

Abstract
The BCL2/IGH translocation is a hallmark of follicular lymphoma and germinal center B‐cell type diffuse large B‐cell lymphoma. Although a strong determinant of these histological subtypes, this translocation is insufficient by itself for lymphomagenesis, so that other genetic alterations are required. To clarify how the BCL2 translocation contributes to the development of specific lymphoma subtypes, we used chimeric mouse models and a bone marrow transplantation system to examine the biological features of BCL2‐overexpressing B cells. These cells showed a cell‐autonomous differentiation preference for follicular B cells. Their cell cycle progression was enhanced in wild‐type but not in Eμ‐BCL2 transgenic mice, indicating that the low proliferative activity of B cells in Eμ‐BCL2 transgenic mice is partly due to their specific microenvironment, which is caused by the abnormal B cells themselves. Moreover, in vitro experiments demonstrated that Eμ‐BCL2 + B cells have reduced responsiveness to terminal differentiation stimulation. According to these results, we hypothesize that B cells that have undergone BCL2/IGH translocation might possibly be forced to localize in follicles, and accumulate genetic abnormalities by being subjected to recurrent stimulation. Our findings lead us to propose that B cells carrying the BCL2/IGH translocation comprise a distinctive cell population that leads to the development of germinal center B‐cell type lymphoma. (Cancer Sci 2009; 100: 2361–2367)
The BCL2/IGH translocation t(14;18)(q32;q21) is one of the most common cytogenetic abnormalities in lymphoid malignancy and is a hallmark of follicular lymphoma (FL) and diffuse large B‐cell lymphoma (DLBCL) of germinal center B‐cell type.( 1 , 2 ) This translocation juxtaposes the BCL2 gene with the immunoglobulin heavy chain (IGH) locus, which then leads to deregulated expression of the anti‐apoptotic Bcl‐2 protein. Some healthy individuals bear B cells that carry the BCL2 translocation at a low frequency, but the translocation is insufficient by itself to induce lymphomagenesis, which means that other gene alterations are required.( 3 , 4 ) Eμ‐BCL2 transgenic (Tg) mice possess several times the number of B‐lineage cells compared to the number in wild‐type (WT) mice, but only 5–15% of these mice develop disease in the first year of life.( 5 , 6 )
The translocation is assumed to be generated by an error during physiological VDJ rearrangement of the IGH gene in early B‐cell development,( 7 ) but it is not known why this translocation is such a strong determinant of the histological subtypes of lymphomas of germinal center B‐cell origin. We hypothesized that B cells with BCL2/IGH translocation might already have some properties that determine the subsequent lymphoma subtypes, and that these characteristics would be highlighted when they were present in normal individuals as a minor population. We used transgenic mouse models and a bone marrow transplantation system to examine BCL2 expressing B‐cell kinetics. They exhibited a unique differentiation preference and their distinctive cell features provide some clues for elucidating the initiating event of lymphoma development triggered by BCL2/IGH translocation. The developmental processes of lymphomas initiated by the BCL2/IGH translocation will be discussed in this report.
Materials and Methods
Mice. Eμ‐BCL2‐22 Tg mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA).( 8 ) Transgenic mice expressing enhanced GFP driven by the CAG promoter were kindly provided by Dr M. Okabe, Osaka University.( 9 ) Both groups of Tg mice had a C57BL/6 background, and were bred and maintained under specific pathogen‐free conditions in the animal care facility at Kyoto University. Mice between 7 and 12 weeks of age were used for the experiments. All experiments were performed under the approved protocols of the Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University.
Generation of lentiviral vectors and transplantation. Recombinant B‐cell‐specific lentiviral vector Eμmar‐L.CD19‐GFP was provided by Drs T. Moreau and C. Tonnelle, Institut Paoli‐Calmettes, Marseille, France.( 10 , 11 ) Flag‐tagged murine BCL2 and its mutants were inserted between the BamHI and XhoI sites of the vector. The packaging plasmid (pCAG‐HIVgp) and the VSV‐G‐ and Rev‐expressing plasmid (pCMV‐VSV‐G‐RSV‐Rev) were provided by Dr H. Miyoshi, RIKEN Bioresource Center, Tsukuba, Japan. Lentiviral infection of bone marrow (BM) cells and transplantation was performed as described previously.( 12 ) Briefly, BM cells were harvested from tibiae and femora of B6 mice 4 days after intravenous administration of 5‐fluorouracil and osmotically lysed red blood cells. The BM cells were then prestimulated with stem cell factor, interleukin (IL)‐3 and IL‐6 for 24 h, spin‐infected with the virus in the presence of cytokines and polybrene, and transplanted intravenously into lethally irradiated (9 Gy) recipient mice.
Flow cytometry, TNP‐Ficoll binding assay, and immunohistochemistry. For the flow cytometry assay, splenocytes were analyzed on a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) using antibodies against B220, CD21, CD23, IgM, IgD (eBioscience, San Diego, CA, USA), TNP, and CD138 (BD Pharmingen, San Diego, CA, USA). For the TNP‐Ficoll binding assay, the mice were injected intravenously with 100 μg TNP30‐Ficoll (Biosearch Technologies, Novato, CA, USA), and TNP‐bound splenic B cells were analyzed. For immunohistochemistry, spleens were snap‐frozen in OCT compound (Tissue‐Tek, Tokyo, Japan), and cryostat sections were immunostained with the following antibodies:( 13 ) anti‐B220 (RA3‐6B2; BD Pharmingen), FDC‐M2 (ImmunoKontact, Abingdon, UK), anti‐CD3ε (KT3; AbD), and MOMA‐1 (AbD Serotec, Raleigh, NC, USA).
BrdU incorporation analysis. Bone marrow mononuclear cells from the CAG‐GFP/Eμ‐BCL2 double Tg mice were extracted by gradient centrifugation and transferred into WT and Eμ‐BCL2 Tg mice without pretreatment. For this procedure, only B cells expressing BCL2 transgene were engrafted. Ten days later, feeding of the recipient mice was started with 0.8 mg/ml BrdU (Sigma, St. Louis, MO, USA) in their drinking water for the periods indicated, and BrdU incorporation into splenic B cells was assessed with the BrdU flow kit (BD Pharmingen).
Somatic hypermutation analysis. Splenic B cells from the CAG‐GFP/Eμ‐BCL2 double Tg mice were collected by negative selection using SpinSepB (Veritas, Tokyo, Japan), equally divided, and transferred into WT and Eμ‐BCL2 Tg mice. The recipient mice were immunized with intraperitoneal injection of 1 × 105 sheep red blood cells on days 3 and 10 after cell transfer, and sacrificed on day 13 and B220 + GFP + splenocytes were sorted with the FACSAria cell sorter (BD Biosciences). After extraction of DNA from the sorted cells, the 3′‐flank of the VDJH rearrangements that involve members of the VHJ558 family were PCR amplified and analyzed for somatic hypermutation.( 14 )
In vitro stimulation of B cells, real‐time quantitative reverse transcriptiion–polyymerase chain reaction (RT‐PCR), and Western blotting. Splenic B cells of WT and Eμ‐BCL2 Tg mice were collected and stimulated with lipopolysaccharide (LPS) (2.5 μM) or anti‐IgM antibody (10 μM) for 4 days, and analyzed on a FACSCalibur (BD Biosciences). For real‐time quantitative RT‐PCR, RNA was extracted from the cells stimulated for 2 days, and analyzed by means of TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA). FK506 (Sigma) and PD98059 (Cell Signaling, Danvers, MA, USA) were used for the inhibition of the NF‐AT and ERK signaling, respectively.
Results
Deregulated expression of BCL2 causes distortion in differentiation, cell size, and immunological activity of B cells. First, we noted that splenic B cells of Eμ‐BCL2 Tg mice showed increased follicular and reduced marginal zone subsets (Fig. 1a). They also exhibited a reduction in cell volume compared to that of WT mice, which was similar to a previously reported finding concerning BCL2‐overexpressing T cells in lckpr‐Bcl‐2 mice.( 15 ) To determine whether these features are also actually caused by B cells with deregulated Bcl‐2 protein in normal individuals, we established a lentiviral vector system with the CD19 promoter to express BCL2 or its mutants in mouse B cells. Murine bone marrow cells were transduced with the lentiviral vectors and transplanted into irradiated mice so that a minor population of B cells would express deregulated BCL2 under the control of the CD19 promoter. Recipient splenocytes were analyzed 2 months after transplantation by gating of GFP, which was coexpressed by the vectors. BCL2‐containing B cells showed increased follicular and reduced marginal zone subsets, indicating that the B cell differentiation distortion in Eμ‐BCL2 Tg mice was caused most likely by the deregulated Bcl‐2 protein in B cells (Fig. 1b). The same phenotypic shift was seen in B cells containing the BCL2‐Y28F mutant, which retains its anti‐apoptotic function,( 16 , 17 ) but the anti‐apoptosis defective mutant BCL2‐G142A (corresponding to BCL2‐G145A in humans( 18 )) did not noticeably affect the B‐cell phenotype, suggesting that the anti‐apoptotic function of Bcl‐2 plays a major role in the distortion of B cell differentiation. In addition, BCL2‐ and BCL2‐Y28F‐containing B cells were smaller than those containing BCL2‐G142A or mock vectors. To assess the immunological activity of the marginal zone B‐cell subset in BCL2‐expressing B cells, we examined their T‐cell independent response to TNP‐Ficoll injected intravenously. The results showed that the number of TNP‐Ficoll binding cells was significantly reduced in BCL2‐containing B cells (Fig. 1c), thus demonstrating that the marginal zone B cell population in BCL2‐containing B cells was reduced in both the phenotypic and functional analyses. These observations suggest that the deregulated expression of BCL2 in B cells leads to distorted differentiation in conjunction with a preference toward follicular B cells and a reduction in cell size, and that these effects are caused by the anti‐apoptotic activity of deregulated Bcl‐2.
Figure 1.
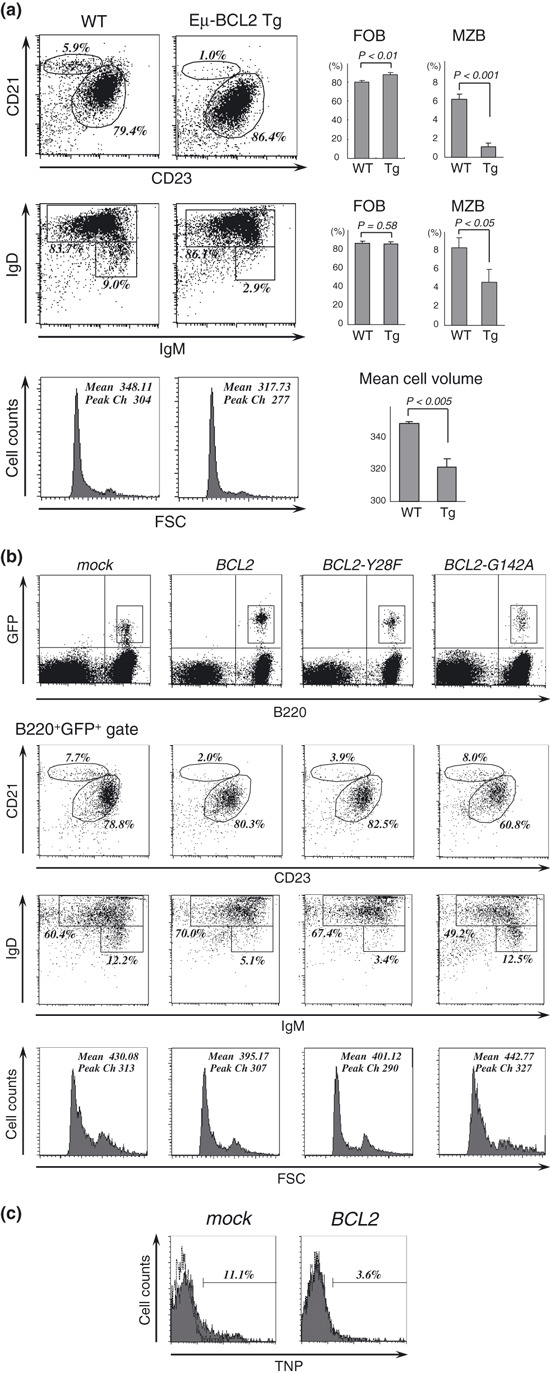
BCL2‐overexpressing B cells have a specific differentiation preference toward follicular B cells. (a) Flow cytometric analysis of the splenic B cells derived from wild‐type and Eμ‐BCL2 Tg mice. B220+ cells were gated and analyzed for marginal zone B cells (CD23lowCD21hi and IgMhiIgDlow subsets) and follicular B cells (CD23hiCD21int and IgM+IgDhi subsets). The numbers in each panel represent the percentages of the gated cells, and the bar graphs the mean percentage of each population (n = 3), with error bars representing the SDs. Forward scatter histograms of the B220+ gated cells illustrate relative cell size, with the mean fluorescence intensity and peak channels indicated. (b) Flow cytometric analysis of the splenic B cells that expressed BCL2 and its mutant genes, which were delivered by lentiviral vectors. GFP+B220+ cells were gated and analyzed for marginal zone and follicular B cell populations as shown in (a). Representative dot plots of at least three independent experiments are shown. Forward scatter histograms of the GFP+B220+ cells are shown below, with the mean fluorescence intensity and peak channels indicated. (c) TNP‐Ficoll binding assay. BCL2 or mock vector‐introduced mice were injected intravenously with TNP‐Ficoll. Thirty minutes later, GFP+ splenic B cells were analyzed for TNP‐Ficoll capturing populations. The dashed line on each histogram represents an isotype control antibody.
B cells with deregulated BCL2 expression are more efficiently stimulated in a WT than in a Eμ‐BCL2 Tg setting. We were able to demonstrate that BCL2‐deregulated B cells have a differentiation preference toward follicular B cells, but low occurrence rates of B‐cell tumors in Eμ‐BCL2 Tg mice indicated that the differentiation preference is not a sufficient condition for the lymphoma development of BCL2‐deregulated B cells. The low transformation frequency of B cells in Eμ‐BCL2 Tg mice has been partly attributed to their propensity to reside in the G0 phase of the cell cycle.( 19 , 20 ) We next wanted to identify differences between the host animals in order to focus on the microenvironment of BCL2‐deregulated B cells. To this purpose, we generated BCL2 + GFP + B cells by crossing Eμ‐BCL2 Tg with CAG‐GFP Tg mice and extracting their bone marrow mononuclear cells by means of gradient centrifugation. These cells were then transferred equally into WT and Eμ‐BCL2 Tg mice. Ten days after the transfer, splenocytes were extracted from the recipient mice, and GFP + BCL2 + B cells and GFP − endogenous B cells were separately analyzed for their blastic cell populations (Fig. 2A). The percentage of blastic cells, that is activated follicular B cells, in the GFP + BCL2 + cells that were transferred into the WT mice was twice as high as in those transferred into the Eμ‐BCL2 Tg mice, suggesting that the microenvironment of the latter mice was somewhat altered.
Figure 2.
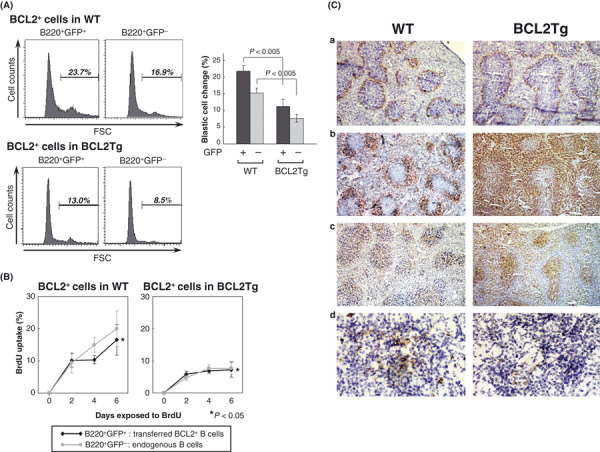
The altered B cell microenvironment in Eμ‐BCL2 Tg mice. (A) Forward scatter histograms showing the differences in cell size of the CAG‐GFP/Eμ‐BCL2 + B cells that were transferred into WT and Eμ‐BCL2 Tg mice. Transferred BCL2 + B cells and endogenous B cells were analyzed by gating of B220+GFP+ and B220+GFP− cells, respectively. The bar graph shows the mean percentage of blastic cells in each population (n = 3), with error bars representing the SDs. (B) BrdU incorporation assay. After the transference of CAG‐GFP/Eμ‐BCL2 + bone marrow mononuclear cells, WT and Eμ‐BCL2 Tg mice were fed BrdU in their drinking water for the periods indicated. BrdU incorporation at each time point was plotted as a percentage of total B cells and was analyzed separately for B220+GFP+ and B220+GFP− cells. Figures show the means and SDs of experiments performed in triplicate. (C) Cryostat sections of the spleens from WT and Eμ‐BCL2 Tg mice stained with MOMA‐1 for metallophilic macrophages (a), with B220 for B cells (b), with CD3 for T cells (c) (Nicon Eclipse E600; original magnification, ×40), and with FDC‐M2 for follicular dendritic cells (d) (Nicon Eclipse E600; original magnification, ×400). In Eμ‐BCL2 Tg mice, the number of follicular dendritic cells is manifestly reduced.
For assessment of their cell cycle regulation, the recipient mice were next given BrdU‐containing water to drink followed by evaluation of BrdU uptake into B cells (Fig. 2B). BrdU was consistently incorporated into the GFP + BCL2 + B cells transferred into the WT mice, while their cell cycle progression tended to be slower than that of normal endogenous B cells. In contrast, BrdU uptake was suppressed in the GFP + BCL2 + B cells transferred into the Eμ‐BCL2 Tg mice, while endogenous GFP − BCL2 + B cells showed a similarly slowed‐down cell cycle. These results indicate that BCL2‐overexpressing B cells proliferate in follicles in healthy individuals, while they mostly remain quiescent in an Eμ‐BCL2 Tg setting. Immunohistochemical analysis of the splenic architecture of WT and Eμ‐BCL2 Tg mice demonstrated that the follicular dendritic cells of Eμ‐BCL2 Tg mice were markedly defective (Fig. 2Cd) although the follicles themselves were hyperplastic (Fig. 2Ca–c). The lack of follicular dendritic cells in Eμ‐BCL2 Tg mice is assumed to be caused by the reduction in the marginal zone B‐cell subset because marginal zone B cells reportedly play an important role in the development of follicular dendritic cells.( 21 ) Thus, it is probable that follicular B cells do not properly proliferate in Eμ‐BCL2 Tg mice due to the lack of a mature dendritic cell network.
B‐cell mutagenetic activity is reduced in the Eμ‐BCL2 Tg microenvironment. We hypothesized that a germinal center with a better organization would more actively introduce genetic alterations in B cells as a result of the efficient induction of activation‐induced cytidine deaminase (AID). To evaluate the mutagenetic potential of the germinal center reaction in WT and Eμ‐BCL2 Tg mice, we compared the somatic hypermutation frequency of the IGH gene of GFP + BCL2 + B cells that were transferred into WT and Eμ‐BCL2 Tg mice. After two intraperitoneal injections of sheep red blood cells into the recipient mice, GFP + BCL2 + B cells were sorted from the spleens and their genomic DNA was analyzed for mutations in the 3′‐flank of VDJH rearrangements involving members of the VHJ558 family.( 14 ) The frequency of mutation was found to be higher in the cells transferred into WT mice than in those transferred into Eμ‐BCL2 Tg mice, although the difference did not reach statistical significance, possibly because of the limited observation period (data shown in Fig. S1). These results indicate that B cells carrying the BCL2/IGH translocation in normal individuals are more predisposed to acquire genetic abnormalities than was predicted from previous studies of Eμ‐BCL2 Tg mice.
Eμ‐BCL2+ B cells show diminished responsiveness to terminal differentiation stimulation. B‐cell BCL2 expression is physiologically suppressed in germinal centers so that a large number of low‐affinity B cells is eliminated. B cells with irrelevant BCL2 expression are considered to have an advantage for surviving this selection procedure. To determine the fate of BCL2‐overexpressing B cells after stimulation, we assessed the terminal differentiation capacity of B cells derived from Eμ‐BCL2 Tg and WT mice in vitro. We found that Eμ‐BCL2 + B cells showed a diminished responsiveness to both LPS and anti‐IgM antibody stimulation (Fig. 3Aa–b). Especially noteworthy is that terminal differentiation was almost completely blocked in Eμ‐BCL2 + B cells under anti‐IgM stimulation, whereas their surface IgM expression level was comparable to that of WT B cells (Fig. 3Ac). One of the signaling pathways activated in response to B‐cell receptor (BCR) engagement is the calcium‐dependent activation of NF‐AT, which is a requirement for proper B cell activation.( 22 , 23 ) As overexpression of Bcl‐2 is reported to sequester calcineurin and inhibit its activity,( 15 , 24 ) we assumed that the unresponsiveness of Eμ‐BCL2 + B cells to anti‐IgM stimulation might be caused by the inhibition of the calcineurin‐dependent signaling that occurs downstream of BCR. We therefore stimulated WT B cells in the presence of the calcineurin inhibitor FK506, and their differentiation was found to be suppressed (Fig. 3Aa). In real‐time RT‐PCR analysis, downregulation of the BCL6 and PAX5 genes and upregulation of PRDM1(Blimp‐1) gene expression were observed in response to LPS and anti‐IgM stimulation in normal B cells, while the expression of these genes was less altered in Eμ‐BCL2 + B cells and FK506‐pretreated WT B cells (Fig. 3B).
Figure 3.
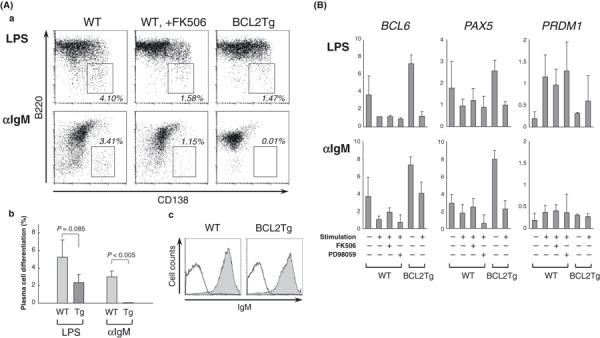
Reduced responsiveness of BCL2‐overexpressing B cells during plasma cell differentiation. (A) (a) Plasma cell differentiation of the WT and Eμ‐BCL2 + B cells after a 4‐day stimulation with lipopolysaccharide (LPS) or anti‐IgM antibody in vitro. WT B cells were also stimulated in the presence of FK506 (middle panels). (b) The mean percentages of B cells that differentiated into plasma cells are shown by the columns, with error bars indicating the SDs (n = 3). (c) Histograms demonstrate surface IgM expression of WT and Eμ‐BCL2 + B cells. (B) Real‐time RT‐PCR analysis of the WT and Eμ‐BCL2 + B cells for the quantification of gene transcripts of BCL6, PAX5, and PRDM1. The cells were stimulated with LPS or anti‐IgM for 2 days, and with FK506 (1 μM) or ERK inhibitor PD98059 (20 μM) in the samples shown. The values represent the relative expression levels normalized to GAPDH. Data are the means and SDs of three independent experiments.
These results lead us to assume that the suppressed terminal differentiation that occurs in response to BCR ligation in Eμ‐BCL2 + B cells is caused at least in part by the inhibition of the calcineurin–NF‐AT pathway.
Discussion
Deregulated BCL2 expression confers not only anti‐apoptotic function but also differentiation preference. One of the major characteristics of human B‐cell lymphoid neoplasms is that particular chromosomal translocations are closely related to certain lymphoma subtypes derived from specific developmental stages.( 25 ) Chromosomal translocation is supposed to render B cells tumorigenic at a stage when the target gene expression is not physiologically active.( 26 ) However, this does not provide a full explanation for the close correlation between chromosomal translocation and the cell origin of lymphomas, because chromosomal translocation is merely the primary event, and additional genetic abnormalities are required for tumor generation. This leads to the hypothesis that B cells might linger at a particular differentiation stage while incurring additional DNA damage. Our findings are novel in demonstrating that the BCL2 translocation not only confers anti‐apoptotic characteristics to B cells, but also several distinctive biological features, including a differentiation preference that potentially leads to the development of lymphoma of specific histological subtypes. The influence of chromosomal translocation upon B cell differentiation is similarly implicated in transgenic mouse models of API2‐MALT1 translocation, which is specific to extranodal marginal zone lymphoma of mucosa‐associated lymphoid tissue (MALT lymphoma). Although the introduction of this transgene alone is not sufficient for lymphoma development, it results in the expansion of marginal zone B cell populations.( 27 )
Differentiation distortion of BCL2‐expressing B cells. The surviving population of B cells in Eμ‐BCL2 Tg mice reportedly comprises mostly low‐affinity germinal center and memory B cells.( 28 ) As high‐affinity germinal center B cells are considered to have priority for differentiation into plasma cells,( 29 , 30 ) it is not surprising that BCL2‐deregulated B cells with low‐affinity antibody expression have little chance to differentiate into plasma cells when competing with normal B cells. Our results showing low responsiveness of BCL2‐overexpressing B cells upon LPS or anti‐IgM stimulation may indicate the existence of an additional mechanism for the reduction in terminal differentiation capacity. We do not believe that our data contradict that of previous reports of the presence (or increase in some studies) of plasma cells in Eμ‐BCL2 Tg mice, as the terminal differentiation defects that we observed were partial, and there was an overwhelming number of B cells in Eμ‐BCL2 Tg mice to compensate for these defects.
Accumulation of genetic alterations for BCL2‐overexpressing B cells. In this study, we have demonstrated that the B cells carrying the BCL2/IGH translocation display a cell‐autonomous differentiation preference toward follicular B cells and low terminal differentiation responsiveness, as well as anti‐apoptotic activity. We hypothesize that they would re‐circulate as memory B cells and accumulate genetic abnormalities while they repeatedly pass through germinal centers (Fig. 4). In t(14;18)+ B cells in healthy individuals, this translocation is carried by an expanding population of memory B cells, which constitutes a potent pre‐malignant niche.( 31 ) Recently, a mutation analysis of the switch μ region of the der(14)(14;18) of FL samples indicated that different subclones often arise from a pool of pretumoral cells that are less clonally evolved.( 32 ) Our study results support the notion of these pre‐malignant BCL2/IGH + B cell niches and shed light on their unique life cycle in normal individuals.
Figure 4.
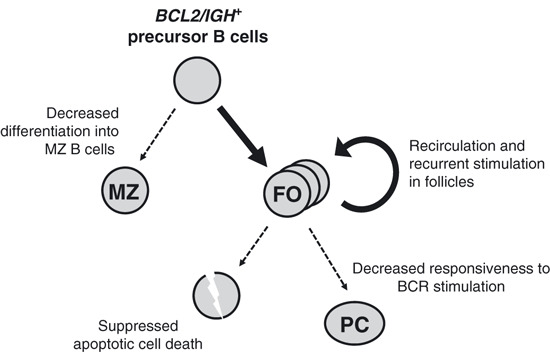
A proposed model of the development and outcome of B cells carrying the BCL2/IGH translocation in normal individuals. Deregulated BCL2‐expressing B cells show suppressed differentiation to marginal zone B cells and differentiation preference toward follicular B cells. The follicular B cells are suppressed in apoptotic cell death and are less responsive to terminal differentiation stimuli, resulting in accumulation of follicular B cells. The cells have been shown to undergo somatic mutation upon antigen stimulation in a normal environment, rendering these cells into a premalignant niche. FO, follicular B cells; MZ, marginal zone B cells; PC, plasma cells.
Significance of the microenvironment surrounding BCL2‐overexpressing B cells during the development of lymphoma. Because of the cell cycle retarding effect of BCL2, the oncogenetic role of BCL2 deregulation has often been assessed in combination with the genetic abnormalities that drive cell proliferation such as c‐MYC or Moloney murine leukemia virus in mouse model.( 33 , 34 ) However, our findings suggest that the retarded cell cycle progression of BCL2‐overexpressing B cells in normal individuals could be restored. This indicates that lower frequency of lymphoma development in Eμ‐BCL2 transgenic mouse may be caused by defects in its microenvironment.
BCL2/IGH translocation is found not only in follicular lymphoma but also in approximately half of germinal center B‐cell (GCB)‐type DLBCL. The mechanism for developing different lymphoma subtypes from the same origin of BCL2/IGH + germinal center B cells has not been elucidated yet, but several previous reports suggest that it could be partly explained by the difference of microenvironment.
In VavP‐BCL2 transgenic mice, BCL2 is expressed in cells of all hematopoietic lineages, and they are reported to develop follicular lymphoma after preceding germinal center hyperplasia.( 35 ) In contrast, Eμ‐BCL2 transgenic mice develop more aggressive B‐cell lymphomas in a low frequency. One of the largest differences between these two transgenic mice is the quantity of immune cells other than B cells. The number of T cells, especially of CD4+ T cells, was shown to be considerably higher in the spleen of VavP‐BCL2 mice compared to that in the Eμ‐BCL2 mice, and the authors hypothesized that the increased survival capacity of B and T cells fostered prolonged germinal center reactions, and potentially led to the generation of follicular lymphoma.
On the other hand, B cells outnumber other blood cells in Eμ‐BCL2 transgenic mice, suggesting that aggressive lymphomas could develop more independently of surrounding immune cells than follicular lymphoma. This hypothesis might also be in line with the histological appearance of follicular lymphoma and DLBCL: follicular lymphoma tissue contains many T cells and follicular dendritic cells, whereas DLBCL typically shows diffuse spreading of malignant B cells. The importance of tumor‐infiltrating immune cells in the pathogenesis of follicular lymphoma is also indicated in the experiment of Dave et al. ( 36 )
In other words, the divergence of the development of follicular lymphoma and GCB‐DLBCL from BCL2/IGH + B cells might be determined in part by the intensity of interaction of B cells with surrounding T cells and other immune cells, and this may potentially be influenced by the presence of antigen stimulation or adhesion molecules expressed on B cells.
As human lymphoid neoplasms often arise in a locus‐ and/or stage‐specific manner, we believe that not only the genetic abnormalities but also the microenvironment of pretumoral cells should be mimicked in order to faithfully reproduce tumors in animal models. For this reason, we propose that chimeric mouse models like ours are more suitable than simple transgenic models for the study of lymphomagenesis, especially when the specific microenvironment plays a role in the oncogenetic process.
Supporting information
Fig. S1. Somatic hypermutation analysis of the BCL2 + B cells that were transferred into WT and Eμ‐BCL2 Tg mice. Splenic B cells were sorted from CAG‐GFP/Eμ‐BCL2 double Tg mice and were divided into two equal parts before being transferred into WT and Eμ‐BCL2 Tg mice. The recipient mice were immunized with sheep red blood cells on days 3 and 10 of the cell transfer, and splenocytes were extracted and GFP+ B cells sorted on day 13. DNA was extracted from GFP+ B cells followed by analysis of the mutation frequencies of the region that flanks the rearranged JH4 genes.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank Dr Masaru Okabe of Osaka University for providing us with the CAG‐GFP Tg mice, Dr Thomas Moreau and Dr Cécile Tonelle of the Institut Paoli‐Calmettes, and Dr Hiroyuki Miyoshi of the RIKEN Bioresource Center for providing us with the vectors used in the lentiviral vector experiment. We are also grateful to Mr Yoshinobu Toda of Kyoto University for performing the histological examination. This work was supported by grants‐in‐aid from the Ministry of Education, Culture, Sports, Science, and Technology (nos. 18790650, 20790673) and partly from the Ministry of Health, Labour and Welfare of Japan.
References
- 1. Huang JZ, Sanger WG, Greiner TC et al. The t(14;18) defines a unique subset of diffuse large B‐cell lymphoma with a germinal center B‐cell gene expression profile. Blood 2002; 99: 2285–90. [DOI] [PubMed] [Google Scholar]
- 2. Iqbal J, Sanger WG, Horsman DE et al. BCL2 translocation defines a unique tumor subset within the germinal center B‐cell‐like diffuse large B‐cell lymphoma. Am J Pathol 2004; 165: 159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Summers KE, Goff LK, Wilson AG, Gupta RK, Lister TA, Fitzgibbon J. Frequency of the Bcl‐2/IgH rearrangement in normal individuals: implications for the monitoring of disease in patients with follicular lymphoma. J Clin Oncol 2001; 19: 420–4. [DOI] [PubMed] [Google Scholar]
- 4. Yasukawa M, Bando S, Dolken G et al. Low frequency of BCL‐2/J H translocation in peripheral blood lymphocytes of healthy Japanese individuals. Blood 2001; 98: 486–8. [DOI] [PubMed] [Google Scholar]
- 5. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high‐grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 1991; 349: 254–6. [DOI] [PubMed] [Google Scholar]
- 6. Strasser A, Harris AW, Cory S. Eμ‐bcl‐2 transgene facilitates spontaneous transformation of early pre‐B and immunoglobulin‐secreting cells but not T cells. Oncogene 1993; 8: 1–9. [PubMed] [Google Scholar]
- 7. Bakhshi A, Wright JJ, Graninger W et al. Mechanism of the t(14;18) chromosomal translocation: structural analysis of both derivative 14 and 18 reciprocal partners. Proc Natl Acad Sci U S A 1987; 84: 2396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strasser A, Whittingham S, Vaux DL et al. Enforced BCL2 expression in B‐lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A 1991; 88: 8661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett 1997; 407: 313–9. [DOI] [PubMed] [Google Scholar]
- 10. Moreau T, Bardin F, Imbert J, Chabannon C, Tonnelle C. Restriction of transgene expression to the B‐lymphoid progeny of human lentivirally transduced CD34+ cells. Mol Ther 2004; 10: 45–56. [DOI] [PubMed] [Google Scholar]
- 11. Moreau T, Barlogis V, Bardin F et al. Development of an enhanced B‐specific lentiviral vector expressing BTK: a tool for gene therapy of XLA . Gene Ther 2008; 15: 942–52. [DOI] [PubMed] [Google Scholar]
- 12. Tsuzuki S, Hong D, Gupta R, Matsuo K, Seto M, Enver T. Isoform‐specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1 . PLoS Med 2007; 4: e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Victoratos P, Lagnel J, Tzima S et al. FDC‐specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity 2006; 24: 65–77. [DOI] [PubMed] [Google Scholar]
- 14. Jolly CJ, Klix N, Neuberger MS. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res 1997; 25: 1913–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Linette GP, Li Y, Roth K, Korsmeyer SJ. Cross talk between cell death and cell cycle progression: BCL‐2 regulates NFAT‐mediated activation. Proc Natl Acad Sci U S A 1996; 93: 9545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang DC, O’Reilly LA, Strasser A, Cory S. The anti‐apoptosis function of Bcl‐2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J 1997; 16: 4628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janumyan YM, Sansam CG, Chattopadhyay A et al. Bcl‐xL/Bcl‐2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J 2003; 22: 5459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin B, Kolluri SK, Lin F et al. Conversion of Bcl‐2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 2004; 116: 527–40. [DOI] [PubMed] [Google Scholar]
- 19. McDonnell TJ, Nunez G, Platt FM et al. Deregulated Bcl‐2‐immunoglobulin transgene expands a resting but responsive immunoglobulin M and D‐expressing B‐cell population. Mol Cell Biol 1990; 10: 1901–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams JM, Harris AW, Strasser A, Ogilvy S, Cory S. Transgenic models of lymphoid neoplasia and development of a pan‐hematopoietic vector. Oncogene 1999; 18: 5268–77. [DOI] [PubMed] [Google Scholar]
- 21. Cinamon G, Zachariah MA, Lam OM, Foss FW Jr., Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immun 2008; 9: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev 2007; 7: 690–702. [DOI] [PubMed] [Google Scholar]
- 23. Gallo EM, Cante‐Barrett K, Crabtree GR. Lymphocyte calcium signaling from membrane to nucleus. Nat Immun 2006; 7: 25–32. [DOI] [PubMed] [Google Scholar]
- 24. Shibasaki F, Kondo E, Akagi T, McKeon F. Suppression of signalling through transcription factor NF‐AT by interactions between calcineurin and Bcl‐2. Nature 1997; 386: 728–31. [DOI] [PubMed] [Google Scholar]
- 25. Kuppers R. Mechanisms of B‐cell lymphoma pathogenesis. Nat Rev Cancer 2005; 5: 251–62. [DOI] [PubMed] [Google Scholar]
- 26. Seto M. Genetic and epigenetic factors involved in B‐cell lymphomagenesis. Cancer Sci 2004; 95: 704–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baens M, Fevery S, Sagaert X et al. Selective expansion of marginal zone B cells in Eμ‐API2‐MALT1 mice is linked to enhanced IκB kinase γ polyubiquitination. Cancer Res 2006; 66: 5270–7. [DOI] [PubMed] [Google Scholar]
- 28. Smith KG, Light A, O’Reilly LA, Ang SM, Strasser A, Tarlinton D. bcl‐2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody‐forming cells. J Exp Med 2000; 191: 475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phan TG, Paus D, Chan TD et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med 2006; 203: 2419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med 2006; 203: 1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roulland S, Navarro JM, Grenot P et al. Follicular lymphoma‐like B cells in healthy individuals: a novel intermediate step in early lymphomagenesis. J Exp Med 2006; 203: 2425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruminy P, Jardin F, Picquenot JM et al. Sμ mutation patterns suggest different progression pathways in follicular lymphoma: early direct or late from FL progenitor cells. Blood 2008; 112: 1951–9. [DOI] [PubMed] [Google Scholar]
- 33. Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl‐2. Nature 1990; 348: 331–3. [DOI] [PubMed] [Google Scholar]
- 34. Shinto Y, Morimoto M, Katsumata M et al. Moloney murine leukemia virus infection accelerates lymphomagenesis in Eμ‐bcl‐2 transgenic mice. Oncogene 1995; 11: 1729–36. [PubMed] [Google Scholar]
- 35. Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. VavP‐Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 2004; 103: 2276–83. [DOI] [PubMed] [Google Scholar]
- 36. Dave SS, Wright G, Tan B et al. Prediction of survival in follicular lymphoma based on molecular features of tumor‐infiltrating immune cells. N Eng J Med 2004; 351: 2159–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Somatic hypermutation analysis of the BCL2 + B cells that were transferred into WT and Eμ‐BCL2 Tg mice. Splenic B cells were sorted from CAG‐GFP/Eμ‐BCL2 double Tg mice and were divided into two equal parts before being transferred into WT and Eμ‐BCL2 Tg mice. The recipient mice were immunized with sheep red blood cells on days 3 and 10 of the cell transfer, and splenocytes were extracted and GFP+ B cells sorted on day 13. DNA was extracted from GFP+ B cells followed by analysis of the mutation frequencies of the region that flanks the rearranged JH4 genes.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item