

Abstract
Protein gene product 9.5 (PGP9.5) is a neurospecific peptide that removes ubiquitin from ubiquitinated proteins and prevents them being targeted for degradation by proteosomes. Its expression is a potential marker of non‐small lung cancer, invasive colorectal cancer and esophageal squamous cell carcinoma. Gallbladder (GB) cancer is the most common malignant tumor of the biliary tract and is usually associated with gallstone disease, a late diagnosis, unsatisfactory treatment and a poor prognosis. To understand the role of PGP9.5 in GB cancer, we examined the methylation status of its promoter and its expression in surgical biopsy samples. Formalin‐fixed, paraffin‐embedded tumors and non‐neoplastic GB tissues (22 carcinomas, eight adenomas, 26 normal epithelia) were collected from patients who had undergone surgical resection. The methylation status of the promoter region of the PGP9.5 gene was determined by methylation‐specific polymerase chain reaction, and the expression of PGP9.5 was examined by immunohistochemistry using tissue microarrays. PGP9.5 promoter was methylated in 84.6% (22/26) of normal GB epithelium, 37.5% (3/8) of adenomas and 27.2% (6/22) of carcinomas. Most tumors with an unmethylated promoter exhibited positive staining for PGP9.5 in epithelial and neoplastic cells, but no PGP9.5 expression was observed in normal epithelia or in tumor tissues with a methylated promoter. No correlation was found between promoter hypomethylation of PGP9.5 and clinicopathological findings (i.e. age, sex, histological type or grade, N‐status, invasion depth or tumor stage) whereas PGP9.5 hypomethylation was found to be inversely correlated with the presence of a gallstone (P = 0.015). These results suggest that PGP9.5 promoter hypomethylation could be a reliable marker for GB cancer and that DNA hypomethylation might play an important role in re‐expression of the PGP9.5 gene in GB cancer. (Cancer Sci 2006; 97: 1205–1210)
Abbreviations:
- 5‐AzadC
5‐aza‐2′‐deoxycytidine
- AJPBD
anomalous junction of the pancreaticobiliary duct
- ESCC
esophageal squamous cell carcinoma
- GB
gallbladder
- MSP
methylation‐specific PCR
- PCR
polymerase chain reaction
- PGP9.5
protein gene product 9.5
- RT
reverse transcription.
Gallbladder cancer is the most common malignant lesion of the biliary tract and the fifth most common malignant neoplasm of the digestive tract, although it demonstrates pronounced geographic and sex‐related variations.( 1 , 2 ) The etiology of this tumor is complex, but there is a strong association between it and the presence of gallstones related to chronic inflammation.( 1 ) Unfortunately, GB cancer is an aggressive disease with a poor prognosis because of its inherent biology and its often advanced stage at diagnosis. In addition, the symptoms and signs of GB cancer are vague and non‐specific, and thus it is difficult to diagnose clinically. Therefore, an understanding of the biological features of GB carcinogenesis is needed to improve its prognosis.
Information on the molecular changes involved in GB carcinogenesis is limited.( 3 ) Considerable evidence indicates that mutations of the dominant oncogene (K‐ras) and tumor suppressor genes (p53, p16 and FHIT) may be involved.( 4 , 5 , 6 , 7 ) In addition, genome‐wide and specific chromosome arm allelotyping analyses demonstrate that a loss of heterozygosity at multiple sites in the genome is frequent in this neoplasm.( 8 , 9 , 10 ) Recently, several groups showed that multiple genes, including SHP, 3‐OST‐2, CDH13, CDH1, CHFR, DAP‐kinase, hMLH, p16, RASSF1A, RUNX3, APC, MGMT, RARβ2, p73 and Reprimo, are hypermethylated in GB tumor tissues,( 11 , 12 , 13 , 14 ) although considerable geographic differences are apparent in methylation patterns of candidate tumor suppressor genes, indicating that gene hypermethylation is a frequent epigenetic event in GB cancer. Moreover, epigenetic transcriptional silencing by promoter hypermethylation is believed to be a common feature in human cancer.( 15 , 16 ) Hence the delineation of epigenetic alterations that occur in GB cancer may be important for the development of molecular markers of early detection, prognosis and cancer treatment.
The PGP9.5 is a member of the ubiquitin carboxyl‐terminal hydrolase family and is involved in the processing of ubiquitin precursors and of ubiquitinated proteins.( 17 ) Ubiquitination of cellular proteins and ubiquitin‐dependent proteolysis regulate the degradation of cell cycle regulation proteins, such as the cyclins, p53 and p27Kip1,( 18 ) suggesting their involvement in cellular carcinogenesis. During recent years, many reports have indicated that the expression of PGP9.5 is related to tumor progression and that this may be useful as a potential marker for various human cancers, such as non‐small cell lung cancer,( 19 , 20 , 21 ) invasive colorectal cancer,( 22 ) pancreatic cancer,( 23 ) medullary thyroid carcinoma,( 24 ) ESCC,( 25 ) myeloma( 26 ) and neuroblastoma.( 27 ) Interestingly, a previous methylation analysis of the promoter region of PGP9.5 showed that a virtual absence of methylated CpG sites in lung cancer cell lines is consistent with relatively high PGP9.5 expression, and that dense methylation contributes to the lack of its expression in HeLa cells,( 28 ) suggesting that PGP9.5 expression is regulated by promoter methylation. Here, we analyzed the methylation status of CpG islands in the promoter region of human PGP9.5 in resected primary GB cancers and searched for correlations between this and the clinicopathological features of this disease.
Materials and Methods
Clinical samples. Primary tumor samples (n = 30) and corresponding non‐neoplastic GB tissues (n = 13) were obtained from GB cancer patients who had been treated with curative resectional surgery at the Department of Surgery in the Kyungpook National University Hospital between 1997 and 2003. In addition, non‐neoplastic GB tissues (n = 13) were obtained surgically from patients with gallstones and without tumor. The histopathologies of neoplastic and non‐neoplastic tissue were reviewed by a single experienced gastrointestinal pathologist. There were 22 carcinomas and eight adenomas. The mean ages of patients with GB carcinomas and adenomas were 65.5 ± 10.1 and 53.1 ± 12.7 years (P = 0.03), respectively, and the frequencies of female patients with GB carcinomas and adenomas were 63.6 and 62.5%, respectively. The histological subtypes of GB carcinomas were eight papillary, three adenosquamous, one mucinous, one neuroenodocrine carcinoma and nine non‐specified. Five tumors were confined within muscle layer (T1b) and six tumors invaded beyond serosa (T3). The other 11 tumors invaded deep into the subserosal layer (T2). The clinical stages of the tumors according to the International Union Against Cancer (UICC) sixth edition were stage IA (5/22), stage IB (3/22), stage IIB (11/22) and stage IV (3/22). All patients with GB carcinomas, except three with liver metastasis, had potentially curative resection. The histological types of GB adenomas were four papillary, three tubular and one pyloric gland type adenoma. Eleven carcinoma and three adenoma patients had a gallstone. This study was approved by the institutional review board of Kyungpook National University Hospital.
Microdissection and DNA extraction. Histological sections were microdissected manually to obtain 70% neoplastic cellularity. Genomic DNA was extracted from microdissected tumors and non‐neoplastic tissues as described previously.( 29 )
Cell culture and 5‐AzadC treatment. Human biliary tract cancer cell lines (SNU‐478, SNU‐869 and SNU‐1196) were obtained from the Korean Cell Line Bank (Seoul, Korea). All cells were cultured in RPMI‐1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat‐inactivated fetal bovine serum (Hyclon, Logan, UT, USA) at 37°C in a humidified atmosphere containing 5% CO2. SNU‐869 and SNU‐1196 cells were treated continuously with 5‐AzadC at different concentrations (1–10 µM) for 3 days; culture media were changed daily.
Total RNA isolation and RT‐PCR. Total RNA was extracted from the human biliary tract cancer cell lines with TRIzol (Invitrogen) following the manufacturer's instructions. The structural integrity of total RNA was confirmed by electrophoresis in 1.2% agarose‐formaldehyde gels. Residual genomic DNA was digested with RNase‐free DNase (Invitrogen). First‐strand cDNA was reverse‐transcribed from 2 µg of total RNA in a total volume of 20 µL using oligo(dT) and a SuperScript preamplification kit (Invitrogen). The resulting cDNA was amplified by PCR using primers specific to target genes. The forward and reverse primers used were: PGPF, 5′‐CATGGCGCCAGTTCAGAGGAC‐3′ and PGPR, 5′‐GTGGACAGCTGTGCACTCTG‐3′. The PCR protocol began with a 12‐min preincubation at 95°C, followed by 30 amplification cycles (45 s at 95°C, 30 s at 58°C, and 1 min at 72°C) and a final 5‐min incubation at 72°C. Amplified products were separated on a 1.2% agarose gel, visualized using ethidium bromide and photographed.
Bisulfite treatment and MSP. DNA methylation patterns in the CpG island of the PGP9.5 gene were determined by chemical modification of unmethylated, but not of methylated, cytosines to uracils, and subsequent PCR using primers specific for either methylated or modified unmethylated DNA.( 30 ) Briefly, 1 µg of genomic DNA was denatured with NaOH and modified with sodium bisulfite. The DNA samples were then purified with using Wizard DNA purification resin (Promega, Madison, WI, USA), again treated with NaOH, precipitated with ethanol and resuspended in distilled water. We used a modified two‐step nested MSP to detect PGP9.5 hypermethylation. The PCR conditions used for step one were as follows: 95°C for 10 min; 40 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s; a final extension for 5 min at 72°C. The PCR conditions used for step two were similar with the exception of the annealing temperature (54°C) and the cycle numbers used (25). All PCR amplifications were carried out using reagents supplied in GeneAmp DNA Amplification Kits using AmpliTaq Gold as the polymerase (PE Applied Biosystems, Foster City, CA, USA) on a PTC‐100 thermocycler (MJ Research, Watertown, MA, USA). PCR products obtained from step one were diluted 1:250 and subjected to the second step of MSP. The primers used for step one were as described previously.( 28 ) The primers used for selectively amplifying unmethylated or methylated alleles of the PGP9.5 gene were: forward unmethylated, 5′‐GGTTTGGTTGTATTATTTTGT‐3′ reverse unmethylated, 5′‐ACTACATCTTACAAACACCCA‐3′ forward methylated, 5′‐GGTTCGGTCGTATTATTTCGC‐3′ and reverse methylated, 5′‐ACTACATCTTACGAACGCCCG‐3′. CpGenome Universal methylated and unmethylated DNA (Chemicon, Temecula, CA, USA) was used as a positive control for the methylated and unmethylated genes, respectively. PCR products (10 µL) were analyzed on 2% agarose gels, stained with ethidium bromide and visualized under ultraviolet light. Each MSP was repeated at least once to confirm results.
Construction of GB cancer tissue microarrays and immunohistochemistry. Tissue microarrays included 22 carcinomas, five adenomas and eight non‐neoplastic GB tissues; they were constructed as described previously.( 31 ) Briefly, a cylindrical core sample from a selected region in a donor block was acquired using a thin‐walled 2 mm inner diameter (i.d.) needle. The acquired tissue was then transferred into the recipient block using a solid steel wire, which fitted closely in the needle. After constructing the array block, multiple 5‐µM sections were cut with a microtome. H&E‐stained sections were used for histological confirmation that samples were of tumor tissues in the arrays.
Protein gene product 9.5 expression was examined immunohistochemically as described previously.( 25 ) Histological sections of 5 µM were treated with xylene and dehydrated in an alcohol series. Endogenous peroxidase was blocked with phosphate buffer containing 2% H2O2. Microwave treatment was carried out for 4 min in Antigen Retrieval Glyca solution (Biogenex, San Ramon, CA, USA). After blocking with normal goat serum, array slides were incubated with polyclonal rabbit antiserum against PGP9.5 (Biogenesis, Sandown, NH, USA) at 1:1000 dilution for 1 h at room temperature. Vectastain ABC and DAB substrate kits (Vector Laboratories, Burlingame, CA, USA) were used to visualize antibody binding, and the sections were counterstained with hematoxylin. Immunohistochemical staining for PGP9.5 was interpreted by an experienced pathologist unaware of clinical data. Tumor sections showing >30% cell staining in five high‐power fields were classified as positive for staining.
Statistical analysis. Differences between the frequencies of PGP9.5 methylation in GB carcinomas, adenomas and non‐neoplastic lesions were analyzed using the χ2 or Fisher's exact test. All statistical analyses were carried out using SPSS software (SPSS Inc., Chicago, IL, USA). P‐values < 0.05 were regarded as being statistically significant.
Results
Promoter methylation analysis of PGP9.5 in neoplastic and non‐neoplastic GB tissues. The methylation status of the promoter region of the PGP9.5 gene was determined using a two‐step nested MSP in order to increase sensitivity for the detection of hypermethylation. Step one primers flanked the CpG islands of the PGP9.5 gene. Hence, these nested primers recognize the bisulfite‐modified template but do not discriminate between methylated and unmethylated alleles. PCR products of step one were diluted and subjected to a second step of MSP that incorporated one set of primers for PGP9.5 (labeled as unmethylated [U] or methylated [M]) that were designed to recognize the bisulfite‐induced modifications of unmethylated cytosines. A complete CpG island was found in the 400‐bp 5′ flanking region of the human PGP9.5 gene, coinciding with the 5′ end of minimal promoter (GenBank accession no. X17377). Representative examples of the MSP analysis are illustrated in Fig. 1. Unmethylated bands were detected in 100% of samples (i.e. both non‐malignant and malignant tissues), whereas methylated bands were detected strongly in normal GB epithelium but absolutely not in GB carcinoma. These methylation statuses were confirmed by bisulfite‐sequencing analysis in a subset of cases (data not shown). The frequencies of PGP9.5 methylation were: 84.6% (22/26) of normal GB epithelia, 37.5% (3/8) of adenomas, and 27.2% (6/22) of carcinomas (P < 0.05). Although hypomethylation of PGP9.5 in carcinoma was higher than that in adenoma, no significant difference in methylation frequencies was observed between the two. In addition, hypomethylation of the PGP9.5 gene in carcinomas was not found to be associated with age, sex, histological subtype, differentiation, tumor size, invasion depth or staging (data not shown). In carcinomas, complete hypomethylation was detected without gallstones (11/11), whereas 45.5% (5/11) of neoplastic tissues with gallstones showed hypomethylation of the PGP9.5 promoter (Fig. 2) (P = 0.015).
Figure 1.
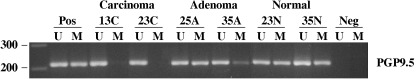
Representative methylation analysis of the protein gene product 9.5 (PGP9.5) promoter in neoplastic and non‐neoplastic gallbladder tissues by methylation‐specific polymerase chain reaction (MSP). MSP was carried out using unmethylation‐specific (U) and methylation‐specific (M) primers. Universal methylated and unmethylated DNA was used as a positive control for the M and U forms. Water was used as a negative control. A, adenoma; C, carcinoma; Lane M, amplified product obtained using M primers; Lane U, amplified product obtained using U primers; N, non‐neoplastic tissue; Neg, negative control; Pos, positive control.
Figure 2.
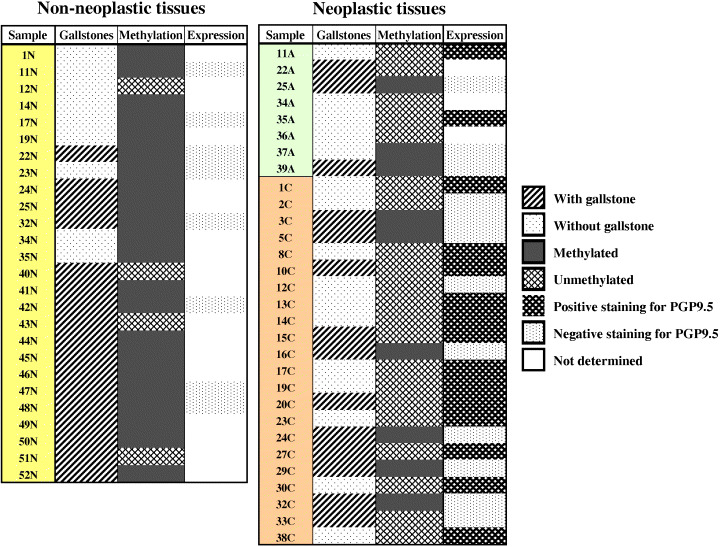
Summary of methylation pattern and immunohistochemical expression of protein gene product 9.5 (PGP9.5) gene in resected neoplastic and non‐neoplastic gallbladder tissue samples. A, adenomas; C, carcinomas; N, normal epithelia.
PGP9.5 protein expression in neoplastic and non‐neoplastic GB tissues. We next examined the status of PGP9.5 expression in resected GB cancers using an immunohistochemical approach. In a section of non‐neoplastic GB tissues (Fig. 3A), the majority of normal epithelial cells were negative for PGP9.5, whereas PGP9.5 stained positively in nerve tissues (arrow), which served as a positive internal control. A few isolated cells scatted throughout the lamina propria were also positive for PGP9.5. These positive cells were reminiscent of neuroendocrine cells, based on their morphologies and patterns of distribution. In addition, fibroblasts and myofibroblasts located within connective tissue were also positive. Unmethylated adenomas and carcinomas showed strong PGP9.5 expression in the epithelium and neoplastic cells of the lamina propria (Fig. 3B,C). In all neoplastic cells, PGP9.5 expression was detected in both cytoplasm and nucleus. Surrounding leukocytes also showed nuclear staining. In methylated adenoma and carcinoma, the cytoplasmic and nuclear expressions of PGP9.5 were markedly reduced in tumor cells versus surrounding leukocytes (Fig. 3D,E). Of the 18 GB cancer specimens analyzed showing PGP9.5 unmethylation, 13 (72.2%) exhibited positive PGP9.5 staining. However, no expression was observed in nine methylated adenomas and carcinomas (Fig. 2).
Figure 3.
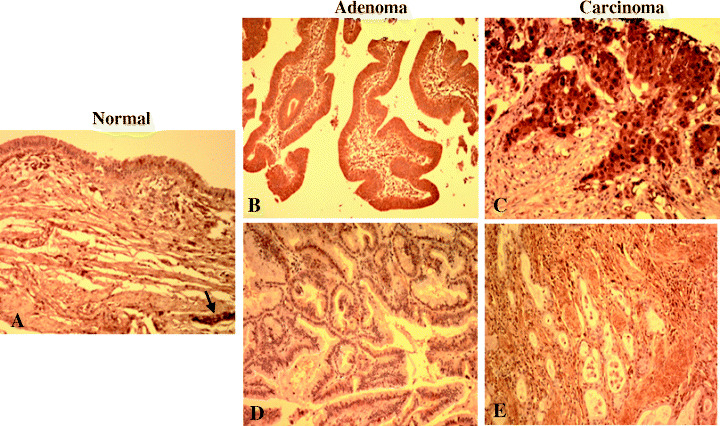
Immunohistochemical analysis of protein gene product 9.5 (PGP9.5) in formalin‐fixed, paraffin‐embedded sections of neoplastic and non‐neoplastic gallbladder tissues. (A) Normal gallbladder. PGP9.5 expression was seen mainly in peripheral nerve tissue (arrow). In unmethylated (B) adenoma and (C) carcinoma, surface epithelial cells, neoplastic cells and inflammatory cells in the lamina propria were found to express PGP9.5 in the cytoplasm and nuclei. In methylated (D) adenoma and (E) carcinoma, the cytoplasmic and nuclear expression of PGP9.5 was markedly reduced in tumor cells versus surrounding leukocytes.
Inverse correlation between PGP9.5 methylation and expression. To examine the potential role of DNA methylation in the regulation of PGP9.5 expression, we examined the levels of PGP9.5 expression and the methylation status of PGP9.5 promoter in human biliary tract cancer cell lines (SNU‐478, SNU‐869 and SNU‐1196). RT‐PCR and MSP analysis showed that PGP9.5 mRNA was present exclusively in SNU‐478 cells containing an unmethylated promoter, whereas PGP9.5 mRNA was not found in SNU‐869 and SNU‐1196 cells, which possess the methylated promoter (Fig. 4). Next, to further investigate whether promoter methylation could repress PGP9.5 gene expression, PGP9.5‐non‐expressing cells (SNU‐869 and SNU‐1196) were treated with the methyltransferase inhibitor 5‐AzadC for 3 days. Semi‐quantitative RT‐PCR then revealed that vehicle alone failed to induce PGP9.5 mRNA transcripts, whereas 5‐AzadC treatment induced the re‐expression of PGP9.5 mRNA (Fig. 5). Dose–response studies showed no apparent difference between the amounts of mRNA induced by 5‐AzadC at 1, 5 and 10 µM. These results suggest that demethylation of the PGP9.5 promoter is the underlying molecular mechanism responsible for the induction of PGP9.5 mRNA.
Figure 4.
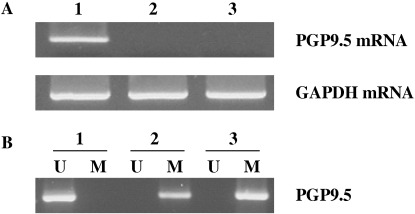
Expression of protein gene product 9.5 (PGP9.5) mRNA and methylation status of the PGP9.5 gene in human biliary tract cancer cell lines. (A) Total RNA was isolated from three human biliary tract cancer cell lines (SNU‐478, SNU‐869, SNU‐1196) and first‐strand cDNA was reverse‐transcribed and amplified using specific primers. Amplified products (10 µL) were run on 1.2% agarose gels and appeared at positions corresponding to expected base pair lengths. (B) Genomic DNA was isolated from all three cell lines and modified with sodium bisulfite, following methylation‐specific polymerase chain reaction using unmethylation‐specific (U) and methylation‐specific (M) primers. 1, SNU‐478 cells; 2, SNU‐869 cells; 3, SNU‐1196 cells.
Figure 5.
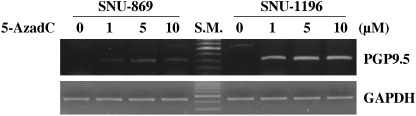
Re‐expression of protein gene product 9.5 (PGP9.5) after 5‐aza‐2′‐deoxycytidine (5‐AzadC) treatment. SNU‐869 and SNU‐1196 cells were treated for 3 days with the indicated concentration of 5‐AzadC, and reverse transcription–polymerase chain reaction analysis was then carried out on isolated total RNA. Note that PGP9.5 mRNA was re‐expressed in these cell lines after treatment with 5‐AzadC. glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control.
Discussion
The present study shows that CpG sites in the promoter region of the PGP9.5 gene are hypomethylated in GB cancer to increase its expression. In addition 5‐AzadC treatment efficiently induced PGP9.5 gene expression in PGP9.5 non‐expressing SNU‐869 and SNU‐1196 cells. These results suggest that PGP9.5 promoter hypomethylation could be a reliable marker for GB cancer, and that DNA hypomethylation might play an important role in restoration of PGP9.5 gene expression in GB cancers. The present study is the first to demonstrate that the malignant transformation of GB epithelia could be associated with PGP9.5 gene re‐expression for lost of methylation and protein expression.
Gallbladder cancer is a relatively uncommon malignancy with a poor survival rate.( 1 ) Despite recent improvements in diagnostic imaging and molecular techniques, most GB cancers are diagnosed at an advanced stage. At present, only surgical excision of all apparent malignancy is associated with an improved 5‐year survival rate. Therefore, new sensitive and specific markers for GB cancer are urgently needed to facilitate the early detection of this cancer among susceptible populations. In an effort to identify such biomarkers and to better understand the biology of this cancer, we analyzed the methylation status of the PGP9.5 gene in 30 patients and assessed its correlation with clinicopathological features. The findings of the present study suggest that the PGP9.5 gene may be a potential molecular target for early detection and therapy.
The importance of DNA methylation in the transcriptional silencing of cancer‐associated genes is being increasingly recognized.( 16 ) A variety of tumor suppressor, growth regulatory, apoptosis and mismatch repair genes are inactivated by promoter region hypermethylation in malignant neoplasms. The genome‐wide DNA hypomethylation found in various human cancers occurs primarily in normally methylated repetitive sequences.( 32 ) DNA hypomethylation at unique sequences could also increase the expression of cancer‐promoting genes.( 32 ) In fact, a correlation between site‐specific hypomethylation and transcriptional activation has been observed for genes MAGE,( 33 ) S100A4,( 34 ) synuclein γ ( 35 ) and others.( 32 , 36 ) In addition, the extent of hypomethylation appears to correlate with tumor grade and prognosis in certain cancers.( 37 ) However, our understanding of the global pattern of hypomethylation at multiple loci is limited to the few genes analyzed to date. Taken together, our data suggest the possibility that promoter‐specific hypomethylation resulting in restoration of affected gene and protein expression could be a crucial epigenetic event in human cancers.
Available evidence regarding the function of PGP9.5 in cancer is contradictory. PGP9.5 has been found to be silenced by hypermethylation in certain tumor types, such as pancreatic, ESCC and head and neck squamous cell carcinoma,( 38 , 39 , 40 ) which suggests that this molecule could act as a tumor suppressor. Conversely, numerous studies have found that PGP9.5 is overexpressed in various types of primary cancers as the result of or as the cause of transformation.( 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 ) Although PGP9.5 is known to be a dual function protein, that is, as a hydrolase to generate free ubiquitin and as a ligase to produce multi‐ubiquitin chains,( 17 , 41 ) both activities are involved in regulating ubiquitin levels. Moreover, the accumulation of ubiquitin has been documented in various types of primary cancers.( 42 , 43 ) Based on these observations, our finding that the PGP9.5 gene was re‐expressed in GB carcinoma and adenoma through hypomethylation suggests that its hypomethylation is tumor specific and that it is more likely to be an oncogene than a tumor suppressor gene. Additionally, plasminogen activator inhibitor‐1, which is involved in cancer growth and metastasis,( 44 , 45 ) was reported to be overexpressed in a PGP9.5‐expressed ESCC cell line.( 46 ) In lung cancer cells (H1299), PGP9.5 was found to be involved in the cytoplasmic degradation of p27Kip1 via its interaction and nuclear localization with JAB1, a Jun activation domain binding protein.( 47 ) Moreover, reduced p27Kip1 expression was found to be correlated with cancer development and poor survival.( 48 ) Interestingly, the cellular localization pattern of PGP9.5 protein in growing H1299 cells is similar to our finding that PGP9.5 was detected in the nucleus and cytoplasm of all neoplastic cells. Therefore, although the exact nature of the cellular redistribution of PGP9.5 between nucleus and cytoplasm remains to be determined, PGP9.5 appears to advance the tumorigenic pathway in cancer cells. Furthermore, it would be interesting to investigate the relationship between PGP9.5 and p27Kip1 expression in GB cancer.
It is surprising to note that hypomethylation of the PGP9.5 promoter occurred more frequently in GB cancers that were not associated with gallstones. Although the pathogenic mechanisms of GB cancers are not fully understood at the molecular level, most GB cancers are thought to be caused by mechanical (e.g. a stone) or chemical (e.g. regurgitation of pancreatic juice) irritation. It was reported in a previous study that hypermethylation of multiple genes seems to be associated with inflammatory changes of gallbladder epithelium.( 12 , 13 ) Because gallstones give rise to cholecystitis and are found frequently in GB cancers, we speculated that chronic inflammatory stimulus caused by gallstone could have an influence on hypermethylation of the PGP9.5 gene. Another factor linked to lack of PGP9.5 methylation in tumors with the absence of gallstones may be AJPBD. It is relatively frequent in East Asia and is known to play an important role in the development of GB cancers.( 49 ) K‐ras mutations are rare and p53 mutations occur early during the multistage pathogenesis of gallstone‐associated GB cancers, whereas AJPBD‐associated GB cancers are characterized by K‐ras mutations and a relatively late onset of p53 mutations,( 4 , 6 ) suggesting two pathways involved in the pathogenesis of GB cancers. Although the data regarding the presence of AJPBD are limited, because we do not carry out endoscopic retrograde cholangiopancratography routinely in GB cancer, we assume that pancreatic juice reflux with consequent hyperplasia of the GB epithelium influences hypomethylation of the PGP9.5 gene, as does the K‐ras mutation.
Acknowledgments
This work was supported by the Regional Technology Innovation Program of the Ministry of Commerce, Industry and Energy (grant no. RTI04‐01‐01), by a Biomedical Research Institute grant from Kyungpook National University Hospital (2005), and by the Brain Korea 21 Project in 2006.
References
- 1. Misra S, Chaturvedi A, Misra NC, Sharma ID. Carcinoma of the gallbladder. Lancet Oncol 2003; 4: 167–76. [DOI] [PubMed] [Google Scholar]
- 2. Lazcano‐Ponce EC, Miquel JF, Munoz N et al. Epidemiology and molecular pathology of gallbladder cancer. CA Cancer J Clin 2001; 51: 349–64. [DOI] [PubMed] [Google Scholar]
- 3. Wistuba II, Gazdar AF. Gallbladder cancer: lessons from a rare tumour. Nat Rev Cancer 2004; 4: 695–706. [DOI] [PubMed] [Google Scholar]
- 4. Wistuba II, Sugio K, Hung J et al. Allele‐specific mutations involved in the pathogenesis of endemic gallbladder carcinoma in Chile. Cancer Res 1995; 55: 2511–15. [PubMed] [Google Scholar]
- 5. Ajiki T, Fujimori T, Onoyama H et al. K‐ras gene mutation in gall bladder carcinomas and dysplasia. Gut 1996; 38: 426–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanada K, Itoh M, Fujii F, Tsuchida A, Ooishi H, Kajiyama G. K‐ras and p53 mutations in stage I gallbladder carcinoma with an anomalous junction of the pancreaticobiliary duct. Cancer 1996; 77: 452–8. [DOI] [PubMed] [Google Scholar]
- 7. Wistuba II, Ashfaq R, Maitra A, Alvarez H, Riquelme E, Gazdar AF. Fragile histidine triad gene abnormalities in the pathogenesis of gallbladder carcinoma. Am J Pathol 2002; 160: 2073–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hidaka E, Yanagisawa A, Sakai Y, Seki M, Kitagawa T, Setoguchi T. Losses of heterozygosity on chromosomes 17p and 9p/18q may play important roles in early and advanced phases of gallbladder carcinogenesis. J Cancer Res Clin Oncol 1999; 125: 439–43. [DOI] [PubMed] [Google Scholar]
- 9. Wistuba II, Tang M, Maitra A et al. Genome‐wide allelotyping analysis reveals multiple sites of allelic loss in gallbladder carcinoma. Cancer Res 2001; 61: 3795–800. [PubMed] [Google Scholar]
- 10. Wistuba II, Maitra A, Carrasco R et al. High resolution chromosome 3p, 8p, 9q and 22q allelotyping analysis in the pathogenesis of gallbladder carcinoma. Br J Cancer 2002; 87: 432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tozawa T, Tamura G, Honda T et al. Promoter hypermethylation of DAP‐kinase is associated with poor survival in primary tract carcinoma patients. Cancer Sci 2004; 95: 736–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. House MG, Wistuba II, Argani P et al. Progression of gene hypermethylation in gallstone disease leading to gallbladder cancer. Ann Surg Oncol 2003; 10: 882–9. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi T, Shivapurkar N, Riquelme E et al. Aberrant promoter hypermethylation of multiple genes in gallbladder carcinoma and chronic cholecystitis. Clin Cancer Res 2004; 10: 6126–33. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi T, Suzuki M, Shigematsu H et al. Aberrant methylation of Reprimo in human malignancies. Int J Cancer 2005; 115: 503–10. [DOI] [PubMed] [Google Scholar]
- 15. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000; 16: 168–74. [DOI] [PubMed] [Google Scholar]
- 16. Jones PA, Baylin SB. The fundamental role of epigentic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 17. Wilkinson KD, Lee KM, Deshpande S, Duerksen‐Hughes P, Boss JM, Pohl J. The neuron‐specific protein PGP9.5 is a ubiquitin carboxyl‐terminal hydrolase. Science 1989; 246: 670–3. [DOI] [PubMed] [Google Scholar]
- 18. Spataro V, Norbury C, Harris AL. The ubiquitin‐proteosome pathway in cancer. Br J Cancer 1998; 77: 448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hibi K, Liu Q, Beaudry GA et al. Serial analysis of gene expression in non‐small‐cell lung cancer. Cancer Res 1998; 58: 5690–4. [PubMed] [Google Scholar]
- 20. Hibi K, Westra WH, Borges M, Goodman S, Sidransky D, Jen J. PGP9.5 as a candidate tumor marker for non‐small‐cell lung cancer. Am J Pathol 1999; 155: 711–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sasaki H, Yukiue H, Moriyama S et al. Expression of protein gene product 9.5 is correlated with T‐status in non‐small‐cell lung cancer. Jap Clin Oncol 2001; 31: 532–5. [DOI] [PubMed] [Google Scholar]
- 22. Yamazaki T, Hibi K, Takase T et al. PGP9.5 as a marker for invasive colorectal cancer. Clin Cancer Res 2002; 8: 192–5. [PubMed] [Google Scholar]
- 23. Tezel E, Hibi K, Nagasaka T, Nakao A. PGP9.5 as a prognostic factor in pancreatic cancer. Clin Cancer Res 2000; 6: 4764–7. [PubMed] [Google Scholar]
- 24. Takano T, Miyauchi A, Matsuzuka F et al. PGP9.5 mRNA could contribute to molecular‐based diagnosis of medullary thyroid carcinoma. Eur J Cancer 2004; 40: 614–18. [DOI] [PubMed] [Google Scholar]
- 25. Takase T, Hibi K, Yamazaki T et al. PGP9.5 overexpression in esophageal squamous cell carcinoma. Hepato-Gastroenterology 2003; 50: 11–13. [PubMed] [Google Scholar]
- 26. Otsuki T, Yata K, Takata‐Tomokuni A et al. Expression of protein gene product 9.5 (PGP9.5)/ubiquitin‐C‐terminal hydrolase 1 (UCHL‐1) in human myeloma cells. Br J Haemat 2004; 127: 292–8. [DOI] [PubMed] [Google Scholar]
- 27. Yanagisawa TY, Sasahara Y, Fujie H et al. Detection of the PGP9.5 and tyrosine hydroxylase mRNAs for minimal residual neuroblastoma cells in bone marrow and peripheral blood. Tohoku J Exp Med 1998; 184: 229–40. [DOI] [PubMed] [Google Scholar]
- 28. Bittencourt Rosas SL, Caballero OL, Dong SM, De Costa Carvalho MG, Sidransky D, Jin J. Methylation status in the promoter region of the human PGP9.5 gene in cancer and normal tissues. Cancer Lett 2001; 170: 73–9. [DOI] [PubMed] [Google Scholar]
- 29. Moskaluk CA, Kern SE. Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997; 150: 1547–52. [PMC free article] [PubMed] [Google Scholar]
- 30. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kononen J, Bubendorf L, Kallioniemi A et al. Tissue microarray for high‐throughput molecular profiling of tumor specimens. Nat Med 1998; 4: 844–7. [DOI] [PubMed] [Google Scholar]
- 32. Enrlich M. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21: 5400–13. [DOI] [PubMed] [Google Scholar]
- 33. Jang SJ, Soria JC, Wang L et al. Activation of melanoma antigen tumor antigens occurs early in lung carcinogenesis. Cancer Res 2001; 61: 7959–63. [PubMed] [Google Scholar]
- 34. Rosty C, Ueki T, Argani P et al. Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am J Pathol 2002; 160: 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gupta A, Godwin AK, Vanderveer L, Lu A, Liu J. Hypomethylation of the synuclein gene CpG island promotes its aberrant expression in breast carcinoma and ovarian carcinoma. Cancer Res 2003; 63: 664–73. [PubMed] [Google Scholar]
- 36. Sato N, Maitra A, Fukushima N et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res 2003; 63: 4158–66. [PubMed] [Google Scholar]
- 37. Lin CH, Hsieh SY, Sheen IS et al. Genome‐wide hypomethylation in hepatocellular carcinogenesis. Cancer Res 2001; 61: 4238–43. [PubMed] [Google Scholar]
- 38. Sato N, Fukushima N, Maitra A et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high‐throughput microarrays. Cancer Res 2003; 63: 3735–42. [PubMed] [Google Scholar]
- 39. Tokumaru Y, Yamashita K, Osada M et al. Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res 2004; 64: 5982–7. [DOI] [PubMed] [Google Scholar]
- 40. Mandelker DL, Yamashita K, Tokumaru Y et al. PGP9.5 promoter hypermethylation is an independent prognostic factor for esophageal squamous cell carcinoma. Cancer Res 2005; 65: 4963–868. [DOI] [PubMed] [Google Scholar]
- 41. Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT. The UCH‐L1 gene encodes two opposing enzymatic activities that affect α‐synuclein degradation and Parkinson's disease susceptibility. Cell 2002; 111: 209–18. [DOI] [PubMed] [Google Scholar]
- 42. Ishibashi Y, Takada K, Joh K, Ohkawa K, Aoki T, Matsuda M. Ubiquitin immunoreactivity in human malignant tumors. Br J Cancer 1991; 63: 320–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishibashi Y, Hanyu N, Suzuki Y et al. Quantitative analysis of free ubiquitin and multi‐ubiquitin chain in colorectal cancer. Cancer Lett 2004; 211: 111–17. [DOI] [PubMed] [Google Scholar]
- 44. Hibi K, Kodera Y, Ito K, Akiyama S, Shirane M, Nakao A. Plasminogen activator inhibitor‐1 is a downstream mediator of the PGP9.5‐related oncogenic pathway in esophageal squamous cell carcinoma. Anticancer Res 2004; 24: 3731–4. [PubMed] [Google Scholar]
- 45. Reuning U, Magdolen V, Wilhelm O et al. Multifunctional potential of the plasminogen activation system in tumor invasion and metastasis. Int J Oncol 1998; 13: 893–906. [DOI] [PubMed] [Google Scholar]
- 46. Bajou K, Noel A, Gerard RD et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nature Med 1998; 4: 923–8. [DOI] [PubMed] [Google Scholar]
- 47. Caballero OL, Resto V, Patturajan M et al. Interaction and colocalization of PGP9.5 with JAB1 and p27Kip1. Oncogene 2002; 21: 3003–10. [DOI] [PubMed] [Google Scholar]
- 48. Slingerland J, Pagano MJ. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183: 10–17. [DOI] [PubMed] [Google Scholar]
- 49. Kimura K, Ohto M, Saisho H et al. Association of gallbladder carcinoma and anomalous pancreaticobiliary ductal union. Gastroenterology 1985; 89: 1258–65. [DOI] [PubMed] [Google Scholar]