

Abstract
Testis derived transcript (TES) is a candidate tumor suppressor gene located at the human chromosome 7q31, and its function in ovarian cancer is still unknown. Using ovarian cancer cell lines and tissue samples, we demonstrated that both loss of heterozygosity and hypermethylation of the TES gene occurred in ovarian cancer at high frequencies, and there were significant correlations between TES expression and hypermethylation or loss of heterozygosity. We also detected methylation in ovarian cancer cell line A2780 after treatment with 5‐aza‐2‐deoxycytidine. The expression level of TES was enormously up‐regulated, then caused changes to the biological behaviors of A2780 cells: cell growth properties were greatly impaired, colony formatting abilities were suppressed to very low levels, and the apoptosis rate was highly raised compared to the control group. Our findings suggest that the TES gene functions as a tumor suppressor gene and is frequently silenced by hypermethylation and loss of heterozygosity in ovarian cancers.
(Cancer Sci 2010; 101: 1255–1260)
Nowadays, we realize that tumors are a complex process made up of multiple steps. In past decades, a great number of genes was proved to be involved in the development and progression of tumors. In recent years, several genes were identified to be able to suppress tumor growth (as tumor suppressor genes), such as P53,( 1 ) phosphatase and tensin homologue,( 2 ) and p16,( 3 ) highlighting the optimistic prospect of research into these genes. A great many studies showed that alterations of tumor suppressor genes like loss of heterozygosity( 4 , 5 ) and promoter hypermethylation( 6 , 7 ) were detected in almost every type of cancers.
Ovarian cancer is the third most frequent female cancer type and the leading cause of death by cancer among women in China. More than 70% of patients were already in the late stage on diagnosis, and lost the chance to have an operation, which then required a great deal of money and medical resources.
It has also been demonstrated that some tumor suppressor genes play a notable role in ovarian cancer; the testis derived transcript (TES) gene is one of them. The TES gene is located at 7q31, within the fragile chromosomal region FRA7G.( 8 , 9 ) It spans about 48 kb encompassing seven exons. There are three isoforms of human TES, which differ from each other in the 3‐untranslated region (3‐UTR).( 10 ) At its COOH terminal, the TES protein has three zinc‐binding domain present in Lin‐11, Isl‐1 and Mec‐3 domains which play a very important role in focal adhesion targeting.( 11 ) TES was proved to be a putative tumor suppressor gene;( 12 ) in the past few years, much evidence has indicated TES anticancer functions in the mechanisms of tumorigenesis, angiogenesis, and metastasis. Frequent loss of heterozygosity at 7q31 happened in a variety of malignances, implying that loss of gene expression within this region had a relationship with cancer. Moreover, studies also reported a great rate of TES hypermethylation in glioblastomas and other malignances, leading to the TES expression decline or total loss.
Although there some research reported TES anticancer functions in other malignancies, we still didn’t get information about its role in ovarian cancer. In this study, we investigated both loss of heterozygosity and hypermethylation in 41 cases of ovarian cancer. The results showed that TES expression had significant correlations with loss of heterozygosity and hypermethylation. Then we up‐regulated TES in ovarian cancer cell line A2780 with 5‐aza‐2‐deoxycytidine (5‐aza‐dC) and apparent changes of cell biological behaviors were detected, further implying that TES is a tumor suppressor gene and is often silenced in ovarian cancer because of either hypermethylation or loss of heterozygosity.
Materials and Methods
Cell lines and 5‐aza‐dC treatment. Epithelial ovarian cancer cell lines (SKOV3 and A2780) were routinely cultured in RPMI‐1640 growth medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS at 37°C, 5% CO2. 5‐aza‐dC was dissolved in double‐distilled H2O and filtered with a 0.22‐μm filter membrane. 5 × 105 cells were plated into a 25‐cm2 flask with RPMI‐1640. Twenty‐four hours later, cells were treated with 5 μm 5‐aza‐dC, and the medium was changed every 3 days. Total RNA was prepared with Trizol reagent (Invitrogen) 10 days later in accordance with the manufacturer’s instructions. Five‐hundred ng RNA was used for quantitative RT‐PCR on Stepone Plus (Applied Biosystems, Foster City, CA, USA). TES expression was evaluated with the following primers: forward, 5‐CCTTCAAAGTGCCATGAGTTGTCTC‐3; reverse, 5‐TTCATACTCAGTTTGCAGCAATAGC‐3. PCR conditions were as follows: 95°C for 30 s, 35 cycles of 95°C for 5 s, and 60°C for 32 s. GAPDH was used as the endogenous control.
Flow cytometry analysis of cell apoptosis. Cell apoptosis was analyzed by an Annexin V‐FITC/propidium iodide (PI) kit according to the manufacturer’s instructions. Briefly, harvested cells were resuspended in 100 μL Annexin V–FITC binding buffer and adjusted to about 1 × 106/mL, then 5 μL Annexin V‐FITC and 10 μL PI (20 μg/mL) were added, following 15‐min incubation in the dark. Flow cytometry was conducted on a FACS caliber (BD Biosciences, Hercules, CA, USA).
Cell proliferation assay. 3 × 103 A2780 cells at approximately 80% confluence were plated per well into 96‐well plates and incubated overnight, then cells were treated with different concentrations of 5‐aza‐dC separately. The cell growth curve was assessed using an 3‐(4,5‐dimethylthiazol)‐2,5‐diphenyl tetrazolium (MTT) assay according to the manufacturer’s instructions. The results were expressed as the absorbance at 490 nm at the indicated time points.
Colony formation assay. 0.5 × 103 A2780 cells were plated per well into six‐well plates, and separated into two groups according to different 5‐aza‐dC concentrations. The culture medium was changed routinely and colony numbers were counted by inverted microscope 2 weeks later.
Western blotting. To investigate TES expression change after 5‐aza‐dC treatment, Western blotting was performed in a A2780 cell line. Briefly, 50 μg of protein samples were loaded onto SDS‐PAGE gels and electroblotted to polyvinylidene difluoride membranes. The membranes were incubated with primary anti‐TES (Abcam, Cambridge, MA, USA), followed by reaction with a secondary antibody. β‐Actin was used as the endogenous control.
Patients and samples. Samples were collected from 41 patients diagnosed with ovarian cancer in Qilu Hospital (Shandong, China) between June 2008 and June 2009; all patients provided consent and approval was obtained from the ethics committee. One mL whole blood was extracted from each patient before the operation and tissues were obtained after surgical resection, then immediately stored at −80°C. All malignant cases were classified and graded according to the criteria of the International Federation of Obstetrics and Gynecology (FIGO). An additional 12 normal ovarian tissues were collected as controls.
DNA extraction. Genomic DNA was extracted according to the standard phenol/chloroform extraction. In brief, 25 mg tissue or 200 μL whole blood with Proteinase K (20 mg/mL) was incubated overnight at 50°C in a waterbath. DNA was extracted by phenol/chloroform twice, then precipitated with 100% ethanol and at the end dissolved in TE solution. For paraffin‐embedded tissues used in loss of heterozygosity analysis, 8‐μm tissue slides with 2‐h dehydration by xylene were prestained with Toluidine blue, and the tumor cell zone was removed with a tiny needle on an inverted microscope.
Microsatellite analysis of loss of heterozygosity. Five microsatellite markers flanking chromosome 7q31 (D7S2502, D7S2543, D7S486, D7S2460, D7S522) were chosen from Genebank for the PCR‐based analysis of loss of heterozygosity. One μg total DNA was used to perform PCR under the following conditions: denaturing at 95°C for 3 min, 30 cycles of 95°C for 15 s; and annealing of each primer for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. Ten μL of PCR products was separated by 8% denatured polyacrylamide gels (300 V for 2–3 h) and then the gels were silver‐stained. Two experienced researchers separately analyzed the staining results.
Methylation‐specific PCR (MS‐PCR). One μg of each DNA sample was bisulfite modified using the CpGenome DNA modification kit (Chemicon, Temecula, CA, US) according to the manufacturer’s instructions. The locations of CpG islands within the TES gene promoter have been described previously. Primers and PCR conditions for MSP( 13 ) are available on request. DNA treated by M. Sss. I was used as a positive control.
Immunohistochemistry. Protocols for immunohistochemistry staining were described previously.( 14 ) Sections stained without anti‐TES were used as negative control. For evaluation of TES protein, classification standards were as follows: negative, totally no staining or <10% tumor cells showed positive staining; and positive, ≥10% tumors cells were positively stained.
Statistical analysis. The software SPSS version 16.0 (SPSS, Chicago, IL, USA) was used for statistical analysis. The appropriate χ2‐test was chosen to analyze the association between two categorical variables. The level of statistical difference was defined at 0.05.
Results
TES protein expression in ovarian cancer. We immunohistochemically assayed TES protein levels in 41 ovarian cancer samples and 12 normal ovarian tissues. All the normal samples stained positively for TES antibody; however, loss of TES protein was identified in 56.1% (23/41) of ovarian cancer samples (Fig. 1).
Figure 1.
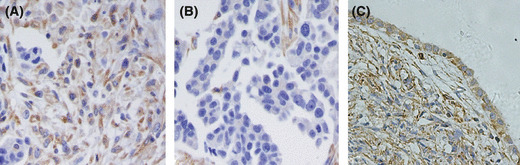
Testis derived transcript (TES) expression in normal ovary and ovarian cancer tissues. (A) Positive staining of TES protein in ovarian cancer. (B) TES expression was lost in ovarian cancer. (C) Normal ovarian tissue was stained positively.
Frequent TES hypermethylation in ovarian cancer. We observed hypermethylation in 36.5% of ovarian cancer tissues (15/41); in contrast, none of the 12 normal ovary tissues was hypermethylated (Fig. 2). We also detected hypermethylation in the A2780 cell line. There was a significant correlation between hypermethylation and TES protein level (P = 0.001; Table 1).
Figure 2.

Methylation analysis of testis derived transcript in ovarian cancers. C, cancer samples; M, methylated; M. Sss I, positive control; N, normal ovary tissues; U, unmethylated. (DNA modificated by CpG methyltransferase M. Sss I.)
Table 1.
Relationship between TES expression and methylation
| TES expression | P‐value | ||
|---|---|---|---|
| Negative | Positive | ||
| Methylation | |||
| Negative | 7 | 19 | 0.001 |
| Positive | 12 | 3 | |
TES, testis derived transcript.
Allelic loss of TES in ovarian cancer. Twenty‐six of all 41 cases were informative for loss of heterozygosity analysis. We detected TES locus loss in 34.6% (9/26) of ovarian cancers (at least one marker was lost) (Fig. 3). When we examined the relationship between TES expression and the frequency of loss of heterozygosity, eight of nine cases with loss of heterozygosity presented lack of TES protein expression (Table 2). Previous studies had reported a correlation between methylation and loss of heterozygosity; however, our results showed that only one case presented both hypermethylation and loss of heterozygosity (Table 3). There wasn’t an obvious correlation between methylation and loss of heterozygosity (P = 0.833; Table 3).
Figure 3.
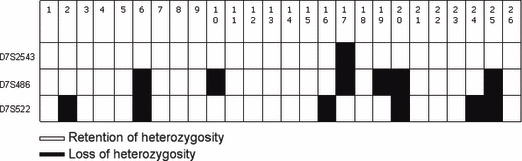
Loss of heterozygosity in ovarian cancer.
Table 2.
Relationship between TES expression and locus loss
| TES locus loss | P‐value | ||
|---|---|---|---|
| ROH | |||
| TES expression | |||
| Negative | 8 | 8 | 0.037 |
| Positive | 9 | 1 | |
ROH, retention of heterozygosity; TES, testis derived transcript.
Table 3.
Relationship between TES locus loss and methylation
| TES locus loss | P‐value | ||
|---|---|---|---|
| ROH | |||
| Methylation | |||
| Negative | 13 | 8 | 0.833 |
| Positive | 4 | 1 | |
ROH, retention of heterozygosity; TES, testis derived transcript.
TES expression in ovarian cancer cell lines. Quantitative RT‐PCR and Western blotting were preformed to evaluate the expression changes after treatment with 5‐aza‐dC. The results indicated an enormous up‐regulation in both transcriptional (more than 45‐fold) and translational levels in the A2780 cell line after treatment with 5 μm 5‐aza‐dC, but no obvious change was detected in SKOV3 cells (4, 5). In addition, by quantitative RT‐PCR, we found no changes in the 1‐μm group compared to the 0‐μm group for both cell lines (data not shown).
Figure 4.
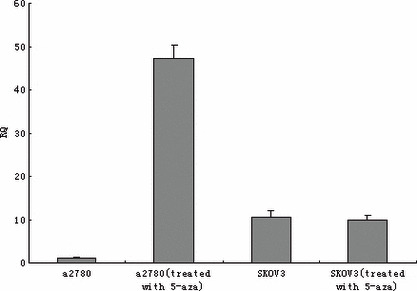
RT‐PCR analysis of testis derived transcript with or without 5‐aza‐2‐deoxycytidine treatment in A2780 and SKOV3 cell lines.
Figure 5.
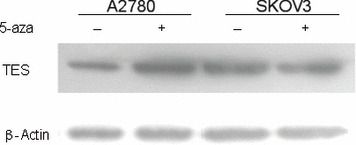
Western blot of testis derived transcript (TES) in SKOV3 and A2780 cell lines before and after 5‐aza‐2‐deoxycytidine treatment. β‐Actin was used as an endogenous control.
Cell growth analysis. We used 5 μm 5‐aza‐dC for treating the two cell lines. In A2780 cells, significant difference was observed in cell growth curve of the two groups. Compared to the 0‐μm group, the growth properties of A2780 cells in the 5‐μm group were obviously impaired, indicated by the much lower absorbance at 490 nm (P = 0.013; Fig. 6A). However, as for the SKOV3 cell line, no apparent changes were detected between the two groups (P = 0.796; Fig. 6B).
Figure 6.
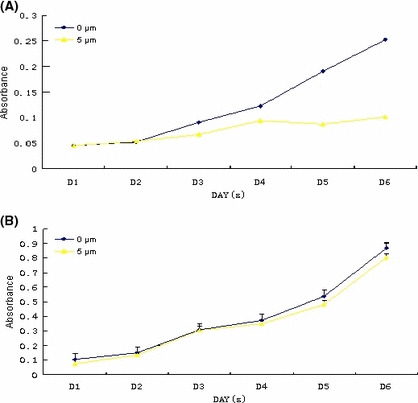
Cell proliferation curve with or without 5‐aza‐2‐deoxycytidine. (A) A2780 cell line; (B) SKOV3 cell line.
Colony formation assay. Colony formation assay showed strong differentiation between the 0‐μm and 5‐μm groups for A2780 cells, but not SKOV3 cells. The A2780 cells treated with 5 μm 5‐aza‐dC lost the capacity to grow into large colonies, compared with the cells in 0‐μm group; moreover, the colonies in the 5‐μm group were more dispersive. However, we didn’t detect obvious changes in SKOV3 cells after treatment with 5‐aza‐dC (Fig. 7).
Figure 7.
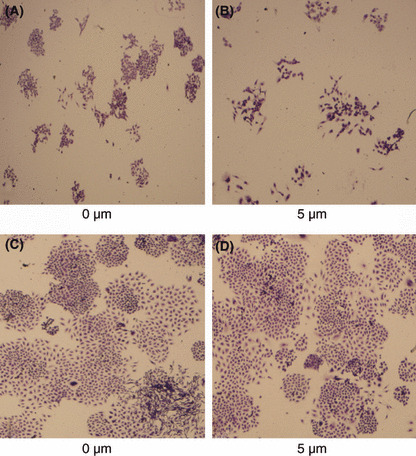
Colony formation assay with or without 5‐aza‐2‐deoxycytidine. (A,B) A2780 cell line; (C,D), SKOV3 cell line.
Cell apoptosis assay. The two cell lines treated with 5 μm 5‐aza‐dC were assayed for cell apoptosis. In the A2780 control group (no 5‐aza‐dC was given), the total rate of apoptosis was very slight, just 0.99 (early apoptotic rate, 0.3; late apoptotic rate, 0.69). However, many more apoptotic cells were detected in A2780 cells treated with 5‐aza‐dC: the rate raised to 3.31 (early apoptotic rate, 1.38; late apoptotic rate, 1.93). As for the SKOV3 cell line, the apoptotic conditions didn’t vary much before and after treatment with 5‐aza‐dC (1.59 vs 1.43; Fig. 8).
Figure 8.
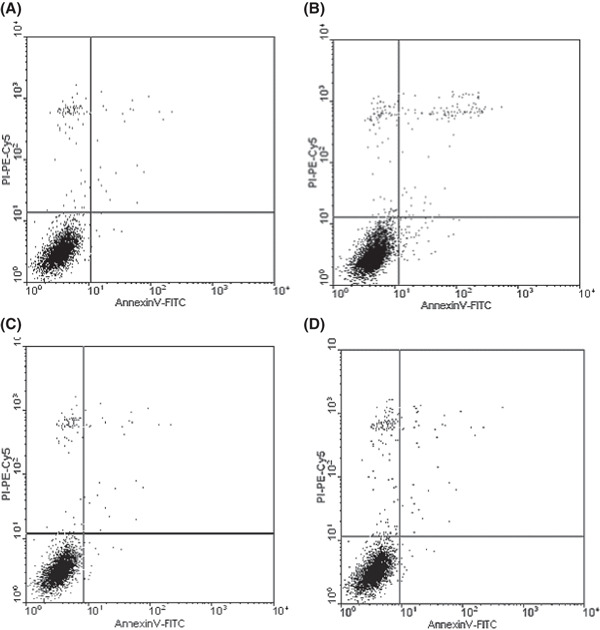
Cell apoptosis induced by treatment with 5 μm 5‐aza‐2‐deoxycytidine. (A) Apoptosis of A2780 cells in the group without 5‐aza‐dC treatment (early apoptotic rate, 0.3; late apoptotic rate, 0.69); (B) apoptosis of A2780 cells in the 5 μm 5‐aza‐2‐deoxycytidine group (early apoptotic rate, 1.38; late apoptotic rate, 1.93); (C) apoptosis of SKOV3 cells in the group without 5‐aza‐dC treatment (early apoptotic rate, 0.44; late apoptotic rate, 0.99); (D) apoptosis of SKOV3 cells in the 5 μm 5‐aza‐dC group (early apoptotic rate, 0.57; late apoptotic rate, 1.02).
Discussion
Because we have not yet found a valid approach for the early detection of ovarian cancer, much research focuses on early diagnosis and cure. In this field, studies of tumor suppressor genes might show us a bright future. Epigenetic and genetic alterations of tumor suppressor genes like promoter hypermethylation, loss of heterozygosity, and mutation occur frequently in various malignances, down‐regulating the expressions of these genes, and giving tumors a chance.
To date, nearly 100 articles have reported loss of heterozygosity in ovarian cancer at chromosome arms 4p, 5q, 7q, 8p, 9p, 10q, 12q, 13q, 14q, 16q, 17p, 17q, and so on.( 15 , 16 ) Tumor suppressor genes located in these sites were proven to be low expressed or even non‐expressed, such as phosphatase and tensin homologue,( 17 ) breast cancer 1,( 18 )cyclin‐dependent kinase inhibitor 2A,( 19 ) and adenomatous polyposis coli,( 20 ) providing evidence for the antitumor character of these genes. Loss of heterozygosity at 7q31 was common in breast carcinoma (40–83%),( 21 , 22 ) prostate cancer (more than 80%),( 23 ) and renal cell carcinoma (30%).( 24 ) In addition, loss of heterozygosity was also found in pancreatic carcinoma,( 25 ) colonic carcinoma,( 26 ) and many other malignancies.( 27 , 28 , 29 , 30 ) Recently, more and more studies have indicated that TES may act as a tumor suppressor gene in tumorigenesis,( 31 ) metastasis,( 11 ) and prognosis.( 32 , 33 ) In this study, when we tested the frequency of loss of heterozygosity by microsatellite analysis, 34.6% cases presented at least one marker lost. As to our results, eight of nine cases indicated loss of heterozygosity with a decline of TES expression. In contrast, TES protein level was normal in nine of 17 retention of heterozygosity cases; there is a statistical difference (P = 0.037).
DNA methylation is considered to be another mechanism for the second hit for Knudson’s hypothesis.( 34 ) It is an enzymatic process to add the methyl group at the fifth carbon of cytosines of the dinucleotide 5′‐CpG‐3′ sequence which is plentiful within nearly all promoters. Cytosine methylation can function on regulating the expression of endogenous genes, silencing transposons, and controlling the stability of the genome.( 35 ) In recent years, studies on promoter hypermethylation of the tumor suppressor genes have reported great achievement. Hypermethylation of DAPK,( 36 ) OPCML,( 37 ) CDH13,( 38 ) and GATA4( 39 ) were reported to be frequent in ovarian cancer. Up to now, only two previous studies have detected methylation of the TES gene. In the first study, hypermethylation of the TES gene was detected in all 30 tumor‐derived cell lines,( 12 ) including the A2780 cell line, which was also proved to be methylated in our study. In the second study of glioblastomas, TES promoter methylation existed in 58% cases and this alteration was more common in primary than secondary glioblastomas.( 40 ) Both the studies showed high frequency of hypermethylation in the TES gene, implying its potential role in suppressing tumor activities. Consistent with these studies, in our study, hypermethylation was another cause for TES silence in ovarian cancer: more than 35% of cases were observed hypermethylated, and in contrast, methylation happened in none of the 12 normal tissues. There was a significant difference between the ovarian malignant and non‐malignant groups. We also investigated the relationship between TES expression and hypermethylation. In the normal TES expression group, three in 22 cases presented hypermethylation, whereas 12 of 19 cases in the low TES expression group were methylated, indicating the important influence of hypermethylation on TES protein level regulation.
By Knudson’s hypothesis, inactivation of tumor suppressor genes was caused by two hits: the first is usually a mutation at one of the alleles, and the second may occur by mutations, deletions, or some other mechanisms. The analysis for loss of heterozygosity and hypermethylation is a useful approach to identify tumor suppressor genes involved in tumorigenesis. But recent studies had suggested that some genes may be just one‐hit genes, meaning that just one alteration can impair the expression of these genes. A previous study presented evidence that the TES gene is also a one‐hit gene. In TES‐knockout mice, semi(+/−) or total loss(−/−) of TES caused many more high risks for tumorigenesis (ORs were 38.9 and 27.6, respectively).( 31 ) Interestingly, in our study we also observed the phenomenon that there was just one case in which both loss of heterozygosity and hypermethylation occurred. Further research needs to be undertaken to determine whether the one‐hit mechanism really works on the TES gene.
In a previous study, the impact of TES on cells was studied; after transfecting the full‐length TES coding cDNA into cell lines, TES expression was highly up‐regulated, which resulted in cell growth deduction and less colony formation.( 12 ) Another study proved that re‐expression of TES in T47D cells can induce more apoptosis and impair the capability to form a mass in nude mice.( 41 ) We raised the TES protein level in A2780 cells by treatment with 5‐aza‐dC; high TES expression greatly suppressed the biological behaviors of A2780 cells. As the cell proliferation curve indicated, the growth properties of A2780 cells treated with 5 μm 5‐aza‐dC were much less than cells in the control group, suggesting that by treatment with the appropriate concentration of 5‐aza‐dC, we can prevent ovarian cancer from rapidly growing. In addition, colony formation assay results further indicated that the colonies in the 5‐μm 5‐aza‐dC group were much fewer in number and smaller than those in the control group, and the cells appeared very scattered. Though we were unsure how re‐expressed TES achieved this, the results indicated that TES has the potential to stop the ovarian cancer cells shaping into huge solid tumors. That is also further evidence for the negative functions of TES in malignant tumors. We also tested the apoptotic conditions in cells treated with 5‐aza‐dC and compared to the no‐treatment group, more cells were induced to apoptosis by 5‐aza‐dC. This result implies that TES may cause cell apoptosis after the reversion of its hypermethylation. But 5‐aza‐dC didn’t show obvious influences on SKOV3 cells (in which TES wasn’t methylated) in any of the three assays. Above all, we proved that TES has the potential to slow the proliferation of cancer cells, weaken their capacity to format cell colonies, and can also induce more cells into apoptosis, implying the possibility of using TES as a therapy for ovarian cancer. Future research into in vivo tumorigenesis is required to reveal the functions of TES in ovarian cancer and other types of malignances.
In summary, we firstly investigated highly frequent promoter hypermethylation and loss of heterozygosity of the TES gene in ovarian cancer. There were significant correlations between TES expression and hypermethylation or loss of heterozygosity, which proved that the TES gene was a tumor suppressor gene in ovarian cancer. By restoring the TES expression, both the proliferation and invasive abilities of A2780 cells were enormously suppressed. At the same time, more cells were induced to apoptosis, showing that TES can suppress tumor growth by regulating various biological behaviors of ovarian cancer; this provided more and detailed evidence for the role of the tumor suppressor gene TES in ovarian cancer. According to our results, specific therapy targeting TES might have broad prospects in the future.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
Our project was supported by grants to Beihua Kong from the National Natural Science Foundation of China (no. 30872738). We thank Ying Zhao, Jie Li, ShuHui Hong, XiaoLi Kong, XiaoYan Li, Jiang Zhu, and Ning Zhang for technical support and critical discussions.
References
- 1. Tan TH, Wallis J, Levine AJ. Identification of the p53 protein domain involved in formation of the simian virus 40 large T‐antigen‐p53 protein complex. J Virol 1986; 59: 574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Myers MP, Tonks NK. PTEN: sometimes taking it off can be better than putting it on. Am J Hum Genet 1997; 61: 1234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aguiar RC, Dahia PL, Sill H, Toledo SP, Goldman JM, Cross NC. Deletion analysis of the p16 tumour suppressor gene in phaeochromocytomas. Clin Endocrinol (Oxf) 1996; 45: 93–6. [PubMed] [Google Scholar]
- 4. Callahan R, Cropp C, Sheng ZM et al. Definition of regions of the human genome affected by loss of heterozygosity in primary human breast tumors. J Cell Biochem Suppl 1993; 17G: 167–72. [DOI] [PubMed] [Google Scholar]
- 5. Brown MR, Chuaqui R, Vocke CD et al. Allelic loss on chromosome arm 8p: analysis of sporadic epithelial ovarian tumors. Gynecol Oncol 1999; 74: 98–102. [DOI] [PubMed] [Google Scholar]
- 6. Pfeifer GP, Rauch TA. DNA methylation patterns in lung carcinomas. Semin Cancer Biol 2009; 19: 181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dworkin AM, Spearman AD, Tseng SY, Sweet K, Toland AE. Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam Cancer 2009; 8: 339–46. [DOI] [PubMed] [Google Scholar]
- 8. Huang H, Qian C, Jenkins RB, Smith DI. Fish mapping of YAC clones at human chromosomal band 7q31.2: identification of YACS spanning FRA7G within the common region of LOH in breast and prostate cancer. Genes Chromosomes Cancer 1998; 21: 152–9. [DOI] [PubMed] [Google Scholar]
- 9. Huang H, Qian J, Proffit J, Wilber K, Jenkins R, Smith DI. FRA7G extends over a broad region: coincidence of human endogenous retroviral sequences (HERV‐H) and small polydispersed circular DNAs (spcDNA) and fragile sites. Oncogene 1998; 16: 2311–9. [DOI] [PubMed] [Google Scholar]
- 10. Tatarelli C, Linnenbach A, Mimori K, Croce CM. Characterization of the human TESTIN gene localized in the FRA7G region at 7q31.2. Genomics 2000; 68: 1–12. [DOI] [PubMed] [Google Scholar]
- 11. Coutts AS, MacKenzie E, Griffith E, Black DM. TES is a novel focal adhesion protein with a role in cell spreading. J Cell Sci 2003; 116: 897–906. [DOI] [PubMed] [Google Scholar]
- 12. Tobias ES, Hurlstone AF, MacKenzie E, McFarlane R, Black DM. The TES gene at 7q31.1 is methylated in tumours and encodes a novel growth‐suppressing LIM domain protein. Oncogene 2001; 20: 2844–53. [DOI] [PubMed] [Google Scholar]
- 13. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang Q, Nakamura M, Nakamura Y et al. Two‐hit inactivation of FHIT by loss of heterozygosity and hypermethylation in breast cancer. Clin Cancer Res 2002; 8: 2890–3. [PubMed] [Google Scholar]
- 15. Sato T, Saito H, Morita R, Koi S, Lee JH, Nakamura Y. Allelotype of human ovarian cancer. Cancer Res 1991; 51: 5118–22. [PubMed] [Google Scholar]
- 16. Cliby W, Ritland S, Hartmann L et al. Human epithelial ovarian cancer allelotype. Cancer Res 1993; 53: 2393–8. [PubMed] [Google Scholar]
- 17. Kurose K, Zhou XP, Araki T, Cannistra SA, Maher ER, Eng C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am J Pathol 2001; 158: 2097–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ford D, Easton DF, Stratton M et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62: 676–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herman JG, Merlo A, Mao L et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995; 55: 4525–30. [PubMed] [Google Scholar]
- 20. Allan GJ, Cottrell S, Trowsdale J, Foulkes WD. Loss of heterozygosity on chromosome 5 in sporadic ovarian carcinoma is a late event and is not associated with mutations in APC at 5q21‐22. Hum Mutat 1994; 3: 283–91. [DOI] [PubMed] [Google Scholar]
- 21. Bieche I, Champeme MH, Matifas F, Hacene K, Callahan R, Lidereau R. Loss of heterozygosity on chromosome 7q and aggressive primary breast cancer. Lancet 1992; 339: 139–43. [DOI] [PubMed] [Google Scholar]
- 22. Zenklusen JC, Bieche I, Lidereau R, Conti CJ. (C‐A)n microsatellite repeat D7S522 is the most commonly deleted region in human primary breast cancer. Proc Natl Acad Sci U S A 1994; 91: 12155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zenklusen JC, Thompson JC, Troncoso P, Kagan J, Conti CJ. Loss of heterozygosity in human primary prostate carcinomas: a possible tumor suppressor gene at 7q31.1. Cancer Res 1994; 54: 6370–3. [PubMed] [Google Scholar]
- 24. Shridhar V, Sun QC, Miller OJ, Kalemkerian GP, Petros J, Smith DI. Loss of heterozygosity on the long arm of human chromosome 7 in sporadic renal cell carcinomas. Oncogene 1997; 15: 2727–33. [DOI] [PubMed] [Google Scholar]
- 25. Achille A, Biasi MO, Zamboni G et al. Chromosome 7q allelic losses in pancreatic carcinoma. Cancer Res 1996; 56: 3808–13. [PubMed] [Google Scholar]
- 26. Zenklusen JC, Thompson JC, Klein‐Szanto AJ, Conti CJ. Frequent loss of heterozygosity in human primary squamous cell and colon carcinomas at 7q31.1: evidence for a broad range tumor suppressor gene. Cancer Res 1995; 55: 1347–50. [PubMed] [Google Scholar]
- 27. Kuniyasu H, Yasui W, Yokozaki H et al. Frequent loss of heterozygosity of the long arm of chromosome 7 is closely associated with progression of human gastric carcinomas. Int J Cancer 1994; 59: 597–600. [DOI] [PubMed] [Google Scholar]
- 28. Zenklusen JC, Rodriguez LV, LaCava M, Wang Z, Goldstein LS, Conti CJ. Novel susceptibility locus for mouse hepatomas: evidence for a conserved tumor suppressor gene. Genome Res 1996; 6: 1070–6. [DOI] [PubMed] [Google Scholar]
- 29. Atkin NB, Baker MC. Chromosome 7q deletions: observations on 13 malignant tumors. Cancer Genet Cytogenet 1993; 67: 123–5. [DOI] [PubMed] [Google Scholar]
- 30. Pedersen B, Ellegaard J. A factor encoded by 7q31 suppresses expansion of the 7q‐ clone and delays cytogenetic progression. Cancer Genet Cytogenet 1994; 78: 181–8. [DOI] [PubMed] [Google Scholar]
- 31. Drusco A, Zanesi N, Roldo C et al. Knockout mice reveal a tumor suppressor function for Testin. Proc Natl Acad Sci U S A 2005; 102: 10947–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang W, Weng DS, Pan ZZ et al. [Expression and clinical significance of TESTIN in primary gastric cancer]. Ai Zheng 2008; 27: 984–8. [PubMed] [Google Scholar]
- 33. Gunduz E, Gunduz M, Beder L et al. Downregulation of TESTIN and its association with cancer history and a tendency toward poor survival in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg 2009; 135: 254–60. [DOI] [PubMed] [Google Scholar]
- 34. Jones PA. DNA methylation and cancer. Oncogene 2002; 21: 5358–60. [DOI] [PubMed] [Google Scholar]
- 35. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457–63. [DOI] [PubMed] [Google Scholar]
- 36. Collins Y, Dicioccio R, Keitz B, Lele S, Odunsi K. Methylation of death‐associated protein kinase in ovarian carcinomas. Int J Gynecol Cancer 2006; 16 (Suppl 1): 195–9. [DOI] [PubMed] [Google Scholar]
- 37. Sellar GC, Watt KP, Rabiasz GJ et al. OPCML at 11q25 is epigenetically inactivated and has tumor‐suppressor function in epithelial ovarian cancer. Nat Genet 2003; 34: 337–43. [DOI] [PubMed] [Google Scholar]
- 38. Makarla PB, Saboorian MH, Ashfaq R et al. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin Cancer Res 2005; 11: 5365–9. [DOI] [PubMed] [Google Scholar]
- 39. Wakana K, Akiyama Y, Aso T, Yuasa Y. Involvement of GATA‐4/‐5 transcription factors in ovarian carcinogenesis. Cancer Lett 2006; 241: 281–8. [DOI] [PubMed] [Google Scholar]
- 40. Mueller W, Nutt CL, Ehrich M et al. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene 2007; 26: 583–93. [DOI] [PubMed] [Google Scholar]
- 41. Sarti M, Sevignani C, Calin GA et al. Adenoviral transduction of TESTIN gene into breast and uterine cancer cell lines promotes apoptosis and tumor reduction in vivo. Clin Cancer Res 2005; 11: 806–13. [PubMed] [Google Scholar]