

Abstract
Germline mutations of the tumor suppressor gene MEN1 are found not only in typical multiple endocrine neoplasia type 1 (MEN1) but also in its incomplete forms such as familial isolated hyperparathyroidism (FIHP) and apparently sporadic parathyroid tumor (ASPT). No definitive genotype–phenotype correlation has been established between these clinical forms and MEN1 gene mutations. We previously demonstrated that mutant menin proteins associated with MEN1 are rapidly degraded by the ubiquitin–proteasome pathway. To examine whether the intracellular stability of mutant menin is correlated with clinical phenotypes, we developed a method of evaluating menin stability and examined 20 mutants associated with typical MEN1 (17 missense, two in‐frame deletion, one nonsense) and 21 mutants associated with FIHP or ASPT (19 missense, two in‐frame deletion). All tested mutants associated with typical MEN1 showed reduced stability. Some missense and in‐frame deletion mutants (G28A, R171W, T197I, E255K, E274A, Y353del and E366D) associated with FIHP or ASPT were almost as stable as or only slightly less stable than wild‐type menin, while others were as unstable as those associated with typical MEN1. Some stable mutants exhibited substantial biological activities when tested by JunD‐dependent transactivation assay. These findings suggest that certain missense and in‐frame mutations are fairly stable and retain intrinsic biological activity, and might be specifically associated with incomplete clinical phenotypes. The menin stability test will provide useful information for the management of patients carrying germline MEN1 mutations especially when they have missense or in‐frame variants of ambiguous clinical significance. (Cancer Sci 2011; 102: 2097–2102)
Menin is a tumor suppressor protein encoded by MEN1, a gene responsible for multiple endocrine neoplasia type 1 (MEN1), a familial cancer syndrome typically characterized by the development of multiple tumors in the pituitary, parathyroid and enteropancreatic endocrine tissues.( 1 , 2 ) Menin is a nuclear protein having C‐terminal nuclear localizing signal sequences,( 3 ) and exhibits modulatory activity on gene expression such as repression of JunD‐dependent transcription.( 4 ) Diverse roles have been implicated for menin, including cell cycle control, cell differentiation and DNA repair, which are likely to be conferred by interaction with various menin‐binding proteins.( 2 ) Menin is considered to be a scaffold protein tethering such proteins to specific gene loci.( 5 ) Although the mechanism of how menin is involved in gene regulation has been elucidated to some extent, the basis for tissue‐specific tumorigenesis in MEN1 remains unknown.
Heterozygous germline mutations of the MEN1 gene are found in 70–90% of patients clinically diagnosed as MEN1.( 6 ) More than 500 different loss‐of‐function mutations have been identified, which are distributed throughout the gene with no apparent hot spot.( 2 , 6 , 7 ) Germline MEN1 mutations have also been found in a subset of familial isolated hyperparathyroidism (FIHP),( 8 , 9 , 10 ) defined as familial primary hyperparathyroidism not associated with other diseases, and in a few patients clinically diagnosed as having sporadic parathyroid tumor.( 11 ) The latter situation is called apparently sporadic parathyroid tumor (ASPT) because of its heritable, potentially familial nature. Because parathyroid tumor is the most frequent and earliest clinical expression of MEN1, patients with FIHP or ASPT carrying germline MEN1 mutations might develop full manifestations of MEN1. Indeed, identical mutations have been found in both typical MEN1 and FIHP families,( 7 ) indicating that if these mutations are found in FIHP or ASPT, the patients should be treated as those with a predisposition to typical MEN1.
In contrast, it is also recognized that some mutation carriers in FIHP families do not develop endocrine tumors, even at relatively advanced age.( 12 ) Moreover, missense or in‐frame insertion/deletion (indel) mutations, which do not cause protein truncation, account for approximately half of the mutations found in FIHP in contrast to the lower prevalence (approximately 30%) in typical MEN1.( 6 , 7 ) These findings suggest that some missense and in‐frame indel mutations might be low‐penetrance, low‐expressivity mutations specifically associated with incomplete, milder forms of MEN1, and carriers of these mutations might never or less frequently develop the full MEN1 manifestations. However, no clinically relevant genotype–phenotype correlation has been established to date.( 7 , 13 )
A diagnostic DNA test for MEN1 germline mutations is beneficial to the patients with endocrine tumors suggestive of MEN1 and offspring of MEN1 patients.( 2 ) However, it is often difficult to distinguish a disease‐causing mutation from a rare benign polymorphism especially when it is a novel missense mutation found in a patient with incomplete forms of MEN1. We previously demonstrated that menin missense mutants associated with typical MEN1, defined as the occurrence of tumors in at least two of the three main MEN1‐related endocrine tissues, are degraded rapidly by the ubiquitin–proteasome pathway when expressed in culture cells.( 14 ) Our previous study also revealed that a FIHP‐associated mutant E255K is as stable as the wild‐type menin. However, the correlation between the stability of menin mutants and clinical expressions was not clear because the number of mutations examined was small and the mutants were not examined for biological activity. Because the stability of menin mutants might be a useful indicator of clinical outcome, we developed a new immunocytochemical method to precisely evaluate menin stability, and examined more mutants associated with MEN1 and its incomplete forms. We also examined biological activity of some menin variants by JunD‐dependent transactivation experiments.
Materials and Methods
Plasmid construction. Human menin cDNA, donated by Drs M. Ohki and F. Hosoda, National Cancer Center Research Institute, Tokyo, Japan, was ligated to the expression vector pCMV‐Tag2 (Agilent Technologies, Santa Clara, CA, USA) to generate pCMV‐Tag2‐menin, expressing FLAG‐tagged wild‐type menin as described previously.( 14 ) Expression plasmids for menin variants were generated with QuikChange site‐directed mutagenesis kit (Agilent Technologies) and the mutations were confirmed by nucleotide sequencing. The mouse menin cDNA was obtained by PCR amplification using a spleen cDNA library as the template as previously described.( 15 ) The previously reported variant sequences and their corresponding phenotypes were according to the references as follows: P12L, L22R, K119del, H139D, A160P, A242V, A309P, T344R, E363del, W436R and R460X;( 16 ) G28A;( 17 ) D153V and A411P;( 18 ) G156C, F364C and F447L;( 19 ) A160T and D418N;( 20 ) R171W and E366D;( 21 ) V184E;( 9 ) T197I and Y353del;( 22 ) W220L and Y351N;( 23 ) R229L;( 24 ) S253W and E274A;( 11 ) E255K;( 10 ) Q260P;( 25 ) L264P and L267P;( 26 ) P277H;( 27 ) G305D;( 12 ) H317Y;( 14 ) P320R;( 28 ) P320L;( 29 ) L414del;( 30 ) and S555N.( 31 )
The expression vector pCMV‐BICEP‐4 (Sigma, St. Louis, MO, USA), designed to allow translation of two proteins from one bicistronic mRNA, was used for transient co‐expression of N‐terminal FLAG‐tagged and Myc‐tagged menin proteins. Two series of plasmids were constructed: one series expressed FLAG‐tagged wild‐type menin and Myc‐tagged variant menin; and the other series expressed FLAG‐tagged variant menin and Myc‐tagged wild‐type menin. As a control, a plasmid expressing both FLAG‐tagged and Myc‐tagged wild‐type menin was constructed.
Cell culture and transfection. COS7, 293T and WI38VA13 cells were maintained in DMEM containing 10% heat‐inactivated FBS as previously described.( 14 ) Expression plasmids were introduced into culture cells with FuGENE 6 (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s instructions. For degradation experiments, 28 h after transfection with plasmids expressing FLAG‐tagged menin, COS7 cells were treated with 20 μg/mL cycloheximide (CHX) and/or 25 μM MG132 as previously described,( 14 ) incubated for 6 h and then harvested for immunoblotting against the FLAG tag as described below.
Immunoblotting analysis of steady‐state expression levels. Semiconfluent 293T cells were transfected with 0.3 μg pCMV‐BICEP‐4‐based plasmids in a well of a 24‐well collagen‐coated microplate (IWAKI, Tokyo, Japan). Twenty‐eight hours after transfection, whole cell lysates were prepared, separated by 10% SDS‐PAGE and blotted on membranes as previously described.( 14 )
Myc‐tagged menin was detected with peroxidase‐conjugated anti‐c‐Myc monoclonal antibody (SC40HRP; Santa Cruz Biotechnology, Santa Cruz, CA, USA) coupled with ECL‐Plus reagents (GE Healthcare, Waukesha, WI, USA). The blot membranes were then washed in TBS containing 0.1% Tween 20 until the bands became undetectable, and FLAG‐tagged menin was detected with alkaline phosphatase‐conjugated anti‐FLAG monoclonal antibody (A9469; Sigma) coupled with CDP‐Star reagent (New England Biolabs, Ipswich, MA, USA). The membranes were exposed to X‐ray films, and density of the target bands was quantified with the Molecular Image FX densitometer (Bio‐Rad Laboratories, Hercules, CA, USA).
Fluorescent immunocytochemical analysis of steady‐state expression levels. Semiconfluent WI38VA13 cells were transfected with 0.25 μg plasmid in a well of LabTek II 8‐well chamber slides (Nalge Nunc International, Rochester, NY, USA). Forty‐eight hours after transfection, cells were fixed with 4% paraformaldehyde in PBS for 10 min, permealized with 0.1% Triton X‐100 in PBS for 5 min, blocked with 3% BSA in PBS for 10 min, and incubated with the mixture of anti‐FLAG M2 monoclonal antibody FITC conjugate (F4049; Sigma) at 50‐fold dilution and Cy3‐conjugated anti‐c‐myc monoclonal clone 9E10 (C6594; Sigma) at 500‐fold dilution for 1 h. Cells were then washed three times with 3% BSA in PBS and mounted in PBS. All fixing and staining procedures were conducted at room temperature.
Digital photographs were taken with a fluorescence microscope (BX51; Olympus, Tokyo, Japan) equipped with a CCD camera system (DP70; Olympus) and analyzed with WinROOF V6.0 software (Mitani, Tokyo, Japan). The Olympus dichroic mirror/filter unit NIBA and WIG were used for fluorescein and Cy3 fluorescence observation, respectively. The nuclei of transfected cells were recognized by the expression of wild‐type menin. The ratios of the mean numerical value of fluorescence intensity for mutant menin to that for wild‐type menin in each nucleus was calculated, and normalized by the ratio obtained in the cell nuclei transfected with the control plasmid expressing both FLAG‐ and Myc‐tagged wild‐type menin proteins.
JunD‐dependent transactivation repression assay. Semiconfluent 293T cells in a well of a 24‐well collagen‐coated microplate were transfected with 0.08 μg reporter plasmid pAP1‐Luc (Promega, Madison, WI, USA) and 0.05 μg phRG‐B control plasmid (Promega), with or without 0.1 μg JunD‐expression plasmid pcDNA3.1‐JunD (kindly provided by Dr S. K. Agarwal, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA)( 4 ) and 0.1 μg of one of the expression plasmids for FLAG‐tagged wild‐type or mutant menin. Twenty‐eight hours after transfection, cells were collected for reporter assay with a Dual‐Luciferase Reporter Assay System (Promega) as previously described.( 32 ) The lysates were also used for immunoblotting against the FLAG tag.
Results
Degradation of variant menin proteins. Three normal menin proteins (wild‐type and normal polymorphisms R171Q and A541T), five ASPT‐associated mutants and five FIHP‐associated mutants including previously tested E255K and Q260P, and five previously tested MEN1‐associated mutants( 14 ) were examined for degradation in the presence of protein synthesis inhibitor CHX and/or the proteasome inhibitor MG132 (Fig. 1). The normal menin proteins showed only moderate reduction in CHX‐treated cells and little increase in MG132‐treated cells compared with non‐treated cells. In contrast, mutants associated with typical MEN1 disappeared in CHX‐treated cells but increased markedly in MG132‐treated cells, thus reproducing our previous findings.( 14 ) Mutants associated with ASPT or FIHP exhibited variable results. The ASPT‐associated mutants G28A and E274A and FIHP‐associated mutant E255K showed only moderate reduction in CHX‐treated cells and little increase in MG132‐treated cells, indicating that these mutants are resistant to proteasome‐mediated degradation. The other ASPT‐ or FIHP‐associated mutants showed expression patterns similar to those associated with typical MEN1, indicating rapid degradation by the proteasome pathway. These findings indicate that not only E255K, which was the only mutant previously shown to be stable,( 14 ) but also some other menin mutants associated with milder forms of MEN1 are less sensitive to proteasome degradation.
Figure 1.
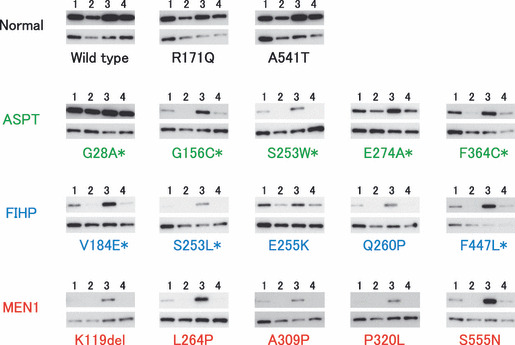
Effects of cycloheximide (CHX) and MG132 on the expression of variant menin. Plasmid expressing FLAG‐tagged menin was transfected into culture cells and detected by immunoblotting against the tag (upper panels) and β‐tubulin (lower panels). Cells were treated with CHX (lane 2), MG132 (lane 3), both (lane 4) or neither (lane 1). Asterisks indicate mutants that were not previously examined.( 14 )
Steady‐state expression levels of variant menin proteins. As previously noted( 14 ) and also shown in Figure 1, the steady‐state expression levels of menin protein appeared to be inversely related to the degradation rate, suggesting their usefulness in predicting the pathological effect of missense and in‐frame indel mutations. However, it was difficult to deduce menin stability by comparing steady‐state protein levels expressed by transfection of simple expression vectors because transfection efficiency was variable among plasmid preparations. In order to compare the steady‐state expression levels of variant and wild‐type menin proteins more precisely, plasmid vectors expressing the bicistronic mRNA encoding both FLAG‐tagged wild‐type menin and Myc‐tagged variant menin were transfected into culture cells, and both proteins were differentially detected by immunoblotting in each sample (Fig. 2).
Figure 2.
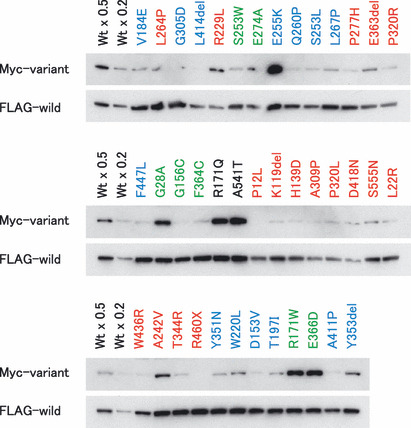
Comparison of steady‐state expression levels of variant menin proteins by immunoblotting. Myc‐tagged variant and FLAG‐tagged wild‐type menin proteins were expressed in culture cells by the transfection of bicistronic expression vectors. Wt × 0.5 and Wt × 0.2 indicate that 0.5 and 0.2 volume of the lysate was loaded on the gel, respectively. Variants associated with typical multiple endocrine neoplasia type 1 (MEN1), familial isolated hyperparathyroidism (FIHP) and apparently sporadic parathyroid tumor (ASPT) are shown in red, blue and green, respectively, while wild‐type and normal polymorphic variants are shown in black. Wt, wild‐type menin.
All menin proteins except for a truncated mutant R460X were detected as a single band of approximately the same size. The signal intensity of the FLAG‐tagged wild‐type menin was variable among plasmids, suggesting variable transfection and blotting efficiencies. Considering the amount of FLAG‐tagged wild‐type menin as the internal control for transfection efficiency, almost all Myc‐tagged mutants associated with typical MEN1 exhibited lower expression levels than Myc‐tagged wild‐type menin and normal polymorphisms. The plasmids expressing FLAG‐tagged variant and Myc‐tagged wild‐type menin proteins also showed similar results (data not shown). These findings confirmed the inverse correlation between steady‐state protein levels and their degradation rates. However, it was difficult to precisely compare the steady‐state expression levels among different plasmids because of the narrow dynamic range of immunoblot analysis.
We then developed a fluorescent immunocytochemical method, which usually provides a wider dynamic range of measurement than immunoblot analysis. The transfected cells were stained simultaneously with fluorescein‐labeled anti‐FLAG antibody and Cy3‐labeled anti‐Myc antibody. The fluorescence intensity was recorded by microscopic digital photography (Fig. 3A). The averages of the ratios of variant to wild‐type menin in each nuclei were calculated and normalized by the average ratio of the control samples transfected with the plasmid expressing both FLAG‐tagged and Myc‐tagged wild‐type menin proteins (Fig. 3B).
Figure 3.
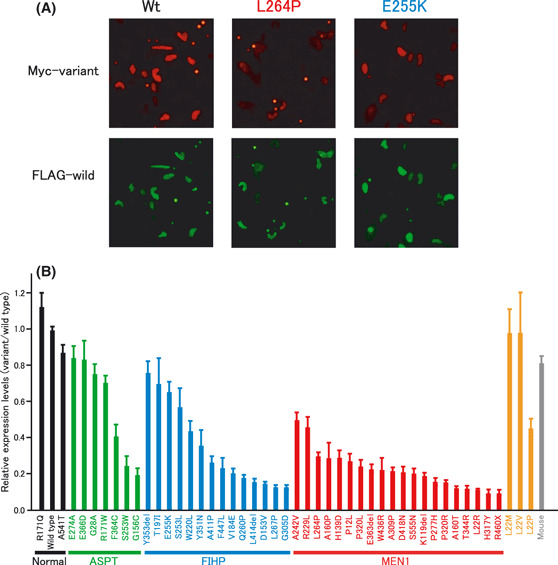
Relative expression levels of variant menin to wild‐type menin examined using the immunocytochemical method. (A) Immunocytochemical detection of each protein in the same nuclei. The small round dots represent fluorescent beads used to determine the dynamic range of signal intensity. (B) Stability of various variant menin proteins. Normal menin (wild type and polymorphisms), apparently sporadic parathyroid tumor (ASPT)‐associated, familial isolated hyperparathyroidism (FIHP)‐associated and multiple endocrine neoplasia type 1 (MEN1)‐associated mutants are shown in black, green, blue and red, respectively. Mutations not reported to be associated with disease and the mouse menin are shown in orange and gray, respectively. The results are placed in order of expression level within each group. The thin bars indicate standard errors of the mean of three independent experiments.
Seven ASPT‐associated, 14 FIHP‐associated and 19 MEN1‐associated missense or in‐frame deletion mutations, and one MEN1‐associated nonsense mutation (R460X) as well as normal polymorphic variants R171Q and A541T were examined for the relative steady‐state expression levels. We also examined three mutations, L22M, L22V and L22P, which have not been reported to be associated with diseases but could be generated by single nucleotide alterations from the wild‐type MEN1 gene, and mouse menin, which has 20 amino acid substitutions and one amino‐acid insertion with respect to human menin.( 33 )
The normal polymorphisms exhibited almost the same expression levels as wild‐type menin, while mutants associated with ASPT, FIHP or typical MEN1 showed variable expression levels in each group (Fig. 3B). All tested mutants associated with typical MEN1 showed reduced expression levels compared with the wild‐type and normal polymorphisms. The MEN1‐associated nonsense mutant R460X was the lowest of all variants examined. Because R460X has no nuclear localization signals,( 3 ) the fluorescence levels measured for this mutant was considered as the background signal of this assay. The relative expression levels of several missense and in‐frame deletion mutants associated with ASPT (E274A, E366D, G28A and R171W) and FIHP (Y353del, T197I and E255K) exhibited almost the same as or only slightly less than those of wild‐type and normal polymorphisms, while others showed low expression levels similar to those associated with typical MEN1. These results from the immunocytochemical analysis are generally consistent with those obtained in the immunoblot analysis (Fig. 2).
We tested four missense mutants at the amino acid position 22, L22M, L22V, L22P and L22R. L22M and L22V showed almost normal expression levels while L22P showed reduced expression, suggesting that the latter is a potentially pathogenic variant. The MEN1‐associated mutant L22R showed the lowest expression among these four variants. The mouse menin exhibited almost normal expression levels despite the many amino acid substitutions compared with the human wild‐type menin, further supporting the correlation of functional integrity and the stability of menin proteins.
Repression of JunD‐mediated transactivation by variant menin. Menin is known to repress JunD‐mediated transactivation.( 4 ) To examine the functional integrity of menin variants, the repressive activity on JunD‐mediated transcription was assayed by using luciferase reporter gene transcribed from the JunD‐dependent promoter.( 4 ) The expression levels of menin proteins were monitored by immunoblotting. The fairly stable mutants E255K and E274A exhibited clear inhibitory activity, although it was significantly less than that of wild‐type menin (Fig. 4). In contrast, the unstable mutant L264P showed no inhibitory activity, as expected from its low protein level. These findings suggest that certain stable menin mutants retain substantial intrinsic biological function and might have some tumor suppressor activity.
Figure 4.
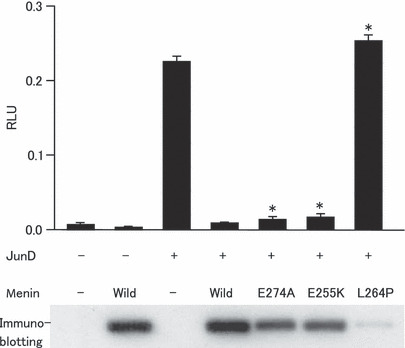
Inhibition of JunD‐mediated transactivation by menin variants. Cells were transfected with or without expression plasmids of JunD and menin variants along with a firefly luciferase reporter gene containing the JunD‐binding promoter sequence. Firefly luciferase activity was normalized with Renilla luciferase activity and the values are expressed as relative luciferase unit (RLU; mean with SE, n = 3). Immunoblot against FLAG tag attached to menin proteins is also shown. Asterisks indicate a statistically significant (P < 0.001) difference from the cells transfected with wild‐type menin and JunD‐expression plasmid.
Discussion
The present study revealed that the stability of missense and in‐frame deletion mutants of menin varies widely within each clinical form, that is, ASPT, FIHP or typical MEN1, and that unstable mutants were found not only in typical MEN1 but also in ASPT and FIHP, while stable mutants were found only in ASPT and FIHP, and not in typical MEN1. Stable mutants E255K and E274A exhibited significantly less than normal but substantial repressive activity on JunD‐dependent transactivation. These findings suggest that some missense and in‐frame indel mutations are fairly stable and retain intrinsic biological activity, and might be partially capable of suppressing tumor development. Although not yet verified, the patients having these stable menin variants might not develop full MEN1 manifestations and represent truly milder forms of MEN1. It is also possible that some stable variants are incidentally identified rare benign polymorphisms and not pathogenic mutations, because subtle reduction of biological activity might not actually cause disease. Thus, the stability test does not allow clear discrimination between rare normal polymorphisms and mutations causing milder forms of MEN1. In contrast, ASPT and FIHP patients and their family members having an unstable menin variant might be those who potentially develop full MEN1 manifestations. The tentative cut‐off value defining unstable mutants might be the stability of the A242V mutant, which was the most stable among the mutants associated with typical MEN1 examined so far. However, because development of tumors, particularly those other than parathyroid tumors, appears to be a stochastic phenomenon even in typical MEN1 families, the cut‐off value will not clearly discriminate between the mutants causing typical MEN1 and those causing truly milder forms. Moreover, it is hypothetically possible that yet‐to‐be‐identified modifier genes might attenuate clinical expression even in families with unstable mutant menin.( 34 ) Nevertheless, patients having unstable missense or indel menin mutations as well as those having truncated mutations should be strictly treated as a mutation carrier of typical MEN1.( 35 ) Because previous studies failed to relate specific MEN1 mutations to a particular clinical phenotype, all mutation carriers have been considered to require full screening of MEN1‐related tumors and lifelong follow up.( 7 , 13 ) The menin stability test will provide useful information for the management of patients with germline MEN1 mutations, especially when the patients have missense or in‐frame indel mutations of ambiguous significance.
We developed a new method to quantify intracellular protein stability. Bicistronic expression vector was used to provide wild‐type menin as the internal control protein for transfection efficiency. The expression levels of wild‐type menin varied among plasmids but did not appear to be correlated with those of co‐expressed menin variants as shown in Figure 2, suggesting that co‐expression of unstable menin mutants did not influence the stability of the internal standard protein. The new method also exploited the merits of the immunocytochemical measurement, which required less time and materials and exhibited a wider dynamic range than the immunoblot analysis. This method enabled measurements of signals only in cell nuclei where menin is considered to function as a transcription regulator. Because fluorochrome‐labeled antibodies recognize N‐terminal tags and nuclear localization sequences exist at the C‐terminal end,( 3 ) the accumulated signals in the nuclei should represent intact proteins. Indeed, the immunoblot analysis revealed one major band of the size expected for intact proteins.
All tested missense and in‐frame indel mutations associated with typical MEN1 were considerably unstable. In contrast, the normal mouse menin was as stable as the human wild‐type menin despite 20 amino acid differences and one amino acid insertion. This is consistent with the observation that the disease‐causing missense mutations are mainly found at the evolutionary conserved amino acid residues.( 2 , 7 , 36 ) Different amino acid substitutions at the same position differently influenced the stability as exemplified by the normal stability of L22M and L22V mutants, which substitute similarly hydrophobic amino acids for leucine, and the reduced stability of L22P, which replaces leucine with the inflexible proline residue. The MEN1‐associated mutant L22R, which introduces a charged residue at the uncharged site, was the least stable mutant at this position. Thus, the loss of stability of menin variants appears to be correlated with the likeliness of causing protein misfolding rather than replacing critical functional residues. Similar unstable mutants degraded by the ubiquitin–proteasome pathway have also been reported in other tumor suppressor genes. Germline missense mutations of the VHL gene are predominantly detected within the region required for the formation of the multiprotein complex including elongins B and C, which stabilize VHL proteins.( 37 ) Missense VHL mutations disrupting elongin binding have been shown to cause rapid VHL degradation.( 37 ) Another example is a subset of naturally occurring mutant NF2 proteins that have been suggested to act as dominant‐negative inhibitors on the wild‐type protein.( 38 ) These mutants, lacking the properly folded sequences required for binding to the plasma membrane and filamentous actin, were shown to be degraded rapidly, suggesting an important role of the ubiquitin–proteasome pathway in eliminating potentially dominant‐negative mutant proteins.( 39 ) These examples and rapid degradation of MEN1‐associated menin mutants imply that misfolding and/or loss of protein binding might lead to ubiquitination and rapid degradation of mutants of certain tumor suppressor proteins including menin. Because menin is a multi‐facet scaffold protein with widely dispersed multiple binding domains, this view is consistent with the fact that disease‐causing missense mutations are distributed, although not evenly, throughout the whole menin molecule. If rapidly degrading menin mutants have potentially dominant‐negative activity like some NF2 mutants, no association of stable menin mutant with typical MEN1 might be explained by negative selection during embryogenesis because total inactivation of menin function has been shown to be embryonic lethal in mice.( 40 ) Further investigations are required to determine whether MEN1‐associated missense menin exhibits a dominant‐negative effect and whether stable missense mutants ever occur in the germline of patients with typical MEN1.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by Grants‐in‐Aid from the Ministry of Health, Labor and Welfare for the third‐term Comprehensive 10‐Year Strategy for Cancer Control and National Cancer Center Research and Development Fund (21‐8‐6, 23‐A‐11 and 20). S. S. was the recipient of the research resident fellowship from the Foundation for Promotion of Cancer Research, Japan.
References
- 1. Chandrasekharappa SC, Guru SC, Manickam P et al. Positional cloning of the gene for multiple endocrine neoplasia‐type 1. Science 1997; 276: 404–7. [DOI] [PubMed] [Google Scholar]
- 2. Tsukada T, Nagamura Y, Ohkura N. MEN1 gene and its mutations: basic and clinical implications. Cancer Sci 2009; 100: 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guru SC, Goldsmith PK, Burns AL et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA 1998; 95: 1630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agarwal SK, Guru SC, Heppner C et al. Menin interacts with the AP1 transcription factor JunD and represses JunD‐activated transcription. Cell 1999; 96: 143–52. [DOI] [PubMed] [Google Scholar]
- 5. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer‐associated target genes. Cancer Cell 2008; 14: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsukada T, Yamaguchi K, Kameya T. The MEN1 gene and associated diseases: an update. Endocr Pathol 2001; 12: 259–73. [DOI] [PubMed] [Google Scholar]
- 7. Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008; 29: 22–32. [DOI] [PubMed] [Google Scholar]
- 8. Shimizu S, Tsukada T, Futami H et al. Germline mutations of the MEN1 gene in Japanese kindred with multiple endocrine neoplasia type 1. Jpn J Cancer Res 1997; 88: 1029–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujimori M, Shirahama S, Sakurai A et al. Novel V184E MEN1 germline mutation in a Japanese kindred with familial hyperparathyroidism. Am J Med Genet 1998; 80: 221–2. [DOI] [PubMed] [Google Scholar]
- 10. Teh BT, Esapa CT, Houlston R et al. A family with isolated hyperparathyroidism segregating a missense MEN1 mutation and showing loss of the wild‐type alleles in the parathyroid tumors. Am J Hum Genet 1998; 63: 1544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uchino S, Noguchi S, Sato M et al. Screening of the MEN1 gene and discovery of germ‐line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res 2000; 60: 5553–7. [PubMed] [Google Scholar]
- 12. Honda M, Tsukada T, Tanaka H et al. A novel mutation of the MEN1 gene in a Japanese kindred with familial isolated primary hyperparathyroidism. Eur J Endocrinol 2000; 142: 138–43. [DOI] [PubMed] [Google Scholar]
- 13. Wautot V, Vercherat C, Lespinasse J et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat 2002; 20: 35–47. [DOI] [PubMed] [Google Scholar]
- 14. Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin‐proteasome pathway. Mol Cell Biol 2004; 24: 6569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maruyama K, Tsukada T, Hosono T et al. Structure and distribution of rat menin mRNA. Mol Cell Endocrinol 1999; 156: 25–33. [DOI] [PubMed] [Google Scholar]
- 16. Agarwal SK, Kester MB, Debelenko LV et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 1997; 6: 1169–75. [DOI] [PubMed] [Google Scholar]
- 17. Balogh K, Hunyady L, Patocs A et al. MEN1 gene mutations in Hungarian patients with multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2007; 67: 727–34. [DOI] [PubMed] [Google Scholar]
- 18. Pannett AAJ, Kennedy AM, Tuner JJO et al. Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol (Oxf) 2003; 58: 639–46. [DOI] [PubMed] [Google Scholar]
- 19. Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjöld M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab 2007; 92: 3389–95. [DOI] [PubMed] [Google Scholar]
- 20. Teh BT, Kytölä S, Farnebo F et al. Mutation analysis of the MEN1 gene in multiple endocrine neoplasia type 1, familial acromegaly and familial isolated hyperparathyroidism. J Clin Endocrinol Metab 1998; 83: 2621–6. [DOI] [PubMed] [Google Scholar]
- 21. Jäger AC, Friis‐Hansen L, Hansen TV et al. Characteristics of the Danish families with multiple endocrine neoplasia type 1. Mol Cell Endocrinol 2006; 249: 123–32. [DOI] [PubMed] [Google Scholar]
- 22. Warner J, Epstein M, Sweet A et al. Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet 2004; 41: 155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hannan FM, Nesbit MA, Christie PT et al. Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab 2008; 4: 53–8. [DOI] [PubMed] [Google Scholar]
- 24. Cardinal JW, Bergman L, Hayward N et al. A report of a national mutation testing service for the MEN1 gene: clinical presentations and implications for mutation testing. J Med Genet 2005; 42: 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kassem M, Kruse TA, Wong FK, Larsson C, Teh BT. Familial isolated hyperparathyroidism as a variant of multiple endocrine neoplasia type 1 in a large Danish pedigree. J Clin Endocrinol Metab 2000; 85: 165–7. [DOI] [PubMed] [Google Scholar]
- 26. Poncin J, Abs R, Velkeniers B et al. Mutation analysis of the MEN1 gene in Belgian patients with multiple endocrine neoplasia type 1 and related diseases. Hum Mutat 1999; 13: 54–60. [DOI] [PubMed] [Google Scholar]
- 27. Perrier ND, Villablanca A, Larsson C et al. Genetic screening for MEN1 mutations in families presenting with familial primary hyperparathyroidism. World J Surg 2002; 26: 907–13. [DOI] [PubMed] [Google Scholar]
- 28. Klein RD, Salih S, Bessoni J, Bale AE. Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 2005; 7: 131–8. [DOI] [PubMed] [Google Scholar]
- 29. Tanaka C, Yoshimoto K, Yamada S et al. Absence of germ‐line mutations of the multiple endocrine neoplasia type 1 (MEN1) gene in familial pituitary adenoma in contrast to MEN1 in Japanese. J Clin Endocrinol Metab 1998; 83: 960–5. [DOI] [PubMed] [Google Scholar]
- 30. Sato M, Matsubara S, Miyauchi A et al. Identification of five novel germline mutations of MEN1 gene in Japanese multiple endocrine neoplasia type 1 (MEN1) families. J Med Genet 1998; 35: 915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giraud S, Zhang CX, Serova‐Sinilnikova O et al. Germ‐line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet 1998; 63: 455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohkura N, Nagamura Y, Tsukada T. Differential transactivation by orphan nuclear receptor NOR1 and its fusion gene product EWS/NOR1: possible involvement of poly(ADP‐ribose) polymerase I, PARP‐1. J Cell Biochem 2008; 105: 785–800. [DOI] [PubMed] [Google Scholar]
- 33. Stewart C, Parente F, Piehl F et al. Characterization of the mouse Men1 gene and its expression during development. Oncogene 1998; 17: 2485–93. [DOI] [PubMed] [Google Scholar]
- 34. Lemos MC, Harding B, Reed AAC et al. Genetic background influences embryonic lethality and the occurrence of neural tube defects in Men1 null mice: relevance to genetic modifiers. J Endocrinol 2009; 203: 133–42. [DOI] [PubMed] [Google Scholar]
- 35. Brandi ML, Gagel RF, Angeli A et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86: 5658–71. [DOI] [PubMed] [Google Scholar]
- 36. Maruyama K, Tsukada T, Honda M et al. Complementary DNA structure and genomic organization of Drosophila menin. Mol Cell Endocrinol 2000; 168: 135–40. [DOI] [PubMed] [Google Scholar]
- 37. Schoenfeld AR, Davidowitz EJ, Burk RD. Elongin BC complex prevents degradation of von Hippel‐Lindau tumor suppressor gene products. Proc Natl Acad Sci USA 2000; 97: 8507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giovannini M, Robanus‐Maandag E, Niwa‐Kawakita M et al. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev 1999; 13: 978–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gautreau A, Manent J, Fievet B, Louvard D, Giovannini M, Arpin M. Mutant products of the NF2 tumor suppressor gene are degraded by the ubiquitin‐proteasome pathway. J Biol Chem 2002; 277: 31279–82. [DOI] [PubMed] [Google Scholar]
- 40. Crabtree JS, Scacheri PC, Ward JM et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci USA 2001; 98: 1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]