0 days in vehicle‐treated animals (Fig. 5c; P < 0.05). These results suggested that orally administrated Ki26894 effectively inhibited TGF‐β signaling and tumor burden in in vivo MDA‐231‐D‐bearing mice.
Discussion
Increased production of TGF‐β in human tumors is correlated with poor prognosis,( 2 ) and conditional overexpression of TGF‐β1 in mice has been shown to accelerate metastasis of mammary tumors in vivo.( 23 ) Recent studies have revealed that Smad signaling plays a critical role in metastasis of breast cancers.( 14 , 15 , 24 ) Dominant‐negative TβR‐II has been shown to reduce metastasis of some breast cancer cells, including MDA‐MB‐231 and MCF10A cells,( 25 ) and systemic expression of inhibitory Smad, Smad7, by adenovirus has been found to inhibit metastasis of JygMC(A) mouse mammary carcinoma cells,( 17 ) suggesting that blocking the biological effects of TGF‐β may be an attractive strategy for prevention of breast cancer metastasis.( 5 ) Use of specific small molecules designed to inhibit the effects of TGF‐β at the level of signaling receptors may be a promising strategy for antagonizing the effects of TGF‐β.( 8 , 9 , 26 , 27 ) In the present study, we characterize the activity of Ki26894 against human breast cancer cells in vitro and in vivo. MDA‐231‐D cells were chosen, because inoculation of cells into the left ventricle of BALB/c nu/nu female mice may be one of the best models for study of bone metastasis of breast cancer. Using MDA‐231‐D cells, we showed that Ki26894 is a potent TβR‐I kinase inhibitor that blocks the in vitro metastatic phenotypes induced by TGF‐β.
We have evaluated the effect of oral administration of Ki26894 on metastasis of MDA‐231‐D cells to bone in an intracardiac injection model. The administration dosage of Ki26894 was determined on the basis of two facts: that the dose did not influence on feed intake, and that serum from the dose‐treated mice effectively decreased TGF‐β‐induced reporter activity (Fig. 5a). Interestingly, we observed widespread skeletal metastasis in the control mice, whereas Ki26894 treatment significantly reduced both the incidence and the extent of bone metastasis detected by X‐ray radiography.
Many steps are involved in metastasis from the primary site to the skeleton, that is, new vessel formation, invasion, arrest in secondary capillary beds, extravasation, response to the bone micro‐environment, and proliferation of cancer cells.( 28 , 29 ) At sites of bone metastasis, PTHrP, IL‐11, and some cytokines produced by breast cancer cells stimulate osteoclast activity via the receptor activator of nuclear factor‐κB (RANK)‐RANK ligand (RANKL) system, and osteoclasts in turn induce the release of bone‐derived growth factors.( 13 , 30 , 31 ) TGF‐β promotes these steps through various mechanisms, including angiogenesis, induction of EMT, suppression of immune cell activity, and promotion of cancer cell survival.( 2 , 3 , 4 ) Among these steps, bone metastasis of MDA‐MB‐231 transplanted by cardiac puncture reflects those after the arrest of tumor cells in capillary vessels in bone. Since we demonstrated that invasion in type I collagen‐coated chambers was attenuated by treatment with Ki26894 in vitro, one possible reason for the reduction of incidence of bone metastasis by Ki26894 may be inhibition of TGF‐β‐induced migration and invasion of tumor cells into bone marrow. Another possible mechanism is suppression of bone destruction by osteoclasts, which was promoted by PTHrP and IL‐11 secreted by breast cancer cells. Consistent with this, Ki26894 suppressed TGF‐β‐induced transcription of PTHrP and IL‐11 mRNA in MDA‐231‐D cells in vitro.
The findings of significant reduction of bone metastasis as well as prolongation of survival of MDA‐231‐D‐bearing mice by Ki26894 treatment suggest that this therapeutic approach may potentially be of use in preventing early micrometastasis of breast cancers. During the preparation of this manuscript, Bandyopadhyay et al. (2006) reported that treatment with another TβR‐I kinase inhibitor, TβRI‐I, attenuated the bone metastasis of breast cancer cells.( 32 ) Furthermore, Ge et al. (2006) demonstrated that oral administration of the TβR‐I kinase inhibitor SD‐208 inhibited the growth and metastasis of syngeneic murine mammary carcinoma R3T.( 33 ) Their findings and those of the present study are essentially in agreement. In addition, we have shown that oral administration of Ki26894 prolongs the survival of MDA‐231‐D‐bearing mice, suggesting beneficial effects of Ki26894 or related molecules in preventing bone metastasis of breast carcinoma. Since TGF‐β plays roles in tumor cells as well as host cells of mice, it will be of great interest to examine the effects of Ki26894 on TGF‐β signaling in various cancer cells as well as in tumor micro‐environments.
Acknowledgments
We thank Dr H. Miyoshi (RIKEN, Tsukuba) for the lentivirus system, and Dr M. Matsuura (the Cancer Institute) for statistical analyses. We thank N. Kaneniwa, E. Kobayashi, Y. Yuuki, and S. Sato (the Cancer Institute) for technical assistance. This research was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Miyazono K, Suzuki H, Imamura T. Regulation of TGF‐β signaling and its roles in progression of tumors. Cancer Sci 2003; 94: 230–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Derynck R, Akhurst RJ, Balmain A. TGF‐β signaling in tumor suppression and cancer progression. Nat Genet 2001; 29: 117–29. [DOI] [PubMed] [Google Scholar]
- 3. Dumont N, Arteaga CL. Targeting the TGF‐β signaling network in human neoplasia. Cancer Cell 2003; 3: 531–6. [DOI] [PubMed] [Google Scholar]
- 4. Roberts AB, Wakefield LM. The two faces of transforming growth factor‐β in carcinogenesis. Proc Natl Acad Sci USA 2003; 100: 8621–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biswas S, Criswell TL, Wang SE, Arteaga CL. Inhibition of transforming growth factor‐β signaling in human cancer: targeting a tumor suppressor network as a therapeutic strategy. Clin Cancer Res 2006; 12: 4142–6. [DOI] [PubMed] [Google Scholar]
- 6. Heldin CH, Miyazono K, Ten Dijke P. TGF‐β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997; 390: 465–71. [DOI] [PubMed] [Google Scholar]
- 7. Shi Y, Massagué J. Mechanisms of TGF‐β signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- 8. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF‐β signalling inhibitors for cancer therapy. Nat Rev Drug Discov 2004; 3: 1011–22. [DOI] [PubMed] [Google Scholar]
- 9. Tojo M, Hamashima Y, Hanyu A et al. The ALK‐5 inhibitor A‐83–01 inhibits Smad signaling and epithelial‐to‐mesenchymal transition by transforming growth factor‐β. Cancer Sci 2005; 96: 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arguello F, Baggs RB, Frantz CN. A murine model of experimental metastasis to bone and bone marrow. Cancer Res 1988; 48: 6876–81. [PubMed] [Google Scholar]
- 11. Yoneda T, Sasaki A, Mundy GR. Osteolytic bone metastasis in breast cancer. Breast Cancer Res Treat 1994; 32: 73–84. [DOI] [PubMed] [Google Scholar]
- 12. Yin JJ, Selander K, Chirgwin JM et al. TGF‐β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999; 103: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kang Y, Siegel PM, Shu W et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–49. [DOI] [PubMed] [Google Scholar]
- 14. Kang Y, He W, Tulley S et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA 2005; 102: 13909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deckers M, Van Dinther M, Buijs J et al. The tumor suppressor Smad4 is required for transforming growth factor‐β‐induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006; 66: 2202–9. [DOI] [PubMed] [Google Scholar]
- 16. Tian F, DaCosta Byfield S, Parks WT et al. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res 2003; 63: 8284–92. [PubMed] [Google Scholar]
- 17. Azuma H, Ehata S, Miyazaki H et al. Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC (A) cells. J Natl Cancer Inst 2005; 97: 1734–46. [DOI] [PubMed] [Google Scholar]
- 18. Saito H, Tsunenari T, Onuma E, Sato K, Ogata E, Yamada‐Okabe H. Humanized monoclonal antibody against parathyroid hormone‐related protein suppresses osteolytic bone metastasis of human breast cancer cells derived from MDA‐MB‐231. Anticancer Res 2005; 25: 3817–23. [PubMed] [Google Scholar]
- 19. Shibuya K, Shirakawa J, Kameyama T et al. CD226 (DNAM‐1) is involved in lymphocyte function‐associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J Exp Med 2003; 198: 1829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maeda S, Hayashi M, Komiya S, Imamura T, Miyazono K. Endogenous TGF‐β signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J 2004; 23: 552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang X, Dutton CM, Qi WN, Block JA, Garamszegi N, Scully SP. siRNA mediated inhibition of MMP‐1 reduces invasive potential of a human chondrosarcoma cell line. J Cell Physiol 2005; 202: 723–30. [DOI] [PubMed] [Google Scholar]
- 22. Chen CR, Kang Y, Massagué J. Defective repression of c‐myc in breast cancer cells: a loss at the core of the transforming growth factor‐β growth arrest program. Proc Natl Acad Sci USA 2001; 98: 992–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muraoka‐Cook RS, Kurokawa H, Koh Y et al. Conditional overexpression of active transforming growth factor‐β1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res 2004; 64: 9002–11. [DOI] [PubMed] [Google Scholar]
- 24. Tian F, Byfield SD, Parks WT et al. Smad‐binding defective mutant of transforming growth factor‐β type I receptor enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res 2004; 64: 4523–30. [DOI] [PubMed] [Google Scholar]
- 25. Tang B, Vu M, Booker T et al. TGF‐β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest 2003; 112: 1116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DaCosta Byfield S, Major C, Laping NJ, Roberts AB. SB‐505124 is a selective inhibitor of transforming growth factor‐β type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2004; 65: 744–52. [DOI] [PubMed] [Google Scholar]
- 27. Hjelmeland MD, Hjelmeland AB, Sathornsumetee S et al. SB‐431542, a small molecule transforming growth factor‐β‐receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther 2004; 3: 737–45. [PubMed] [Google Scholar]
- 28. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2: 563–72. [DOI] [PubMed] [Google Scholar]
- 29. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002; 2: 584–93. [DOI] [PubMed] [Google Scholar]
- 30. Lindemann RK, Ballschmieter P, Nordheim A, Dittmer J. Transforming growth factor‐β regulates parathyroid hormone‐related protein expression in MDA‐MB‐231 breast cancer cells through a novel Smad/Ets synergism. J Biol Chem 2001; 276: 46661–70. [DOI] [PubMed] [Google Scholar]
- 31. Roodman GD. Mechanisms of bone metastasis. N Engl J Med 2004; 350: 1655–64. [DOI] [PubMed] [Google Scholar]
- 32. Bandyopadhyay A, Agyin JK, Wang L et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor‐β type I receptor kinase inhibitor. Cancer Res 2006; 66: 6714–21. [DOI] [PubMed] [Google Scholar]
- 33. Ge R, Rajeev V, Ray P et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor‐β type I receptor kinase in vivo . Clin Cancer Res 2006; 12: 4315–30. [DOI] [PubMed] [Google Scholar]