

Abstract
Epidermal growth factor receptor (EGFR) mutations in lung cancer enhance tyrosine kinase activity and increase sensitivity to the EGFR tyrosine kinase inhibitor, gefitinib. Mutation analysis of the EGFR gene is therefore indispensable for predicting gefitinib response. We investigated a CA‐repeat polymorphism in the EGFR gene related to EGFR mutations. Because an increasing number of CA‐repeats at intron 1 of the EGFR gene has been reported to reduce transcription activity, we examined the relationship between EGFR mutations and this CA‐repeat polymorphism. EGFR mutations at exon 19 were closely associated with shorter CA‐repeat length in the shorter allele, but this was not the case for EGFR mutations at exons 18 or 21. Increased intrinsic EGFR mRNA expression in non‐cancerous lung tissues from lung adenocarcinoma patients was also significantly associated with shorter CA‐repeat length. A higher frequency of EGFR mutations at exon 19 was associated with shorter CA‐repeat length only in patients with high levels of EGFR mRNA expression. To determine the phenotypes of cells possessing shorter CA‐repeats, an in vitro study using human bronchial epithelial cells with different CA‐repeat lengths was performed; more rapid cell growth and activated EGF/EGFR signaling were found more often in the cells having both shorter CA‐repeats and increased EGFR mRNA expression. These results suggest that CA‐repeat length in the EGFR gene may be a genetic factor related to cancer in the case of EGFR mutations at exon 19. The mechanism likely involves enhanced intrinsic expression of EGFR mRNA and activated EGF/EGFR signaling that accompany shorter CA‐repeats. (Cancer Sci 2008; 99: 1180–1187)
Lung cancer is the most common form of cancer death among males and the third most common among females in Japan,( 1 ) Overall survival from lung cancer remains unsatisfactory, with a five‐year survival rate of 14%, indicating that this is one of the most difficult cancers to treat.( 2 ) However, the therapeutic strategy has changed since molecular targeting therapies, such as the use of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, became available. Specifically, mutations in the EGFR gene found in lung cancer tissues have been reported to be a predictive marker for clinical response to EGFR tyrosine kinase inhibitor therapy, which may lead to the development of customized therapy.( 3 , 4 , 5 , 6 )
Mutations in the EGFR gene were found in 30–40% of all lung cancer patients in Japan, and were found in over 50% of female non‐smokers with adenocarcinomas.( 7 , 8 , 9 , 10 ) Although the overall rate of response to EGFR tyrosine kinase inhibitors for previously treated patients was 27.5% among Japanese,( 11 ) the frequency of gefitinib responsive patients among those with EGFR mutations was approximately 80%.( 5 , 9 ) Therefore, it is clear that DNA sequence analysis of EGFR in lung cancer is essential for predicting response to EGFR tyrosine kinase inhibitor therapy. In addition, EGFR mutations have been reported to be involved in lung carcinogenesis; an investigation using transgenic mice carrying the EGFR mutants EGFR L858R and EGFR ΔL747–S752 demonstrated that mutant EGFR was required for the development and maintenance of lung adenocarcinomas.( 12 ) Identification of clinical and genetic factors underlying EGFR mutations will be particularly meaningful not only for understanding lung carcinogenesis in non‐smokers but also for making clinical decisions about the use of EGFR tyrosine kinase inhibitors in non‐operative patients.
The EGFR gene is located on chromosome 7p12.1–12.3 in humans, and its expression is regulated by one promoter region and two enhancer regions.( 13 ) The promoter region contains a GC‐rich sequence without the characteristic TATA and CAAT boxes, and multiple transcription start sites exist. At least four Sp1 binding sites and one TC factor binding site are known, and basal transcription is regulated by Sp1.( 14 , 15 ) Two enhancer elements, one located upstream of the promoter (–1429/–1109) and one located downstream at intron 1 (+1788/+2318), function cooperatively.( 16 ) A polymorphic simple sequence repeat with 14–21 CA‐repeats was first demonstrated close to the downstream enhancer by Chi et al.( 17 ) An increased number of CA‐repeats was reported to be associated with decreased EGFR transcription activity: a 2‐fold increase in transcription activity with 16 CA‐repeats compared to that with more than 18 CA‐repeats.( 18 ) An in vitro run‐off assay using a 4050 bp polymerase chain reaction (PCR) product of the EGFR gene showed that the level of EGFR transcription was reduced by 80% with 21 CA‐repeats compared to that with 16 CA‐repeats. Using competitive reverse transcription‐polymerase chain reaction (RT‐PCR), it was demonstrated that pre mRNA expression levels in various cancer cell lines were correlated with the number of CA‐repeats; higher levels of EGFR mRNA were found in cancer cells with lower numbers of CA‐repeats, which is consistent with the in vitro experiments.
The relationship between lung cancer and CA‐repeat polymorphism has been studied in terms of lung cancer risk or clinical phenotype, specifically gefitinib responsiveness. A case‐control study revealed an inverse relationship between lung cancer risk and the number of CA‐repeats in a Caucasian population,( 19 ) although no association with lung cancer risk was reported in a Korean population.( 20 ) A clinical study of 86 patients with advanced non‐small‐cell lung cancer treated with gefitinib in Korea showed that a low number of CA‐repeats was associated with gefinitib responsiveness, although no significant association with EGFR mutation status nor EGFR expression levels was found in cancer tissues.( 21 ) On the other hand, it has been reported that shorter CA‐repeat length is associated with an increased expression of EGFR.( 18 , 22 , 23 , 24 ) Furthermore, shorter CA‐repeat alleles in lung cancer were more likely to be amplified, resulting in more prevalent allelic imbalance at the EGFR locus; EGFR mutations were found to favor shorter CA‐repeat alleles.( 25 ) However, most of these studies were based on cancer cell lines and resected cancer tissues.
We previously reported that EGFR mutations at exons 18, 19 and 21 evidenced different clinical profiles, suggesting that EGFR mutations should be analyzed according to the exons at which they occur.( 10 ) In addition, we think that the relationship between CA‐repeat polymorphism and intrinsic EGFR expression should also be examined in non‐cancerous tissues and cultured normal lung epithelial cells, because EGFR expression in cancer tissues may be modified by various genetic alterations such as mutations and allelic imbalance. Indeed, in lung cancer tissues with the same CA‐repeat status, EGFR mRNA levels differed according to whether the EGFR allele was wild‐type or mutated, and also depended on the EGFR copy number.( 26 )
In the present study, seeking to identify the genetic factors underlying EGFR mutations, we investigated the relationship between the CA‐repeat polymorphism and EGFR mutations according to exon in 154 patients with lung cancer, 70% of which were of pathological stages I and II. We also studied the association between CA‐repeat polymorphism and EGFR mRNA levels in non‐cancerous tissues from 74 lung adenocarcinoma patients being followed in an in vitro study of CA‐repeat length, and cell growth using normal human bronchial epithelial (HBE) cells with different CA‐repeat lengths obtained from 11 lung cancer patients.
Materials and Methods
Tissue and pleural effusion. We studied a total of 154 Japanese patients with lung cancer. We obtained tissue specimens as follows: 123 surgical specimens, 9 transbronchial fiberscopic specimens, 19 pleural effusions from non‐resectable lung cancers, two specimens from metastatic lesions of the brain and skin, and one sample of cells from urine. Study patients were admitted to the Saga Medical School Hospital, Saga, Japan, between 2000 and 2007; 142 patients had not received anticancer chemotherapy or thoracic irradiation, and 12 patients were recurrent cases. Clinical stage was determined according to the criteria of the International Union Against Cancer. Histological subtype and tumor content were confirmed by Hematoxylin and Eosin staining with tumor samples; pleural effusion was assessed as class V by a pathologist. EGFR mutation status and CA‐repeat length were investigated using DNA direct sequencing. All procedures were performed with the informed consent of the patients, and the study was approved by the Saga University Institutional Review Board Committee.
DNA extraction and sequencing analysis. DNA was isolated from freshly frozen lung cancer tissues using a QIAamp DNA mini kit (QIAGEN, Hilden, Germany) following the manufacturer's instructions. Mutations at exons 18, 19, and 21 were determined using PCR‐based direct sequencing in cancer tissues; numbers of CA‐repeats were determined using adjacent, normal (non‐cancerous) tissues. The primers used for EGFR mutations were previously described.( 3 , 10 ) The primer sets used for determination of CA‐repeat were 5′‐CGGCTGTCCGGCCACTGG‐3′ (sense) and 5′‐CAGCTCAAGGTTGGAATTGTGC‐3′ (antisense) (amplicon size: 378 bp). PCR amplification was performed in a 20‐µL volume using Discoverase DHPLC DNA polymerase (Invitrogen Inc., CA, USA) at 95°C for 10 min followed by 40 cycles (each cycle at 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min), with a final extension at 72°C for 10 min. The amplified products were isolated using Microcon YM‐50 (Millipore Inc., MA, USA) and sequenced directly using the Applied Biosystems PRISM dye terminator cycle sequencing method with an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
EGFR mRNA expression. Total RNAs were isolated from both lung cancer tissues and non‐cancerous tissues using ISOGEN reagent (Nippon gene, Japan). Levels of EGFR mRNA were determined by real‐time RT‐PCR. One µg total RNA was applied to RT with MuLV reverse transcriptase (Roche Molecular Systems, NJ, USA) at 37°C for 60 min. The cDNAs obtained were processed by quantitative SYBR Green real‐time PCR. Each 20 µL SYBR Green reaction consisted of 2 µL cDNA, 2 µL 10 × LightCycler‐ DNA Master SYBR Green I (Roche Diagnostics Corporation, IN), and 1 µM each of forward and reverse primers. EGFR specific primers were 5′‐GTCTCTTGCCGGAATGTCAG‐3′ (sense) and 5′‐CTCACCCTCCAGAAGGTTGC‐3′ (antisense) (amplicon size: 67 bp), as previously reported.( 27 ) Quantitative PCR was performed on a Light‐Cycler V3 System (Roche Diagnostics Corporation, IN, USA) with 60 cycles, using three‐stage program parameters for each cycle as recommended by the manufacturer: 2 s at 95°C, 10 s at 62°C, and 15 s at 72°C. A melting curve analysis was run to assess specificity of the amplified PCR products. Quantification focused on the initial exponential phase of amplification above baseline according to the Light‐Cycler software. EGFR mRNA levels were standardized by β‐actin mRNA and log‐transformed as log(EGFR mRNA/β‐actin mRNA) for both cancer and non‐cancerous tissues.
Cell culture and assessment of cell growth. Primary HBE cells were isolated from bronchial mucosal biopsies of eight lung cancer patients with CA 16/15 (repeat number of the longer allele/shorter allele), six patients with CA 20/19, and one patient with CA 8/7. Following isolation, cells were cultured in Keratinocyte Serum‐Free Medium® (Gibco BRL, Life Technologies, Inc., Rockville, MD, USA) containing EGF and bovine pituitary extract at 37°C in 5% CO2 as described previously.( 28 ) Cells were subjected to experiments after three passages. EGFR mRNA detection was performed in the same way as described above for tissue samples.
Western blot analysis. Normal HBE cells were first incubated for 24 h in medium without EGF and bovine pituitary extract, then treated with 100 ng/mL EGF (Sigma, Saint Lous, MS, USA). Whole cell lysates were prepared from cells using lysis buffer containing 50 mM Tris‐HCL pH 8.0, 150 mM NaCl, 5 mM MgCl2, 1% TritonX‐100, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 40 mM sodium fluoride, 1 mM sodium orthovanadate, 1 µg/mL leupeptin, 10 µg/mL aprotinin, and 1 mM phenolmethylsulfonyl fluoride, as reported previously.( 12 ) Protein (50 µg) was separated using a 10% NuPAGE electrophoresis system (NOVEX, San Diego, CA, USA), transferred to a nitrocellulose membrane (Schleicher & Schuell, Inc., Keene, NH, USA), blocked with 5% milk at 4°C overnight, and finally reacted with anti‐EGFR, Erk1/2 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA, antiphospho EGFR, or antiphospho Erk1/2 antibodies (Cell Signaling Technology, Inc., Danvers, MA, USA). An ECL kit (Amersham Corp., Arlington Heights, IL, USA) was used for detection.
Statistical analysis. Baseline characteristics of patients with or without EGFR mutations were compared using the χ 2 test for categorical data and the Student's t‐test for continuous data. A logistic regression model was used to evaluate the contribution of CA‐repeat to EGFR mutations among lung adenocarcinoma patients, considering gender, age, and smoking status as potential confounders. Differences in means of log(EGFR mRNA/β‐actin mRNA) between two groups defined by CA‐repeat number (less than 17 vs 17 or more) in the shorter allele were compared using the Student's t‐test. The effect of CA‐repeat length on EGFR mRNA expression level was evaluated by categorical regression analysis with optimal scaling using alternation least squares with adjustment for age, gender, and smoking status (SPSS Categories version 11.0, SPSS Inc., Chicago, IL, USA). For this analysis, we categorized the dependent variable into seven ranks with an approximately normal distribution: log(EGFR mRNA/β‐actin mRNA) ≤–2.23, –1.61–0.12, 0.06–1.37, 1.41–2.58, 2.68–3.47, 4.12–5.40, and ≥5.75. Association between CA‐repeat length and EGFR mutations at exon 19 was assessed using the ƒÔ( 2 ) test separately by level of log(EGFR mRNA/β‐actin mRNA), which was classified into two groups according to the median (<2.10, ≥2.10). Cell growth was compared between the two groups of CA‐repeat length in normal HBE cells using the Kruskal–Wallis test; doubling times and EGFR mRNA levels were compared using the Mann–Whitney test.
All P‐values presented are two‐tailed; those below 0.05 were considered statistically significant. Analyzes were done using SPSS version 12 (SPSS Inc.).
Results
EGFR mutations and numbers of CA‐repeats. Clinicopathological characteristics of the 154 patients are shown in Table 1. EGFR mutations occurring at exons 18, 19, or 21 were detected in 51 (33%) of the patients. Clear associations of EGFR mutations were observed with gender (P < 0.001), smoking status (smoker vs non‐smoker; P < 0.001), and histological type (squamous cell carcinoma, adenocarcinoma, or other; P = 0.02). EGFR mutations were found in 49 (41%) of 119 adenocarcinoma patients but in only one (4%) of 25 squamous cell carcinoma patients. Distributions of CA‐repeat numbers at intron 1 in the shorter and longer alleles of the EGFR gene among 146 patients (eight patients were non‐informative) revealed two peaks (Fig. 1): CA 19 (39%) and CA 15 (27%) in the shorter alleles and CA 20 (45%) and CA 16 (23%) in the longer alleles.
Table 1.
Characteristics of patients by EGFR mutation status
| EGFR mutations | P | ||
|---|---|---|---|
| Present (n = 51) | Absent (n = 103) | ||
| Age (mean ± SD, years) | 66.5 ± 10.9 | 67.8 ± 9.6 | ns ‡ |
| Gender | |||
| Male | 13 (14%) | 78 (86%) | P < 0.001 |
| Female | 38 (60%) | 25 (40%) | |
| Smoking status | |||
| Smoker | 12 (13%) | 78 (87%) | P < 0.001 |
| Nonsmoker | 39 (61%) | 25 (39%) | |
| Histology | |||
| Adenocarcinoma | 49 (41%) | 70 (59%) | P = 0.02 |
| Squamous cell carcinoma | 1 (4%) | 24 (96%) | |
| Others | 1 (10%) | 9 (90%) | |
| Pathological stage † | |||
| I | 35 (38%) | 58 (62%) | ns |
| II | 3 (21%) | 11 (79%) | |
| III | 8 (33%) | 16 (67%) | |
| IV | 5 (23%) | 17 (77%) | |
Examination for distant metastasis was not done in one case.
Not significant.
EGFR, epidermal growth factor receptor.
Figure 1.

Distribution of CA‐repeat of the shorter allele (a), and the longer allele (b) in lung cancer patients.
EGFR mutations and CA‐repeats. Because most (96%) of the EGFR mutations were found in adenocarcinoma patients, we investigated the relationship between EGFR mutations and CA‐repeat length only in those patients; 113 patients were examined (we were unable to determine both CA‐repeat number and EGFR mutation status in six patients). EGFR mutations at exons 18, 19, and 21 were detected in 3 (3%), 22 (20%), and 23 (20%), respectively, of the adenocarcinoma patients. The mutations at exon 21 were L858R in 21 patients and L861Q in one patient. All mutations at exon 19 were in‐frame deletions with sizes ranging from 9 to 24 base pair.
When study patients were divided into two groups according to the median CA‐repeat length (<17 or ≥17) in the shorter allele, EGFR mutations at exon 19 were more frequently found in the patients with shorter CA‐repeat length (P = 0.02, Table 2). On the other hand, EGFR mutation frequencies at exons 18 and 21 did not significantly differ with CA‐repeat length (Table 2). Logistic regression analysis confirmed the significant contribution of CA‐repeat to EGFR mutations at exon 19 among the lung adenocarcinoma patients (P = 0.02), independently of gender, age, and smoking status (data not shown). In contrast, the same analysis for overall (at exons 18, 19, and 21 combined) EGFR mutations in all 154 lung cancer patients demonstrated statistically significant associations with gender and smoking status, but not with CA‐repeat length, and the analysis for overall EGFR mutation frequency only in lung adenocarcinoma patients failed to demonstrate a statistically significant association with either gender or smoking status (data not shown). An analysis using CA‐repeat length in the longer allele produced results similar to, but not as clear as, those obtained for the shorter allele (data not shown); hereafter, only the results for CA‐repeats in the shorter allele are shown.
Table 2.
Association between EGFR mutations and CA‐repeat in 113 lung adenocarcinoma patients
| EGFR mutation | P | |||
|---|---|---|---|---|
| Present | Absent | |||
| Deletion | Substitution | |||
| Exon 18 | ||||
| CA‐repeat | ||||
| <17 | 0 | 1 (1.8%) | 55 (98.2%) | ns † |
| ≥17 | 0 | 2 (3.5%) | 55 (96.5%) | |
| Exon 19 | ||||
| CA‐repeat | ||||
| <17 | 16 (28.6%) | 0 | 40 (71.4%) | 0.02 |
| ≥17 | 6 (10.5%) | 0 | 51 (89.5%) | |
| Exon 21 | ||||
| CA‐repeat | ||||
| <17 | 0 | 10 (17.9%) | 46 (82.1%) | ns |
| ≥17 | 0 | 13 (22.8%) | 44 (77.2%) | |
Not significant.
EGFR, epidermal growth factor receptor.
EGFR mRNA expression and CA‐repeats in non‐cancerous lung tissues. EGFR mRNA levels in non‐cancer tissues from 74 lung adenocarcinoma patients (excluding 39 non‐informative cases), with adjustment for gender, age, and smoking status, did not differ significantly by CA‐repeat length (P = 0.19, Fig. 2a). However, when patients were divided by age category according to the median age (70 years), increased levels of log(EGFR mRNA/β‐actin mRNA) were associated with shorter CA‐repeat length in the patients below age 70 (P = 0.03, Fig. 2c). Gender and smoking status may also influence the levels of log(EGFR mRNA/β‐actin mRNA) (Fig. 2b,d, respectively). Therefore, we carried out a categorical regression analysis, with adjustment for the confounding variables (age, gender, and smoking status), and found that increased log(EGFR mRNA/β‐actin mRNA) levels were associated with shorter CA‐repeat length (P = 0.02, Table 3). As for cancer tissues, the median values of EGFR mRNA/β‐actin mRNA were 10.7, and 9.8 in longer, and shorter CA‐repeat length, respectively. No statistically significant association was found between CA‐repeat length and EGFR mRNA levels in cancer tissues (data not shown).
Figure 2.
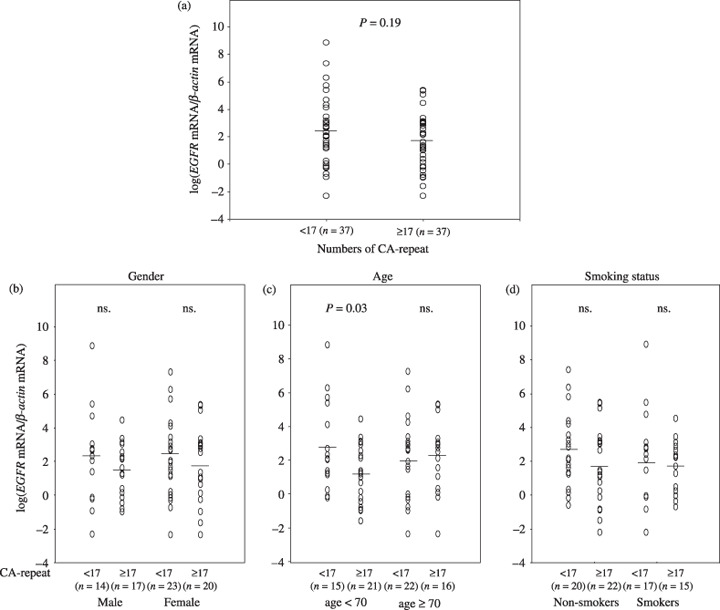
Distribution of log(epidermal growth factor receptor (EGFR) mRNA/β‐actin mRNA) in non‐cancerous tissues among lung adenocarcinoma patients according to number of CA‐repeats (<17 or ≥17) (a), gender (b), age (c), and smoking status (d). ns, not significant.
Table 3.
Categorical regression analysis of log(EGFR mRNA/β‐actin mRNA) in 74 lung adenocarcinoma patients
| Variables | β | P |
|---|---|---|
| Gender (male versus female) | –0.08 | 0.68 |
| Age (<70 versus ≥70 years) | –0.12 | 0.31 |
| Smoking status (non‐smokers versus smokers) | –0.24 | 0.24 |
| CA‐repeat length (<17 versus≥17) | –0.29 | 0.02 |
EGFR, epidermal growth factor receptor.
Further examining the relationship between CA‐repeat length and EGFR mutations at exon 19 according to whether log(EGFR mRNA/β‐actin mRNA) was greater than or less than the median (2.10), we found a significantly increased frequency of EGFR mutations associated with shorter CA‐repeat length only in the patients with higher EGFR mRNA expression (P = 0.045; Table 4). Logistic regression analysis of EGFR mutation frequency at exon 19, taking into account age, gender, and smoking status, also showed a significant contribution of shorter CA‐repeat length in the patients with higher EGFR mRNA expression (P = 0.03, β = –2.6).
Table 4.
EGFR mutations at exon 19 and CA‐repeat length in relation to log(EGFR mRNA/β‐actin mRNA) levels
| CA‐repeat length | P * | |||
|---|---|---|---|---|
| <17 | ≥17 | |||
| log(EGFR mRNA/β‐actin mRNA) < median † | ||||
| Mutations at exon19 | Present | 3 | 2 | 0.65 |
| Absent | 14 | 17 | ||
| log(EGFR mRNA/β‐actin mRNA) ≥ median | ||||
| Mutations at exon19 | Present | 7 | 1 | 0.045 |
| Absent | 13 | 17 | ||
The exact P‐value (two‐sided) based on the Pearson χ2 test.
Median value of log(EGFR mRNA/β‐actin mRNA) in non‐cancerous tissues = 2.10.
EGFR, epidermal growth factor receptor.
CA‐repeat and growth of normal HBE cells. Normal HBE cells with CA 16/15 evidenced faster cell growth than those with CA 20/19 (P = 0.004, Fig. 3a). This is consistent with a shorter doubling time for HBE cells with CA 16/15 (P = 0.006, Fig. 3b). EGFR mRNA levels in HBE cells with CA 16/15 were significantly higher than those in HBE cells with CA 20/19 (P = 0.017, Fig. 3c). In addition, EGFR phosphorylation induced by EGF was enhanced in one clone with CA 16/15 and in another clone with CA 8/7, compared with that found in 3 clones with CA 20/19 (Fig. 4). These results suggest an inverse relationship between CA‐repeat length and cell growth in HBE cells. Enhanced EGF/EGFR signaling was also found in HBE cells with shorter CA‐repeats.
Figure 3.
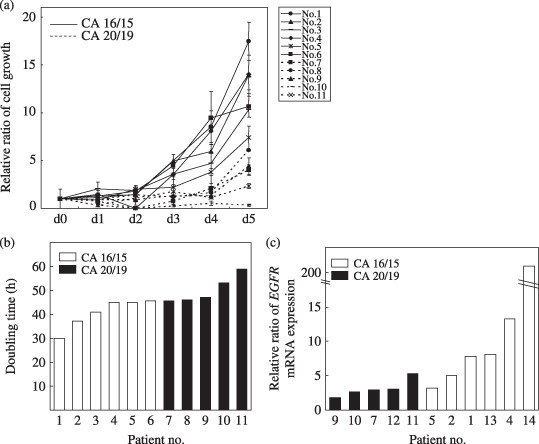
Growth (a) and doubling time (b) of normal human bronchial epithelial (HBE) cells according to number of CA‐repeats. Normal HBE cells were isolated from bronchial mucosal biopsy and subjected to experiments after three passages. Cells (4 × 104 cells/mL) were cultured in 0.5 mL medium, and the number of cells was determined by trypan blue staining after incubation for the indicated period. Relative cell numbers, which were divided by the cell number on day 0, are shown and the values are expressed as means ± SD of triplicate analyzes. (c) Levels of epidermal growth factor receptor (EGFR) mRNA in normal HBE cells as determined using real‐time reverse transcription‐polymerase chain reaction. The relative expression levels of EGFR mRNA were determined after correction for β‐actin mRNA as a control gene. NS, not significant.
Figure 4.
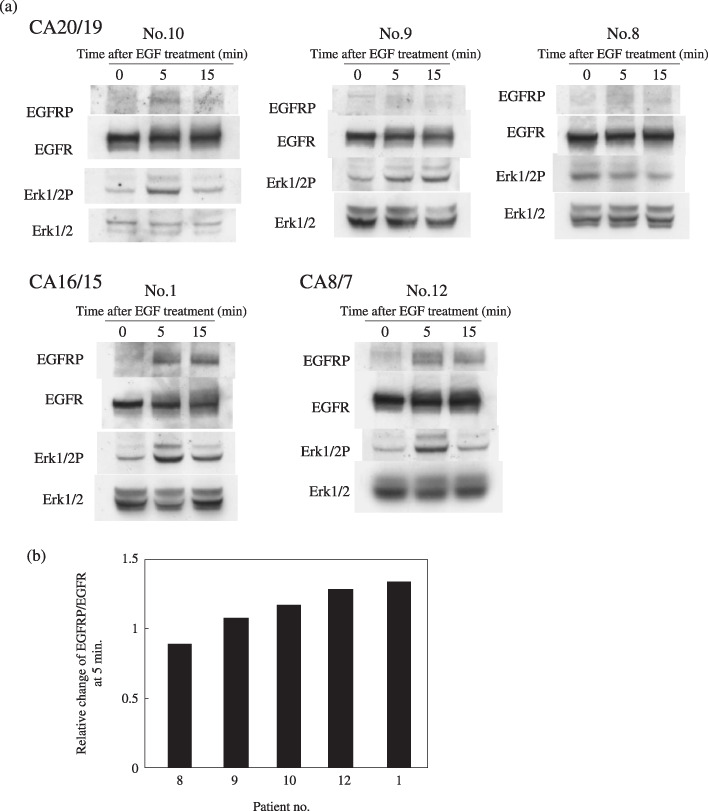
Epidermal growth factor receptor (EGFR) phosphorylation induced by EGF was more apparent in the cells with CA‐repeat lengths in the longer/shorter alleles of 16/15 and 8/7 compared to cells with CA‐repeat lengths of 20/19. Normal human bronchial epithelial (HBE) cells were first incubated for 24 h in medium without EGF and bovine pituitary extract, then treated with 100 ng/mL EGF for the indicated periods. The numbers of CA‐repeats were 20/19 in patients 10, 9, 8, 16/15 in patient 1, and 8/7 in patient 12. Western blot analysis was conducted on whole cell lysates (50 µg) (a). The relative ratio of EGFR phosphorylation was determined as the intensity of EGFR phosphorylation divided with that of EGFR (b).
Discussion
EGFR mutations display different clinicopathological features according to the exons at which they occur. The majority of patients with mutations at exon 21 were female non‐smokers who were diagnosed with adenocarcinomas showing bronchioloalveolar features, whereas patients with mutations at exon 19 included greater proportions of males and current or former smokers and a smaller proportion with bronchioloalveolar features.( 10 ) EGFR mutations at exon 19 were small in‐frame deletions, whereas those at exons 18 and 21 were base substitutions. Our findings that EGFR mutations were associated with gender, smoking status, and histological type, and occurred often in adenocarcinoma patients but rarely in squamous cell carcinoma patients, are consistent with previous reports.( 7 , 8 , 9 , 10 ) Our finding that CA‐repeat length at intron 1 of the EGFR gene displayed bimodal distributions in both the shorter and longer alleles is similar to a result reported in non‐Chinese Asians.( 23 )
The results of the present study may be summarized as follows. First, EGFR mutations at exon 19 were closely associated with shorter CA‐repeat lengths in the shorter allele, but this was not the case with EGFR mutations at exons 18 or 21. This implies that CA‐repeat length is associated with deletion mutations, but not with substitution mutations. Second, the mechanism relating CA‐repeat and EGFR mutations at exon 19 was elucidated by our finding that increased intrinsic EGFR mRNA expression in non‐cancerous tissues was significantly associated with shorter CA‐repeats. Third, the relationship between increased EGFR mutations and shorter CA‐repeat length at exon 19 was apparent in lung adenocarcinoma patients with EGFR mRNA expression levels in the upper 50th percentile, but not in patients with lower EGFR expression levels. Fourth and lastly, our finding that increased cell growth and enhanced EGF/EGFR signaling occurred in HBE cells with shorter CA‐repeat lengths and increased EGFR mRNA expression confirms the relationship between CA‐repeat length and EGFR mRNA expression in the parallel studies mentioned above and provides a clue as to what cell phenotypes might arise from the shorter CA‐repeat. Because EGF/EGFR signaling is enhanced in cells undergoing rapid growth, and we found an inverse relationship between cell growth and CA‐repeat length further related to EGFR mRNA levels, we conclude that EGFR mRNA levels are elevated in cells with shorter CA‐repeat lengths, which results in enhanced EGF/EGFR signaling and therefore more rapid cell growth.
As for the mechanism linking CA‐repeat length and EGFR mRNA expression, it has been postulated that the CA‐repeat length at intron 1 influences DNA bendability and hence the binding of repressor protein.( 29 ) However, we are puzzled as to how increased EGFR expression could be linked to increased EGFR mutations, specifically deletions. Our HBE study demonstrated that shorter CA‐repeat lengths are associated with faster cell growth, which might result from increased EGFR expression and activation of EGF/EGFR signaling. Further study of the cell phenotypes related to shorter CA‐repeat length in association with increased EGFR mutations is needed, though, because there are patients with EGFR mutations and shorter CA‐repeat lengths with no evidence of elevated EGFR mRNA expression, which suggests that other mechanisms may be involved.
It has been reported recently that EGFR is also involved in DNA repair. EGFR binds to DNA‐dependent protein kinase (DNA‐PK) complex and induces the translocation of DNA‐PK complex into the nucleus.( 30 , 31 ) Mutated forms of EGFR abrogate ionizing radiation‐induced nuclear EGFR translocation or binding to DNA‐PK catalytic subunit (DNA‐PKcs), resulting in inhibition of DNA‐PK activity.( 32 ) DNA‐PK is involved in non‐homologous end joining, one of the most important DNA repair systems in mammalians.( 33 ) Because defects in the DNA‐PK complex, Ku proteins, and DNA‐PKcs enhance tumorigenecity in transgenic mice,( 34 , 35 , 36 ) it is possible that EGFR mutations are causally related to cancer development; in fact, transgenic mice carrying the EGFR mutations EGFR L858R and EGFR ΔL747–S752 developed adenocarcinomas of the lung.( 12 ) Taken together, these findings lead us to postulate that shorter CA‐repeat lengths might enhance intrinsic EGFR mRNA expression through altered bendability of the gene, and up‐regulated EGFR might cause enhanced cell growth and attenuated DNA repair capacity – specifically against deletions – through physical interaction with DNA‐PK complex, resulting in increased EGFR mutations in normal lung epithelial cells.
Here we report that an association between EGFR mRNA expression and CA‐repeat length was found only in non‐cancerous tissues. That EGFR mRNA expression in cancer tissues was not highly correlated with that in corresponding non‐cancerous tissues (correlation coefficient 0.34) is consistent with our finding that cancer tissues did not evidence an association between EGFR expression and CA‐repeat length. This may be due to the fact that intrinsic EGFR expression is altered in cancer cells by various factors including mutations as well as allelic imbalance of EGFR, which has also been reported to be associated with CA‐repeat length.( 25 )
As for interethnic differences in CA‐repeat length, lengths of less than 17 in Japanese are less frequent than in Caucasians.( 23 ) However, the frequency of EGFR mutations is higher in Japanese than in other ethnic groups (primarily Caucasians): 30–50% versus 5–20%.( 37 , 38 , 39 ) Mutations of other genes in lung cancer, specifically KRAS and TP53, are more frequent in Caucasians than in Japanese. KRAS and TP53 mutations are known to be caused by smoking, and in particular the G:C‐to‐T:A transversion in TP53 is generally interpreted as a mutagen footprint.( 40 ) However, KRAS and TP53 mutations occur at relatively high rates even among non‐smokers in Japanese (6% and 30%, respectively( 7 )) and at even higher rates in Caucasians (10% and 47.5%, respectively( 40 )). KRAS and EGFR mutations are mutually exclusive, and either type of mutation alone is thought to be sufficient for lung carcinogenesis. These factors relative to mutations in other genes may contribute to interethnic differences in EGFR mutation rates as well as different genetic backgrounds.
In future research, we plan to investigate whether the CA‐repeat polymorphism can serve as an alternative predictive marker for EGFR tyrosine kinase inhibitor response. We also intend to expand our research on the relationship between CA‐repeat and EGFR mutations in lung carcinogenesis.
Acknowledgments
This work was supported by grants from the Ministry of Education, Science, Sports and Culture (19591116, Japan), and the Smoking Research Foundation.
References
- 1. ‘Cancer Statistics in Japan’ Editorial Board . Number of deaths and proportional mortality rates from malignant neoplasms by site in Japan (2005). In: Nomura K, Sobue T, Nakatani H et al ., eds. Cancer Statistics in Japan‐2005. Tokyo: Foundation for Promotion Cancer Research, 2005; 36–9. [Google Scholar]
- 2. Mountain CF, Dresler CM. Regional lymph node classification for lung cancer staging. Chest 1997; 111: 1718–23. [DOI] [PubMed] [Google Scholar]
- 3. Lynch TJ, Bell DW, Sordella R et al . Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 4. Paez JG, Janne PA, Lee JC et al . EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 5. Mitsudomi T, Kosaka T, Endoh H et al . Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non‐small‐cell lung cancer with postoperative recurrence. J Clin Oncol 2005; 23: 2513–20. [DOI] [PubMed] [Google Scholar]
- 6. Shepherd FA, Rosell R. Weighing tumor biology in treatment decisions for patients with non‐small cell lung cancer. J Thorac Oncol 2007; 2: S68–76. [DOI] [PubMed] [Google Scholar]
- 7. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 8. Tokumo M, Toyooka S, Kiura K et al . The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non‐small cell lung cancers. Clin Cancer Res 2005; 11: 1167–73. [PubMed] [Google Scholar]
- 9. Tomizawa Y, Iijima H, Sunaga N et al . Clinicopathologic significance of the mutations of the epidermal growth factor receptor gene in patients with non‐small cell lung cancer. Clin Cancer Res 2005; 11: 6816–22. [DOI] [PubMed] [Google Scholar]
- 10. Sueoka N, Sato A, Eguchi H et al . Mutation profile of EGFR gene detected by denaturing high‐performance liquid chromatography in Japanese lung cancer patients. J Cancer Res Clin Oncol 2007; 133: 93–102. [DOI] [PubMed] [Google Scholar]
- 11. Fukuoka M, Yano S, Giaccone G et al . Multi‐institutional randomized phase II trial of gefitinib for previously treated patients with advanced non‐small‐cell lung cancer (The IDEAL 1 Trial). J Clin Oncol 2003; 21: 2237–46. [DOI] [PubMed] [Google Scholar]
- 12. Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down‐regulation of the receptors. Genes Dev 2006; 20: 1496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ishii S, Xu YH, Stratton RH, Roe BA, Merlino GT, Pastan I. Characterization and sequence of the promoter region of the human epidermal growth factor receptor gene. Proc Natl Acad Sci USA 1985; 82: 4920–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnson AC, Ishii S, Jinno Y, Pastan I, Merlino GT. Epidermal growth factor receptor gene promoter. Deletion analysis and identification of nuclear protein binding sites. J Biol Chem 1988; 263: 5693–9. [PubMed] [Google Scholar]
- 15. Kageyama R, Merlino GT, Pastan I. Epidermal growth factor (EGF) receptor gene transcription. Requirement for Sp1 and an EGF receptor‐specific factor. J Biol Chem 1988; 263: 6329–36. [PubMed] [Google Scholar]
- 16. Maekawa T, Imamoto F, Merlino GT, Pastan I, Ishii S. Cooperative function of two separate enhancers of the human epidermal growth factor receptor proto‐oncogene. J Biol Chem 1989; 264: 5488–94. [PubMed] [Google Scholar]
- 17. Chi DD, Hing AV, Helms C, Steinbrueck T, Mishra SK, Donis‐Keller H. Two chromosome 7 dinucleotide repeat polymorphisms at gene loci epidermal growth factor receptor (EGFR) and pro alpha 2 (I): collagen (COL1A2). Hum Mol Genet 1992; 1: 135. [DOI] [PubMed] [Google Scholar]
- 18. Gebhardt F, Zanker KS, Brandt B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J Biol Chem 1999; 274: 13176–80. [DOI] [PubMed] [Google Scholar]
- 19. Zhang W, Weissfeld JL, Romkes M, Land SR, Grandis JR, Siegfried JM. Association of the EGFR intron 1 CA repeat length with lung cancer risk. Mol Carcinog 2007; 46: 372–80. [DOI] [PubMed] [Google Scholar]
- 20. Lee SJ, Kim KM, Chae MH et al . No association between dinucleotide repeat polymorphism in intron 1 of the epidermal growth factor receptor gene EGFR and risk of lung cancer. Cancer Genet Cytogenet 2007; 172: 29–32. [DOI] [PubMed] [Google Scholar]
- 21. Han SW, Jeon YK, Lee KH et al . Intron 1 CA dinucleotide repeat polymorphism and mutations of epidermal growth factor receptor and gefitinib responsiveness in non‐small‐cell lung cancer. Pharmacogenet Genomics 2007; 17: 313–19. [DOI] [PubMed] [Google Scholar]
- 22. Amador ML, Oppenheimer D, Perea S et al . An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res 2004; 64: 9139–43. [DOI] [PubMed] [Google Scholar]
- 23. Liu W, Innocenti F, Chen P, Das S, Cook EH Jr, Ratain MJ. Interethnic difference in the allelic distribution of human epidermal growth factor receptor intron 1 polymorphism. Clin Cancer Res 2003; 9: 1009–12. [PubMed] [Google Scholar]
- 24. Buerger H, Gebhardt F, Schmidt H et al . Length and loss of heterozygosity of an intron 1 polymorphic sequence of egfr is related to cytogenetic alterations and epithelial growth factor receptor expression. Cancer Res 2000; 60: 854–7. [PubMed] [Google Scholar]
- 25. Nomura M, Shigematsu H, Li L et al . Polymorphisms, mutations, and amplification of the EGFR gene in non‐small cell lung cancers. Plos Med 2007; 4: 715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taron M, Ichinose Y, Rosell R et al . Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib‐treated chemorefractory lung adenocarcinomas. Clin Cancer Res 2005; 11: 5878–85. [DOI] [PubMed] [Google Scholar]
- 27. Xu XC, Lee JJ, Wu TT, Hoque A, Ajani JA, Lippman SM. Increased retinoic acid receptor‐beta4 correlates in vivo with reduced retinoic acid receptor‐beta2 in esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2005; 14: 826–9. [DOI] [PubMed] [Google Scholar]
- 28. Sueoka N, Lee HY, Walsh GL, Hong WK, Kurie JM. Posttranslational mechanisms contribute to the suppression of specific cyclin: CDK complexes by all‐trans retinoic acid in human bronchial epithelial cells. Cancer Res 1999; 59: 3838–44. [PubMed] [Google Scholar]
- 29. Brandt B, Meyer‐Staeckling S, Schmidt H, Agelopoulos K, Buerger H. Mechanisms of egfr gene transcription modulation: relationship to cancer risk and therapy response. Clin Cancer Res 2006; 12: 7252–60. [DOI] [PubMed] [Google Scholar]
- 30. Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA‐dependent protein kinase in mammalian cells. J Biol Chem 1998; 273: 1568–73. [DOI] [PubMed] [Google Scholar]
- 31. Dittmann K, Mayer C, Fehrenbacher B et al . Radiation‐induced epidermal growth factor receptor nuclear import is linked to activation of DNA‐dependent protein kinase. J Biol Chem 2005; 280: 31182–9. [DOI] [PubMed] [Google Scholar]
- 32. Das AK, Chen BP, Story MD, Sato M, Minna JD, Chen DJ, Nirodi CS. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR‐mediated radioprotection in non‐small cell lung carcinoma. Cancer Res 2007; 67: 5267–74. [DOI] [PubMed] [Google Scholar]
- 33. Smith GC, Jackson SP. The DNA‐dependent protein kinase. Genes Dev 1999; 13: 916–34. [DOI] [PubMed] [Google Scholar]
- 34. Mondello C, Rebuzzini P, Dolzan M, Edmonson S, Taccioli GE, Giulotto E. Increased gene amplification in immortal rodent cells deficient for the DNA‐dependent protein kinase catalytic subunit. Cancer Res 2001; 61: 4520–5. [PubMed] [Google Scholar]
- 35. Espejel S, Martin M, Klatt P, Martin‐Caballero J, Flores JM, Blasco MA. Shorter telomeres, accelerated ageing and increased lymphoma in DNA‐PKcs‐deficient mice. EMBO Rep 2004; 5: 503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Difilippantonio MJ, Zhu J, Chen HT et al . DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000; 404: 510–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang SH, Mechanic LE, Yang P et al . Mutations in the tyrosine kinase domain of the epidermal growth factor receptor in non‐small cell lung cancer. Clin Cancer Res 2005; 11: 2106–10. [DOI] [PubMed] [Google Scholar]
- 38. Marchetti A, Martella C, Felicioni L et al . EGFR mutations in non‐small‐cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol 2005; 23: 857–65. [DOI] [PubMed] [Google Scholar]
- 39. Cappuzzo F, Hirsch FR, Rossi E et al . Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non‐small‐cell lung cancer. J Natl Cancer Inst 2005; 97: 643–55. [DOI] [PubMed] [Google Scholar]
- 40. Le Calvez F, Mukeria A, Hunt JD et al . TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res 2005; 65: 5076–83. [DOI] [PubMed] [Google Scholar]