

Abstract
A malfunction in retinoid X receptor (RXR) α due to phosphorylation is associated with the development of hepatocellular carcinoma. However, the precise mechanisms by which phosphorylated RXRα loses its physiological function remain unclear. In the present study we examined whether phosphorylation of RXRα affects its dimeric activity. Fluorescence resonance energy transfer studies and immunoprecipitation assays showed that the physical interaction between RXRα and retinoic acid receptor β was impaired when 293T cells were transfected with phosphomimic mutant RXRα (T82D/S260D), whereas this interaction was activated at a level similar to wild‐type RXRα‐transfected cells when the cells were transfected with an unphosphorylated mutant RXRα (T82A/S260A). Treating the T82A/S260A‐transfected cells with retinoid resulted in a significant increase in the transcriptional activities of the retinoic acid receptor responsive element and RXR responsive element promoters, whereas these transcriptional activities did not increase in the T82D/S260D‐transfected cells. Transfection with T82A/S260A enhanced both the inhibition of cell growth and the induction of apoptosis caused by retinoid, although the T82D/S260D‐transfected cells lost their responsiveness to retinoid. Moreover, transfection with T82A/S260A caused an inhibition of cell growth and a reduction of colony‐forming ability in soft agar in HuH7 human hepatocellular carcinoma cells. These findings suggest that phosphorylation of RXRα abolishes its ability to form homodimers and heterodimers with RXR and retinoic acid receptor β, thus resulting in the loss of cell growth control and the acceleration of cancer development. In conclusion, the inhibition of RXRα phosphorylation and the restoration of its original function as a master regulator of nuclear receptors might therefore be an effective strategy for controlling cancer cell growth. (Cancer Sci 2007; 98: 1868–1874)
Abbreviations:
- 9cRA
9‐cis‐retinoic acid
- CFP
cyan fluorescent protein
- CRBP
cellular retinol‐binding protein
- DMEM
Dulbecco's modified Eagle's medium
- ELISA
enzyme‐linked immunosorbent assay
- Erk
extracellular signal‐related kinase
- FRET
fluorescence resonance energy transfer
- FRETC
corrected fluorescence resonance energy transfer value
- HCC
hepatocellular carcinoma
- LBD
ligand‐binding domain
- MAPK
mitogen‐activated protein kinase
- MEK
mitogen‐activated ERK‐activating kinase
- NFRET
normalized fluorescence resonance energy transfer
- NIH
National Institute of Health
- PCR
polymerase chain reaction
- PPAR
peroxisome proliferator‐activated receptor
- p‐RXR
phosphorylated retinoid X receptor
- RAR
retinoic acid receptor
- RARE
retinoic acid receptor responsive element
- RXR
retinoid X receptor
- RXRE
retinoid X receptor responsive element
- WT
wild type
- YFP
yellow fluorescent protein.
Vitamin A and its analogs, collectively termed retinoids, have a profound effect on cell activities such as cell growth, differentiation, apoptosis, and morphogenesis.( 1 , 2 ) Retinoids exert their biological functions primarily by regulating gene expression through two distinct nuclear receptors, namely, RAR and RXR. Both families of receptors are composed of three subtypes (α, β, and γ). RAR can be activated by all‐trans retinoic acid and 9cRA, whereas RXR can be activated only by 9cRA.( 3 ) Nuclear retinoid receptors are ligand‐dependent transcription factors. After ligand binding, RXR forms homodimers or heterodimers with RAR, which interacts with RXRE or RARE, located in the promoter region of target genes, thereby modulating gene expression.( 4 ) Other nuclear receptors also require RXR as a heterodimer partner in order to exert their function.( 4 ) Therefore, RXR play a fundamental role in controlling normal cell proliferation and metabolism, and act as master regulators of nuclear receptors.
Abnormalities in the expression and function of retinoids and their receptors play an important role in influencing the development of various human malignancies, including HCC. In previous studies, we found that the RXRα protein was phosphorylated anomalously in human HCC tissue as well as in HCC cell lines.( 5 , 6 ) p‐RXRα, the phosphorylated form of RXRα, was resistant to ubiquitination and to proteasome‐mediated degradation in both human HCC tissues and in a human HCC cell line.( 7 ) Moreover, p‐RXRα lost its physiological transactivation activity.( 6 ) These findings suggest that the accumulation of p‐RXRα (i.e. non‐functional RXRα) may interfere with the function of normal RXRα in a dominant‐negative manner, thereby playing a critical role in the development of HCC. However, the precise mechanism by which p‐RXRα affects the development of HCC has not yet been determined.
Recent studies indicate that RXRα is a target for phosphorylation, which could in turn regulate the function of its heterodimeric binding partners. For instance, the phosphorylation of RXRα by MAPK and MAPK kinase 4 inhibits transactivation of the vitamin D3 receptor and RAR, respectively.( 8 ) Because heterodimerization with other members of the nuclear receptor family is a fundamental process by which RXR exerts its pleiotropic effects on numerous biological pathways, the present study examined whether the phosphorylation of RXRα has an effect on its heterodimeric activity with RARβ. FRET is a technique that is useful for measuring the molecular proximity of nuclear receptors following ligand binding in living cells.( 9 ) Therefore, we used FRET and immunoprecipitation western blot assays in order to clarify the interactions between RXRα and RARβ in phosphomimic or unphosphorylated mutant RXRα‐transfected 293T cells. Moreover, we also examined whether the prevention of RXRα phosphorylation inhibits cell growth and colony‐forming activity in soft agar using unphosphorylated mutant RXRα‐transfected HuH7 human HCC cells.
Materials and Methods
Plasmid construction. The WT human RXRα plasmid pRShRXRα was kindly provided by Dr R. M. Evans (Salk Institute, La Jolla, CA, USA).( 10 ) An unphosphorylated form of RXRα mutant, T82A/ S260A, in which threonine 82 and serine 260 were mutated to alanine, and a phosphomimic form of RXRα mutant, T82D/S260D, in which threonine 82 and serine 260 were mutated to aspartate, were generated as described previously.( 6 ) These mutants specifically mimic the unphosphorylated or phosphorylated form of RXRα proteins because the transfection of these mutants could alter the molecular weight of the RXRα protein via ubiquitination‐ dependent mechanisms, thus causing the biological activities.( 6 , 7 ) The structures of these mutant plasmids are shown in Fig. 1.
Figure 1.
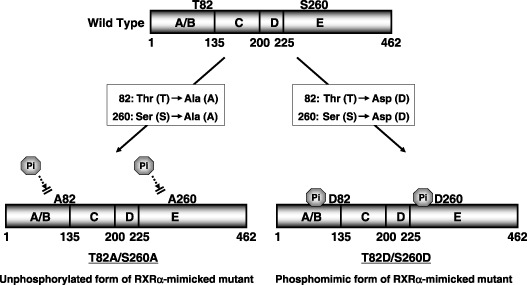
Schematic structure of wild‐type retinoid X receptor (RXR) α and the RXRα mutants (T82A/S260A and T82D/S260D). T82A/S260A, in which threonine 82 and serine 260 were mutated to alanine, is an unphosphorylated form of RXRα. T82D/S260D, in which threonine 82 and serine 260 were mutated to aspartate, is a phosphomimic of RXRα.
Yellow fluorescent protein–RXRα was constructed by subcloning PCR‐amplified RXRα cDNA with BglII sites attached at both ends, into the BglII and BamHI sites of the pCS2‐Venus vector. Venus, a variant of YFP resistant to intracellular pH change, was generously provided by Dr A. Miyawaki (RIKEN, Wako, Japan). cDNA encoding point mutants of RXRα were also subcloned into pCS2‐Venus using the same methods. To generate CFP–RARβ, a DNA fragment encoding full‐length RARβ was obtained by PCR with BamHI sites introduced into both ends, and was cloned into the pECFP‐N1 vector (Clontech, Palo Alto, CA, USA). The YFP–CFP fusion protein with a six‐amino acid linker was constructed by conventional PCR methods and used as a positive control for FRET analysis. As a negative control, the pCS2‐Venus vector was used to express Venus protein as an acceptor.
Cell culture. HEK‐293T (293T) human embryonic kidney epithelial cells were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). HuH7 human HCC cells were obtained from the Japanese Cancer Research Resources Bank (Tokyo, Japan). The cells were maintained in DMEM supplemented with 10% fetal calf serum (DF‐10), penicillin (100 units/mL), and streptomycin (100 µg/mL), in an incubator with humidified air at 37°C with 5% CO2.
Fluorescence resonance energy transfer experiments. The 293T cells (1 × 104) were cotransfected with the YFP–WT RXRα or mutant RXRα (T82D/S260D‐RXRα or T82A/S260A‐RXRα) plasmids plus CFP–RARβ, in six‐well 35 mm‐diameter plates using Lipofectamine Plus reagent (Invitrogen, Tokyo, Japan) according to the manufacturer's instructions.( 5 , 6 ) After 24 h, the medium was changed to DF‐10 containing 1.0 µM 9cRA and 30 µM MG 132, a potent proteasome inhibitor (Calbiochem, Darmstadt, Germany). The cells were then incubated for 5 h and used for FRET measurement. To obtain images of living cells, the culture dishes were observed using an IX70 inverted microscope; the imaging system used was the same as that described previously.( 11 ) FRET quantification between the two fluorophores was carried out using FRETC( 12 ) and NFRET,( 13 ) as described previously.( 11 ) FRETC images are presented in pseudocolor mode, according to a temperature‐based lookup table, with blue (cold) indicating low values and red (hot) indicating high values. For quantitative comparisons, NFRET values for domains containing more than 100 pixels were calculated, and the average for the domain was calculated. All calculations were carried out using the public domain program Image J Version 1.34 (developed at the NIH).
Protein extraction, immunoprecipitation, and western blot analysis. The WT or mutant RXRα‐transfected cells were treated with 9cRA for 24 h and the total cellular protein was extracted, as described previously.( 14 ) For detection of the expression levels of the RXRα and p‐RXRα proteins that heterodimerized with RARβ, immunoprecipitation assays were carried out. For immunoprecipitation, 3 mg protein was incubated with 5 µg anti‐RARβ antibody at 4°C for 1 h. Immune complexes were precipitated by adding 50 µL protein A agarose (Sigma‐Aldrich, St Louis, MO, USA) to the mixture. After incubating overnight at 4°C, the beads were washed three times in lysis buffer, and bound protein was eluted directly into the sample buffer and subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis, as described previously.( 15 ) The anti‐RXRα antibodies (ΔN197 and D‐20) and the anti‐RARβ antibody (C‐19) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The ΔN197 antibody is regarded as a specific antibody for the phosphorylated form of RXRα protein.( 7 , 14 ) Monoclonal antibody against glyceraldehyde‐3‐phosphate dehydrogenase was from Chemicon International (Temecula, CA, USA), and was used as a loading control. The intensities of the blots were quantified using the NIH image software package version 1.61.
Reporter assays for RARE and RXRE. Reporter assays were carried out as described previously.( 6 ) The RARE luciferase reporter plasmid tk‐CRBP II‐RARE‐Luc and the RXRE luciferase reporter plasmid tk‐CRBP II‐RXRE‐Luc were kindly provided by the late Dr K. Umesono (Kyoto University, Kyoto, Japan).( 16 ) The 293T and HuH7 cells (2 × 105) were cotransfected with a combination of WT or mutant RXRα‐expressing plasmids (300 ng/35‐mm dish) with the RARE or RXRE reporter plasmids (750 ng/35‐mm dish) using Lipofectamine Plus reagent, along with pRL‐CMV (Renilla luciferase, 100 ng/35 mm‐dish; Promega, Madison, WI, USA) as an internal standard to normalize the transfection efficiency.( 17 ) After exposing the cells to the transfection mixture for 24 h, the cells were treated with vehicle or 1.0 µM 9cRA for 24 h. The cell lysates were then prepared and the luciferase activity of each cell lysate was determined using a dual‐luciferase reporter assay system (Promega), as described previously.( 17 ) Changes in the firefly luciferase activity were calculated and plotted after normalizing the values to changes in Renilla luciferase activity in the same sample.
Cell growth assay. After the 293T cells (2 × 103) were transfected with WT or mutant RXRα‐expressing plasmids, the cells were treated with vehicle or 1.0 µM 9cRA for 24 h. Cell growth was assayed using the WST‐1 system (Dojindo Laboratories, Tokyo, Japan), according to the manufacturer's instructions, as described previously.( 18 ) WT or mutant RXRα (T82D/S260D or T82A/S260A)‐transfected HuH7 cells (1 × 103) were also treated with 1.0 µM 9cRA for 6 days. Every 24 h, the number of viable cells in replica plates were counted using the Trypan blue dye exclusion method, as described previously.( 18 )
Apoptosis assays. The 293T cells (5.0 × 103) were transfected with WT or mutant RXRα‐expressing plasmids, as described above. The transfected cells were then treated with vehicle or 1.0 µM 9cRA for 24 h, and the cell lysates were used to determine apoptosis. To quantify the induction of apoptosis, a DNA fragmentation assay was carried out using the Cell Death Detection ELISA kit (Roche Diagnostics, Mannheim, Germany). DNA fragmentation of mutant RXRα‐transfected 293T cells was quantified using the ELISA kit, according to the manufacturer's instructions, as described previously.( 19 )
Soft agar colony‐formation assay. Ten millimeters of molten 0.5% Noble Agar (Difco, Lawrence, KS, USA) in DF‐10 medium was first added to plates and allowed to harden to form the bottom agar. WT or mutant RXRα (T82A/S260A)‐transfected HuH7 cells (2 × 104) were suspended in 8 mL DF‐10 medium and then 2 mL of 1.7% molten agar (45°C) was added, and this mixture was immediately laid over the bottom medium. Triplicate plates were incubated at 37°C under 5% CO2, and the number of macroscopic colonies per plate was counted after 3 weeks.
Statistical analysis. The data are expressed as mean ± SD. The statistical significance of the difference in mean values was assessed using one‐way ANOVA, followed by Sheffe's t‐test.
Results
Phosphorylated RXRa loses its heterodimeric activity with RARb. Heterodimerization with other nuclear receptors is an essential event by which RXR exerts its biological function.( 20 ) Therefore, in our initial study, we examined whether RXRα still possessed heterodimeric activity with RARβ after it was phosphomimicked. We used FRET because of its suitability for clarifying the interactions between nuclear receptors following ligand binding in living cells.( 21 , 22 ) Fig. 2 demonstrated that FRET microscopy visualized binding between CFP‐fused RARβ (donor) and YFP‐fused RXRα (acceptor) overexpressed in living 293T cells. FRETC images are presented in pseudocolor, in which the red range indicates a high FRET efficiency (positive control; Fig. 2f) and the blue to black range indicates a low FRET efficiency (negative control; Fig. 2c). The results showed that the FRETC signal detected in the nucleus of YFP–WT RXRα‐ and CFP–RARβ‐co‐expressing 293T cells was highly positive (Fig. 2i). However, when the 293T cells were cotransfected with a phosphomimic mutant RXRα (YFP–T82D/S260D‐RXRα) and CFP–RARβ, the FRETC signal detected in the nucleus was very low, indicating diminished physical interactions between p‐RXRα and RARβ (Fig. 2l). In contrast, when the cells were cotransfected with an unphosphorylated mutant RXRα (YFP–T82A/S260A‐RXRα) and CFP–RARβ, the FRET efficiency was comparable to that in WT RXRα (Fig. 2o). Because FRET efficiency depends on the amount of donor and acceptor capable of interaction, NFRET was calculated to obtain the normalized values for FRET efficiency.( 11 , 13 ) The NFRET values in cells expressing YFP–T82A/S260A‐RXRα/CFP–RARβ were similar to those in the cells expressing YFP–WT‐RXRα/CFP–RARβ and they were significantly higher than in cells expressing YFP–T82D/S260D‐RXRα/CFP–RARβ (Table 1).
Figure 2.
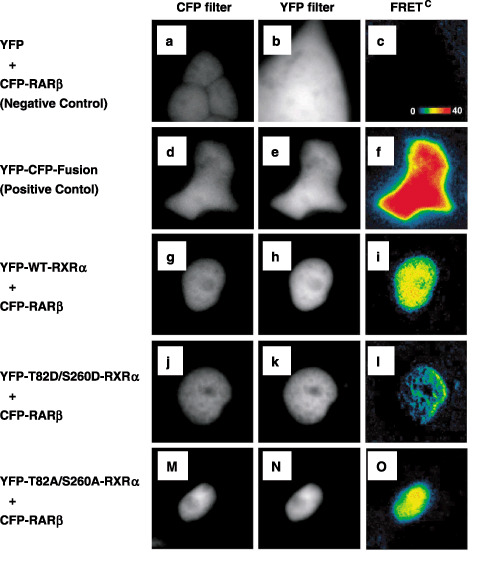
Heterodimerization of RXR α mutants and retinoic acid receptor (RAR) β in living 293T cells as demonstrated by FRET. The 293T cells were cotransfected with (g–i) YFP‐tagged WT‐RXRα, or the RXRα mutants (m–o) T82A/S260A and (j–l) T82D/S260D using CFP‐tagged RARβ plasmids. (d–f) Cells overexpressing the YFP–CFP fusion protein were used as a positive control. (a–c) As a negative control, 293T cells were cotransfected with pCS2‐Venus plasmid and CFP‐tagged RARβ plasmid. Cells were visualized using the donor filter set (CFP filter) and the acceptor filter set (YFP filter). Corrected fluorescence resonance energy transfer values are shown in pseudocolor.
Table 1.
Phosphorylated retinoid X receptor (RXR) α loses its heterodimeric activity with retinoic acid receptor (RAR) β
| FRET pair | NFRET |
|---|---|
| YFP + CFP–RARβ | 1.15 ± 1.16 (negative control) |
| YFP–CFP fusion | 68.31 ± 6.11 (positive control) |
| YFP–RXRα (WT) + CFP–RARβ | 13.33 ± 2.23 |
| YFP–RXRα (T82D/S260D) + CFP–RARβ | 7.29 ± 3.45* |
| YFP–RXRα (T82A/S260A) + CFP–RARβ | 12.49 ± 4.08 |
P < 0.01. Values are mean ± SD. Each normalized fluorescence resonance energy transfer (NFRET) value was obtained for several selected regions containing more than 100 pixels in the individual cell. CFP, cyan fluorescent protein; FRET, fluorescence resonance energy transfer; WT, wild type; YFP, yellow fluorescent protein.
We next examined the physical interaction between mutant RXRα and RARβ using an immunoprecipitation western blot analysis. As shown in Fig. 3, the physical interaction between the RXRα and RARβ proteins was activated when the 293T cells were transfected with WT or unphosphorylated mutant RXRα, whereas this interaction was inhibited when the cells were transfected with a phosphomimic mutant of RXRα. Moreover, no blots were detected when the RARβ‐binding proteins were reacted with phospho‐RARα‐specific antibody. These findings suggest that RXRα loses its heterodimeric activity with RARβ when it is phosphomimicked, whereas the heterodimerization is activated when the cells are transfected with unphosphorylated mutant RXRα. Therefore, these findings provide direct evidence that abnormal phosphorylation of RXRα loses its heterodimeric activity with RARβ.
Figure 3.
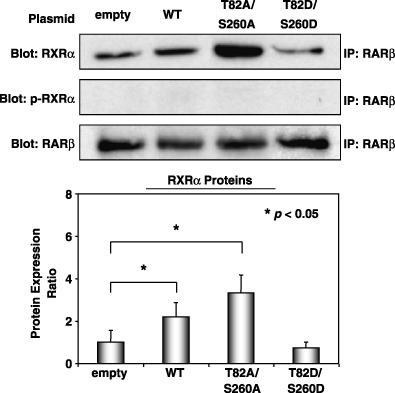
The physical interaction between phosphorylated retinoid X receptor (RXR) α (p‐RXRα) and retinoic acid receptor (RAR) β proteins as demonstrated by immunoprecipitation assay. The wild‐type or mutant RXRα‐transfected 293T cells were treated with 9‐cis‐retinoic acid for 24 h and the total cellular protein was extracted. The expression levels of the RXRα and p‐RXRα proteins that physically interacted with the RARβ protein were determined using an immunoprecipitation western blot analysis. An antibody to glyceraldehyde‐3‐phosphate dehydrogenase was used as a loading control. The intensities of the blots were quantitated by densitometry and the values are shown in a bar graph in the lower panel. Similar results were obtained in a repeat experiment. Asterisks represent significant differences (P < 0.05). Bars, SD.
Overexpression of the phosphomimic mutant RXRa inhibits, but the unphosphorylated mutant RXRa stimulates, the transcriptional activity of the RARE promoter in 293T cells. Using transient transfection luciferase reporter assays, we then examined whether p‐RXRα might inhibit the transcriptional activity of the RARE promoter, or whether overexpression of the unphosphorylated form of RXRα stimulates the transcriptional activity of this promoter (Fig. 4). The results showed that WT RXRα‐transfected 293T cells caused a significant increase in RARE promoter activity when the cells were treated with 9cRA (Fig. 4, columns 2 and 4). In addition, this stimulation was more apparent in T82A/S260A‐RXRα‐transfected cells, where the luciferase activity increased 4.0‐fold in comparison to the control (empty vector) group (Fig. 4, columns 2 and 6). However, transfection with T82D/S260D‐RXRα did not increase the transcriptional activity of this responsive element (Fig. 4, column 8), thus, implying that the phosphorylation of threonine 82 and serine 260 in the RXRα protein might inhibit the ligand‐dependent transcriptional activity.
Figure 4.
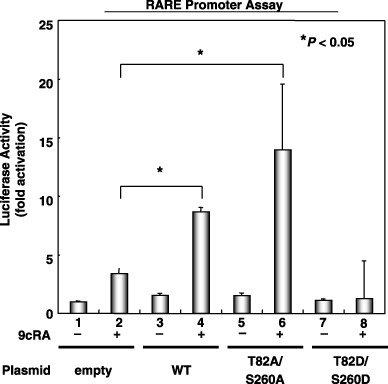
Effects of overexpression of the phosphomimic or unphosphorylated mutants of retinoid X receptor (RXR) α on retinoic acid receptor responsive element (RARE) promoter activity in 293T cells. The 293T cells were cotransfected with wild‐type or mutant RXRα‐expressing plasmids (T82A/S260A or T82D/S260D) using the RARE‐Luc and CMV‐Luc plasmids. The transfected cells were then treated with vehicle or 1.0 µM 9‐cis‐retinoic acid for 24 h and the extracts were examined for luciferase activity. Values are the mean ± SD. Asterisks represent significant differences (P < 0.05). Representative results from three independent experiments are shown.
Overexpression of the unphosphorylated mutant RXRa induces apoptosis and inhibits cell growth, but the phosphomimic mutant RXRa removes these effects in 293T cells. One of the fundamental functions of retinoids is the induction of apoptosis, which, in turn, regulates normal cell proliferation.( 1 ) Therefore, we next examined whether overexpression of phosphomimic or unphosphorylated mutant RXRα affects the induction of apoptosis. When histone‐associated DNA fragmentation was measured using an ELISA assay, the results demonstrated that in the presence of 9cRA, transfection with empty vector, WT RXRα, and T82A/S260A‐RXRα significantly induced apoptosis in 293T cells (Fig. 5a, columns 2, 4, and 6). Moreover, transfection with T82A/ S260A‐RXRα alone also induced apoptosis in these cells without ligand stimulation (Fig. 5a, column 5). However, T82D/S260D‐RXRα‐transfected cells did not induce apoptosis either in the presence or absence of 9cRA (Fig. 5a, columns 7 and 8).
Figure 5.
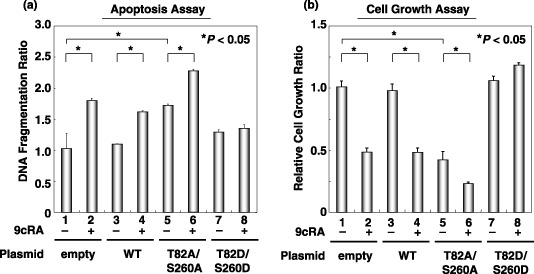
Effects of overexpression of the phosphomimic or unphosphorylated mutants of retinoid X receptor (RXR) α on the induction of apoptosis and the inhibition of 293T cell growth. Wild‐type or mutant RXRα‐transfected cells were treated with vehicle or 1.0 µM 9‐cis‐retinoic acid for 24 h. (a) The cell lysates were then extracted and DNA fragmentation was determined using an ELISA assay. (b) Cell growth in replica plates was examined using the WST‐1 assay system. Values are the mean ± SD. Asterisks represent significant differences (P < 0.05). Representative results from three independent experiments with similar results are shown.
We then examined the effect of the overexpression of mutant RXRα on the growth of 293T cells using the WST‐1 assay. As shown in Fig. 5b, overexpression of T82A/S260A‐RXRα (column 5) caused a significant reduction in cell growth compared with cells transfected with the empty vector (column 1). This reduction was enhanced when the cells were treated with 9cRA (Fig. 5b, column 6). In contrast, there was no inhibition of cell growth in T82D/S260D‐RXRα‐transfected cells in the presence or absence of 9cRA (Fig. 5b, columns 7 and 8). These findings suggest that p‐RXRα‐overexpressing cells are resistant to retinoid‐induced apoptosis and growth inhibition.
Overexpression of unphosphorylated mutant RXRa stimulates the transcriptional activities of both the RARE and RXRE promoters, inhibits cell growth, and reduces colony‐forming ability in soft agar in HuH7 human HCC cells. The possibility that the prevention of phosphorylation of RXRα by transfection with the T82A/S260A mutant inhibits the growth of HuH7 human HCC cells was next examined. We found that in the presence of 9cRA, the promoter activities of both RARE and RXRE were significantly enhanced when the HuH7 cells were transfected with the T82A/S260A mutant (Fig. 6). Transfection with this mutant also caused a remarkable inhibition of cell growth (Fig. 7a), as well as anchorage‐dependent colony formation in soft agar (Fig. 7b) in HuH7 cells, whereas transfection with the T82D/S260D mutant enhanced the growth of cancer cells (Fig. 7a). These findings are consistent with the results of the promoter assay (Fig. 4) and the WST‐1 (Fig. 5b) assay in 293T cells and suggest that prevention of the phosphorylation of RXRα might therefore be an effective strategy to inhibit the growth of HCC cells.
Figure 6.
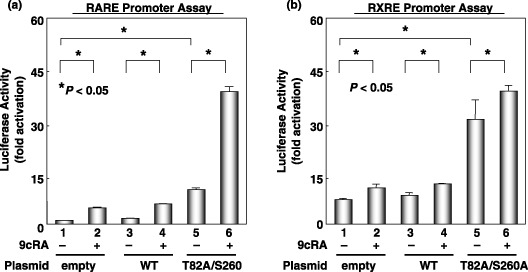
Effects of the overexpression of the unphosphorylated mutant of retinoid X receptor (RXR) α on retinoic acid receptor responsive element (RARE) and RXR responsive element (RXRE) promoter activities in HuH7 cells. The cells were cotransfected with wild‐type or T82A/S260A mutant RXRα‐expressing plasmid using the RARE‐Luc or RXRE‐Luc plasmids. After the transfected cells were treated with vehicle or 1.0 µM 9‐cis‐retinoic acid for 24 h, the extracts were then examined for (a) RARE or (b) RXRE luciferase activities. Values are the mean ± SD. Asterisks represent significant differences (P < 0.05). The results presented are the average of the triplicate findings from three independent experiments.
Figure 7.
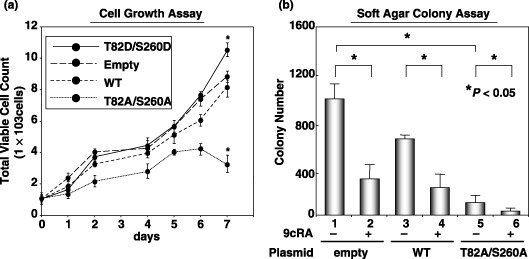
Effects of overexpression of the unphosphorylated or phosphorylated mutants of retinoid X receptor (RXR) α on cell growth and anchorage‐dependent colony‐forming ability in HuH7 cells. (a) Wild‐type (WT) or mutant RXRα (T82A/S260A or T82D/S260D)‐transfected HuH7 cells were treated with 1.0 µM 9‐cis‐retinoic acid for 6 days and the cell growth in replica plates was examined using the trypan blue dye exclusion method. (b) The WT and T82A/S260A mutant RXRα‐transfected HuH7 cells were plated in soft agar and the number of macroscopic colonies per plate was counted after 3 weeks. Values are the mean ± SD. Asterisks represent significant differences (P < 0.05). The results presented are the average of triplicate findings from three independent experiments.
Discussion
Using FRET and immunoprecipitation assays, the present study found that the overexpression of phosphomimic mutant RXRα causes it to lose its ability to form heterodimers with RARβ (2, 3; Table 1). This is the first evidence that post‐translational modification by phosphorylation directly interferes with the dimerization of RXRα in living cells. These results also showed that the downregulation of heterodimeric activity of RXRα due to phosphorylation inhibits the transcriptional activity of the RARE promoters (Fig. 4) and the induction of apoptosis (Fig. 5a), resulting in uncontrolled cell growth and resistance to retinoids (5, 7). These findings may explain our previous observations that RXRα is phosphorylated in human hepatoma tissues as well as in human hepatoma cell lines, and that the accumulation of p‐RXRα interferes with the function of the remaining normal RXRα in a dominant‐negative manner, thereby promoting the growth of hepatoma cells.( 5 , 6 )
Although the precise mechanisms regarding how p‐RXRα loses its dimeric activity with other nuclear receptors remain unclear, recent studies indicate that the phosphorylation of RXRα can regulate the function of its heterodimeric binding partners. For instance, Solomon et al. reported that phosphorylation of RXRα at serine 260 results in the attenuation of ligand‐dependent transactivation by the vitamin D3 receptor–RXR complex in human keratinocytes, thus resulting in the induction of malignant transformation.( 8 ) In addition, the phosphorylation of RXRα at serine 265( 23 ) and at tyrosine 249( 24 ) also inhibits the transcription of retinoid target genes. These reports and our findings using the T82D/S260D mutant (2, 3, 4, 5) share certain similarities in that RXRα phosphorylation, which occurs at specific residues (tyrosine 249, serine 260, and serine 265) located in the omega loop of the LBD, is apparently associated with a malfunction in the retinoid‐dependent signaling pathway. The omega loop, located between helices H1 and H3, is a very flexible and dynamic region that moves substantially during the conformational rearrangement that accompanies ligand binding to the LBD.( 25 ) Therefore, it has been proposed that phosphorylation of the residues in this loop might alter the dynamics of this region and create conformational changes within the LBD, thus disrupting the interactions with co‐regulators and therefore inhibiting the activation of retinoic acid‐responsive genes.( 23 , 26 )
The present study also demonstrates that transfection with unphosphorylated mutant RXRα (T82A/S260A) restores its heterodimeric activity with RARβ (2, 3) and abolishes the resistance to retinoid in 293T cells (4, 5). We also found that the overexpression of T82A/S260A mutant RXRα in human hepatoma HuH7 cells causes an increase in both the RARE and RXRE promoter activities (Fig. 6), but causes a decrease in cellular proliferation and the anchorage‐dependent colony formation of these cancer cells (Fig. 7). These findings suggest that the inhibition of abnormal phosphorylation for RXRα can restore its multidimeric activity with other nuclear receptors, including RXRα itself, because RXRE is a specific responsive element for the RXR homodimer.( 4 ) In HCC cells, but not in normal hepatocytes, the Ras–MAPK signaling pathway is highly activated, and therefore phosphorylating RXRα impairs the functions of this receptor.( 6 ) In addition, aberrant phosphorylation of RXRα is also observed in cell lines of other human adenocarcinomas, such as colorectal cancer( 27 ) and pancreatic cancer (data not shown). We recently reported that the RXRα protein was highly phosphorylated in colorectal cancer tissues as well as in colon cancer cell lines in comparison to their corresponding normal colon epithelial tissue and cell lines.( 27 ) Therefore, the inhibition of RXRα phosphorylation and the restoration of its dimeric activity with other nuclear receptors might be an effective and critical strategy for inhibiting the growth of cancer cells showing aberrant phosphorylation of this protein. Indeed, combining retinoids with vitamin K2, which targets the Ras–MAPK signaling pathway, synergistically inhibits the growth of HCC cells by inhibiting the phosphorylation of RXRα.( 28 ) When phosphorylation of RXRα was inhibited by MEK inhibitor or by transfection with the T82A/S260A mutant, the combination of 9cRA and the PPARγ ligand ciglitazone also synergistically inhibited cell growth and induced apoptosis in colon cancer cells.( 27 ) In conclusion, phosphorylation of RXRα might be a useful molecular target for the prevention and treatment of human malignancies that express anomalous phosphorylation of this protein, including HCC.
Acknowledgments
We thank Drs Y. Komi and H. Tatsukawa (RIKEN, Wako, Japan) for their valuable technical assistance and useful discussions. This work was supported in part by a Grant‐in‐Aid from the Ministry of Education, Science, Sports, and Culture of Japan (no. 18790457 to M. S. and no. 17015016 to H. M.).
References
- 1. Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ, De Luca LM. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis 2000; 21: 1271–9. [PubMed] [Google Scholar]
- 2. Dawson MI. The importance of vitamin A in nutrition. Curr Pharm Des 2000; 6: 311–25. [DOI] [PubMed] [Google Scholar]
- 3. Mangelsdorf DJ, Thummel C, Beato M et al . The nuclear receptor superfamily: the second decade. Cell 1995; 83: 835–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dong D, Noy N. Heterodimer formation by retinoid X receptor: regulation by ligands and by the receptor's self association properties. Biochemistry 1998; 37: 10 691–700. [DOI] [PubMed] [Google Scholar]
- 5. Matsushima‐Nishiwaki R, Shidoji Y, Nishiwaki S, Yamada T, Moriwaki H, Muto Y. Aberrant metabolism of retinoid X receptor proteins in human hepatocellular carcinoma. Mol Cell Endocrinol 1996; 121: 179–90. [DOI] [PubMed] [Google Scholar]
- 6. Matsushima‐Nishiwaki R, Okuno M, Adachi S et al . Phosphorylation of retinoid X receptor alpha at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res 2001; 61: 7675–82. [PubMed] [Google Scholar]
- 7. Adachi S, Okuno M, Matsushima‐Nishiwaki R et al . Phosphorylation of retinoid X receptor suppresses its ubiquitination in human hepatocellular carcinoma. Hepatology 2002; 35: 332–40. [DOI] [PubMed] [Google Scholar]
- 8. Solomon C, White JH, Kremer R. Mitogen‐activated protein kinase inhibits 1,25‐dihydroxyvitamin D3‐dependent signal transduction by phosphorylating human retinoid X receptor α. J Clin Invest 1999; 103: 1729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feige JN, Sage D, Wahli W, Desvergne B, Gelman L. PixFRET, an ImageJ plug‐in for FRET calculation that can accommodate variations in spectral bleed‐throughs. Microsc Res Tech 2005; 68: 51–8. [DOI] [PubMed] [Google Scholar]
- 10. Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, Evans RM. A direct repeat in the cellular retinol‐binding protein type II gene confers differential regulation by RXR and RAR. Cell 1991; 66: 555–61. [DOI] [PubMed] [Google Scholar]
- 11. Takano Y, Adachi S, Okuno M et al . The RING finger protein, RNF8, interacts with retinoid X receptor α and enhances its transcription‐stimulating activity. J Biol Chem 2004; 279: 18 926–34. [DOI] [PubMed] [Google Scholar]
- 12. Jiang X, Sorkin A. Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol Biol Cell 2002; 13: 1522–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J 2001; 81: 2395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsushima‐Nishiwaki R, Okuno M, Takano Y, Kojima S, Friedman SL, Moriwaki H. Molecular mechanism for growth suppression of human hepatocellular carcinoma cells by acyclic retinoid. Carcinogenesis 2003; 24: 1353–9. [DOI] [PubMed] [Google Scholar]
- 15. Shimizu M, Hara A, Okuno M et al . Mechanism of retarded liver regeneration in plasminogen activator‐deficient mice: impaired activation of hepatocyte growth factor after Fas‐mediated massive hepatic apoptosis. Hepatology 2001; 33: 569–76. [DOI] [PubMed] [Google Scholar]
- 16. Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature 1992; 355: 446–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki Y, Shimada J, Shudo K, Matsumura M, Crippa MP, Kojima S. Physical interaction between retinoic acid receptor and Sp1: mechanism for induction of urokinase by retinoic acid. Blood 1999; 93: 4264–76. [PubMed] [Google Scholar]
- 18. Maekawa Y, Yagi K, Nonomura A et al . A tetrazolium‐based colorimetric assay for metabolic activity of stored blood platelets. Thromb Res 2003; 109: 307–14. [DOI] [PubMed] [Google Scholar]
- 19. Trautwein C, Rakemann T, Brenner DA et al . Concanavalin A‐induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 1998; 114: 1035–45. [DOI] [PubMed] [Google Scholar]
- 20. Pfahl M. Signal transduction by retinoid receptors. Skin Pharmacol 1993; 6 (Suppl 1): 8–16. [DOI] [PubMed] [Google Scholar]
- 21. Prufer K, Racz A, Lin GC, Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem 2000; 275: 41 114–23. [DOI] [PubMed] [Google Scholar]
- 22. Feige JN, Gelman L, Tudor C, Engelborghs Y, Wahli W, Desvergne B. Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator‐activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J Biol Chem 2005; 280: 17 880–90. [DOI] [PubMed] [Google Scholar]
- 23. Bruck N, Bastien J, Bour G et al . Phosphorylation of the retinoid X receptor at the omega loop, modulates the expression of retinoic‐acid‐target genes with a promoter context specificity. Cell Signal 2005; 17: 1229–39. [DOI] [PubMed] [Google Scholar]
- 24. Lee HY, Suh YA, Robinson MJ et al . Stress pathway activation induces phosphorylation of retinoid X receptor. J Biol Chem 2000; 275: 32 193–9. [DOI] [PubMed] [Google Scholar]
- 25. Egea PF, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRα ligand‐binding domain bound to its natural ligand: 9‐cis retinoic acid. EMBO J 2000; 19: 2592–01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bastien J, Rochette‐Egly C. Nuclear retinoid receptors and the transcription of retinoid‐target genes. Gene 2004; 328: 1–16. [DOI] [PubMed] [Google Scholar]
- 27. Yamazaki K, Shimizu M, Okuno M et al . Synergistic effects of RXRα and PPARγ ligands to inhibit growth in human colon cancer cells – phosphorylated RXRα is a critical target for colon cancer management. Gut 2007. (in press). [DOI] [PMC free article] [PubMed]
- 28. Kanamori T, Shimizu M, Okuno M et al . Synergistic growth inhibition by acyclic retinoid and vitamin K2 in human hepatocellular carcinoma cells. Cancer Sci 2007; 98: 431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]