

Abstract
Gain‐of‐function mutations of the receptor tyrosine kinase KIT can cause systemic mastocytosis (SM) and gastrointestinal stromal tumors. Most of the constitutively active KIT can be inhibited by imatinib; D816V KIT cannot. In this study, we investigated the activity of triptolide, a diterpenoid isolated from the Chinese herb Tripterygium wilfordii Hook. f., in cells expressing mutant KIT, including D816V KIT. Imatinib‐sensitive HMC‐1.1 cells harboring the mutation V560G in the juxtamembrane domain of KIT, imatinib‐resistant HMC‐1.2 cells harboring both V560G and D816V mutations, and murine P815 cells, were treated with triptolide, and analyzed in terms of growth, apoptosis, and signal transduction. The in vivo antitumor activity was evaluated by using the nude mouse xenograft model. Our results demonstrated that triptolide potently inhibits the growth of both human and murine mast cells harboring not only imatinib‐sensitive KIT mutation but also imatinib‐resistant D816V KIT. Triptolide markedly inhibited KIT mRNA levels and strikingly reduced the levels of phosphorylated and total Stat3, Akt, and Erk1/2, downstream targets of KIT. Triptolide triggered apoptosis by inducing depolarization of mitochondrial potential and release of cytochrome c, downregulation of Mcl‐1 and XIAP. Furthermore, triptolide significantly abrogated the growth of imatinib‐resistant HMC‐1.2 cell xenografts in nude mice and decreased KIT expression in xenografts. Our data demonstrate that triptolide inhibits imatinib‐resistant mast cells harboring D816V KIT. Further investigation of triptolide for treatment of human neoplasms driven by gain‐of‐function KIT mutations is warranted. (Cancer Sci 2009; 100: 1335–1343)
Systemic mastocytosis (SM) is characterized by the abnormal proliferation of mast cells in the bone marrow and spleen, and in other extracutaneous organs.( 1 ) According to World Heath Organization criteria, SM can be divided into four sub‐categories: (1) indolent SM (ISM), (2) SM with associated hematologic clonal non‐mast cell disease (AHNMD),( 2 ) aggressive SM, and (4) mast cell leukemia (MCL).( 3 ) The clinical manifestations of SM depend on the release of mast cell mediators on the tissues involved and from the tissue response to the accumulation of mast cells. ISM can be managed successfully with anti‐mediator drugs. By contrast, no effective treatment exists for aggressive SM, which may further develop into malignant disease, albeit slowly. Treatment of aggressive SM or MCL by the combination of interferon‐α and corticosteroids is only partially effective.( 1 , 3 , 4 ) Recently, cladribine (2‐CDA) has been used to control SM; however, its clinical benefit for patients with mastocytosis remains unclear because of limited long‐term follow‐up data.( 3 ) Therefore, novel effective drugs to treat aggressive SM and MCL are needed.
KIT is a 145‐kDa transmembrane receptor tyrosine kinase of the type III subgroup characterized by five extracellular immunoglobulin‐like domains and a split tyrosine kinase domain. Gain‐of‐function mutations in KIT promote KIT autophosphorylation in the absence of stimulation by stem cell factor,( 4 , 5 , 6 ) which causes activation of its downstream pathways (e.g. Janus kinase/signal transducer and activators of transcription [Jak‐STAT], Ras‐Raf‐mitogen‐activated protein [MAP] kinases, and phosphatidylinositol‐3 [PI3] kinase( 7 )) and leads to uncontrolled cell proliferation and apoptosis resistance.( 8 ) Imatinib has shown anti‐tyrosine kinase activity in the wild type and some KIT mutants.( 5 , 9 ) However, the D816V mutation in KIT is believed to influence the conformation of the activation loop lying at the entrance to the KIT enzymatic pocket, to which imatinib binds.( 7 , 10 , 11 ) Therefore, cells bearing D816V KIT are resistant to imatinib. Unfortunately, approximately 80% of SM patients harbor the activating oncogenic D816V mutation in KIT. Other mutations at codon 816, reported at lower frequency, include D816Y, D816F, and D816H.( 12 ) Recently, PKC412( 9 ) and dasatinib( 13 ) have been shown to kill human mast cells expressing D816V KIT, and the relevant clinical trials are under way. Additionally, downregulating the total level of cellular KIT by flavopiridol which mediates global inhibition of transcription has been an effective approach against imatinib‐resistant KIT‐expressing cells.( 14 , 15 ) However, clinical trials demonstrated the limited activity of flavopiridol as a single agent in the treatment of certain tumors.( 16 )
Since 1972, rigorous attempts have been made to better identify biologically active constituents of the Chinese medicine herb Tripterygium wilfordii Hook f., which has been used for centuries to treat inflammation and autoimmune diseases such as rheumatoid arthritis.( 17 , 18 , 19 ) Triptolide is the key biologically active component that mediates immunosuppressive and anti‐inflammatory activities by inhibiting production of cytokines (e.g. IL‐2, IL‐4, IFN‐γ)( 19 , 20 , 21 ) and NF‐kB‐ and NF‐AT‐mediated transcription.( 22 ) Of note, PG490–88, a water‐soluble semisynthetic compound derived from triptolide, has been investigated in clinical trials as an immunosuppressant to prevent organ transplant rejection.( 23 , 24 , 25 , 26 ) In addition, triptolide inactivates RNA polymerase I and II, and probably renders global transcription arrest.( 27 )
In this study, we hypothesized that triptolide has antineoplastic activity against imatinib‐resistant KIT‐expressing cells by downregulating KIT, and so evaluated its potential translational efficacy against tumor cells expressing mutant KIT, including D816V KIT. Triptolide exerted a strong inhibitory effect in imatinib‐resistant human mast cells by downregulating KIT mRNA level and inducing apoptosis. Furthermore, triptolide inhibited the growth of mast cell lines harboring D816V KIT in xenografted nude mice by inhibiting KIT expression. Our data suggest that triptolide may be a promising agent to overcome imatinib resistance caused by D816V KIT in SM.
Materials and Methods
Cell culture. Imatinib‐sensitive HMC‐1.1 cells harboring the mutation V560G in the juxtamembrane domain of KIT and imatinib‐resistant HMC‐1.2 cells harboring both V560G and D816V mutations( 28 ) were kindly provided by Dr Joseph Butterfield (Mayo Clinic, Rochester, MN, US) and maintained in Iscove's modified Dulbecco's medium (Invitrogen, Guangzhou, China) supplemented with 10% fetal calf serum (Hyclone, Guangzhou, China) at 37°C, 5% CO2. That HMC‐1.1 and HMC‐1.2 differ only by the presence of the D816V KIT mutation makes them ideal models for the study of novel agents against this kinase domain mutation. The murine P815 mastocytoma cell line resistant to imatinib and expressing the D814Y mutation (corresponding to human D816Y)( 29 ) was purchased from the American Type Culture Collection (Manassas, VA, US) and cultured in DMEM supplemented with 10% fetal calf serum and 25 mM HEPES buffer.( 13 ) KBM5 cells were maintained in IMDM supplemented with 10% fetal calf serum. MEF and NHFB (normal human fibroblast) cell lines were cultured in DMEM supplemented with 10% fetal calf serum.( 30 )
Chemicals and antibodies. Triptolide was purchased from Sigma‐Aldrich (Shanghai, China) and prepared as a 20‐mM stock solution in dimethyl sulfoxide (DMSO). Drug stock solution was stored in aliquots at –20°C. Interferon alfa‐2b was from Schering‐Plough (Brinny) Co (Innishannon, Cork, Ireland). Dexamethasone was from Guangzhou Baiyunshan Pharmaceutical Co (Guangzhou, China). Antibodies and their sources were as follows: rabbit polyclonal antibodies against Bax, Mcl‐1 (S‐19), KIT (c‐19), phospho‐KIT on Y568/570, were from Santa Cruz Biotechnology (Santa Cruz, CA, US); antibodies against poly(adenosine diphosphate [ADP]‐ribose) polymerase (PARP), p27Kip1 and p53, Becton‐Dickson Biosciences Pharmingen (San Jose, CA, US); antibodies against phospho‐Erk1/2 (T202/Y204), Erk1/2, Akt, JNK, and XIAP were from Cell Signaling Technology (Beverly, MA, US); mouse monoclonal antibody specific against phosphotyrosine 705 of Stat3 (clone 9E12) and rabbit polyclonal anti‐Stat3, Upstate Technology (Lake Placid, NY, US); mouse anti‐human‐KIT (CD117) monoclonal antibody, R&D Systems (Minneapolis, MN, US); mouse monoclonal antibody against actin, Sigma‐Aldrich (Shanghai, China); rabbit anti‐human RNA polymerase II, phospho‐RNA polymerase II (S5), Bethyl Laboratories (Montgomery, TX, US); and anti‐mouse IgG and anti‐rabbit IgG horseradish peroxidase‐conjugated antibodies, Pierce Biotechnology (Rockford, IL, US).
Cell viability assay. The 3‐(4,5‐dimethyl‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) assay (CellTiter 96Aqueous One Solution reagent, Promega Corp., Shanghai, China) was used to evaluate cell viability as described previously.( 31 ) Briefly, 2 × 105/mL cells in 100 µL were exposed to various concentrations of triptolide for 72 h. Control cells received DMSO with final concentration <0.1%. The absorbance/optical density was read on a 96‐well plate reader at wavelength 490 nm. The drug concentration resulting in 50% inhibition of cell growth (IC50) was determined.
KIT kinase activity assay. HTScan KIT kinase assay kit (#7755, Cell Signaling Technology) was used to evaluate the inhibitory effect of compounds on KIT kinase. This ELISA kinase assay was conducted according to the manufacturer's instructions. In brief, biotin‐KDR peptide substrate (1.5 µM) was phosphorylated at Y996 by 100 ng KIT kinase in the presence of 60 mM HEPES, pH 7.5, 5 mM MgCl2, 5 mM MnCl2, 3 µM Na3VO4, 1.25 mM DTT, 20 µM ATP in 50 µL reaction. Then, reactions were transferred to streptavidin‐coated plates, and then incubated with primary anti‐phospho‐tyrosine monoclonal antibody, and HRP‐conjugated secondary anti‐mouse IgG, respectively. After seven washes, 3,3′,5,5′ tetramethylbenzidine (TMB) was added to develop color, and stopped by 1 M H3PO4. The absorbance was then measured at wavelength 450 nm (A 450 nm) with reference at wavelength 570 nm (A 570 nm).
Cell cycle analysis by flow cytometry. After drug treatment, cells were collected and fixed overnight in 70% cold ethanol at –20°C. The cells were then washed twice in cold PBS and labeled with propidium iodide (PI). Cell cycle distribution, including the proportion of cells in sub‐G1, was determined by use of a FACScalibur flow cytometer equipped with CellQuest software.( 31 )
Measurement of mitochondrial transmembrane potential (ΔΨm). After treatment with triptolide, cells were examined for inner mitochondrial transmembrane potential in mast cells by flow cytometry. Briefly, cells were stained with two probes: MitoTracker Red (chloromethyl‐X‐rosamine [CMXRos]) and MitoTracker Green FM (MTGreen, Invitrogen, Guangzhou, China). Cells were washed in Ca2+‐free PBS, stained with MitoTracker dyes and incubated at 37°C for 1 h in the dark. Then, samples were analyzed with use of FACScalibur flow cytometer with CellQuest software.( 31 , 32 )
Assessment of apoptosis. Apoptosis was evaluated by annexin V‐positive population. Following treatment with triptolide, mast cells were harvested and resuspended in 100 µL of annexin V binding buffer containing the annexin V‐fluoroisothiocyanate and PI (Sigma‐Aldrich, Shanghai, China). The cells were incubated for 15 min at room temperature in the dark. Annexin V‐positive cells were analyzed with use of a FACScalibur flow cytometer with CellQuest software.( 31 , 32 )
SDS‐PAGE and Western blot analysis. Western blot analysis, except for cytochrome c release analysis, involved whole‐cell lysates prepared in radioimmunoprecipitation assay (RIPA) buffer (1 × PBS, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) supplemented with freshly added 10 mM b‐glycerophosphate, 1 mM sodium orthovanadate, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and 1 × Roche complete Mini protease inhibitor cocktail (Roche, Indianapolis, IN, USA).( 31 ) To detect the level of cytochrome c in the cytosol, the cytosolic fraction extraction was prepared as described previously.( 33 , 34 )
Semi‐quantitative RT‐PCR. Total RNA was extracted from 6 × 106 cells with use of Trizol reagent (Invitrogen). After quantification by spectrophotometry, first‐strand complementary DNA (cDNA) was synthesized from 1 µL of total RNA with the use of the RNA PCR Kit (AMV) Ver.3.0 (TaKaRa, Dalian, China) with an Oligo dT‐Adaptor primer. Amplification of cDNA was performed by PCR with gene‐specific primers (Table 1). RT‐PCR products were analyzed on 1.8% agarose gel and visualized and recorded by staining with ethidium bromide.
Table 1.
Sequences of primers
| Gene | Primer sequences (5′ to 3′) | Predicted products (bp) | |
|---|---|---|---|
| KIT | Forward | GCAACACTATAGTATTAAAAAG | 248 |
| Reverse | CCTTTGCAGGACTGTCAAGCA | ||
| c‐Src | Forward | CGACTTTGGGCTGGCTCGGCTCATT | 216 |
| Reverse | ACCTGGTCCAGCACCTCGCGGTTCA | ||
| Lck | Forward | TACCTACGAAGGCTCCAATC | 431 |
| Reverse | ATGTAGAAGCCACCGTTGTC | ||
| Lyn | Forward | GCAGAGGGAATGGCATACATC | 113 |
| Reverse | GCAAGGCCAAAATCTGCAA | ||
| EGFR | Forward | CATTCATGCGTCTTCACCTG | 377 |
| Reverse | CATATCCCCATGGCAAACTC | ||
| GAPDH | Forward | TGGAAATCCCATCACCATCT | 300 |
| Reverse | GTCTTCTGGGTGGCAGTGAT |
EGFR, epidermal growth factor receptor; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
Nude mouse xenograft model. nu/nu BALB/c mice were bred at the animal facility of Sun Yat‐sen University. The mice were housed in barrier facilities with a 12‐h light/12‐h dark cycle, with food and water available ad libitum. Flanks of 6‐ to 8‐week‐old‐male athymic nude mice were inoculated subcutaneously with 3 × 107 HMC‐1.2 cells. Tumors were measured every other day by use of calipers. Tumor volumes were calculated by the following formula: a 2 × b × 0.4, where a is the smallest diameter and b is the diameter perpendicular to a. Five days after inoculation, when tumors reached approximately 50 mm3, the mice were randomly divided into control or experimental groups (10 mice in each group). Triptolide solution was injected intraperitoneally each day as described( 35 ) and on the basis of our preliminary experimental findings. Triptolide was dissolved in tissue‐culture‐grade DMSO before dilution in tissue‐culture medium. The final concentration of DMSO was less than 0.1% v/v. Mice in the control group received the same amount of DMSO in tissue‐culture medium. The body weight, feeding behavior and motor activity of each animal were monitored as indicators of general health. At the end of the experiments (day 21 after administration of triptolide), blood samples were collected for assay of lactate dehydrogenase, and aspartate and alanine aminotransferase activity, and complete blood count. The animals were then euthanazed, and tumor xenografts were immediately removed, weighed, stored and fixed. All animal studies were conducted with the approval of the Sun Yat‐sen University Institutional Animal Care and Use Committee.
Immunohistochemical staining. Formalin‐fixed xenografts of HMC‐1.2 cells were embedded in paraffin and sectioned according to standard techniques. Immunohistochemical staining of xenograft sections (4 µm) involved the MaxVision kit (Maixin Biol, Fuzhou, China) according to the manufacturer's instructions. The primary antibody was monoclonal mouse anti‐human CD117 antibody (KIT) at a 1:150 dilution. An amount of 50 µL MaxVisionTM reagent was applied to each slide. Color was developed with use of 0.05% diaminobenzidine and 0.03% H2O2 in 50 mM Tris‐HCl (pH 7.6), and the slides were counterstained with hematoxylin. A negative control for CD117 was also included for each xenograft specimen by substituting the primary antibody with normal goat serum.
Statistical analysis. All experiments were carried out at least in triplicate, and data are expressed as mean ± standard error (SE), unless otherwise stated. GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA, US) was used for statistical analysis. A P < 0.05 was considered statistically significant.
Results
Triptolide inhibits growth of cells expressing D816V KIT at nanomolar concentrations. HMC‐1.2 cells are resistant to interferon α (IFN‐α), corticosteroids and other conventional therapy. By incubating HMC‐1.2 cells in the presence of IFN‐α‐2b or dexamethasone for 72 h and measuring cell viability by MTS assay, we did find that the IC50 values were 4.19 × 105 IU/mL and 2653 µM respectively (data not shown). In contrast, IFN‐α‐2b significantly inhibited viability of leukemia K562 cells (IC50, 1014 IU/mL); dexamethasone significantly inhibited viability of multiple myeloma MM1S cells (IC50, 0.98 µM) (data not shown). To screen novel effective compounds to kill D816V KIT expressing cells, we investigated whether triptolide was active against mast cells carrying D816V KIT. Exponentially growing HMC‐1.2 cells with both V560G and D816V KIT mutations were exposed to increasing concentrations of triptolide for 72 h. Cell viability, assayed by MTS, was potently inhibited by triptolide in HMC‐1.2 cells in a concentration‐dependent manner, with IC50 at 7 nM (Fig. 1a). Triptolide also showed strong growth inhibitory activity against murine P815 mastocytoma cells expressing D814Y (homologous to human KIT D816Y), with IC50 at 64 nM (Fig. 1b). Growth of control HMC‐1.1 cells harboring only the V560G KIT mutation, which is sensitive to imatinib, was potently inhibited by triptolide, with IC50 at 14 nM (Fig. 1c). Triptolide also inhibited the cell viability of leukemia cell lines KBM5 carrying Bcr‐Abl (IC50: 6.1 nM), Molm13 and MV4–11 carrying FLT‐3‐ITD mutation (IC50: 2.9 and 3.8 nM, respectively), HEL carrying Jak2 V617F mutation (IC50: 42.3 nM), 38 and EOL‐1 carrying mutant PDGFRα (IC50: 10.7 nM) (data not shown). These results suggest that triptolide may also be effective against cells expressing activated protein tyrosine kinases Bcr‐Abl, PDGFRα, FLT‐3 and Jak2.
Figure 1.
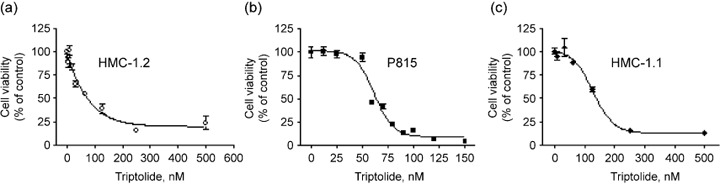
Triptolide inhibits proliferation of human mast cells bearing D816V KIT. HMC‐1.2 (a), P815 (b) and HMC‐1.1 (c) cells were exposed to increasing concentrations of triptolide for 72 h. Cell viability was measured by MTS assay and was plotted as percentage cell viability relative to the control. Triptolide dose‐response curves are shown. The error bars indicate SE.
We also examined the effect of triptolide on normal cells. MEF cells and NHFB cells were treated with increasing concentrations of triptolide for 72 h, cell viability assayed by MTS indicated that triptolide exhibited minimal effects on the growth of these two lines of cells; the IC50 values were 8.8 and 8.6 µM, respectively (data not shown). This implied that triptolide might hold a therapeutic window.
Triptolide inhibits KIT expression at transcription level. Since triptolide inhibits the cell viability in D816V KIT expressing cells, we investigated whether KIT was the target for triptolide. We first asked whether triptolide exerted a directly inhibitory effect on KIT tyrosine kinase activity. To this end, we measured KIT kinase activity with a cell‐free system in the presence of triptolide or KIT inhibitor EXEL‐0862 (positive control).( 31 ) The results showed that submicromolar EXEL‐0862 significantly inhibited the KIT kinase activity in a dose‐dependent manner (Fig. 2a), but triptolide, even at as high as 10 µM concentration, did not exhibit a significant inhibitory effect on KIT kinase activity. Therefore, the results did not support a direct inhibitory effect of triptolide on KIT kinase activity.
Figure 2.
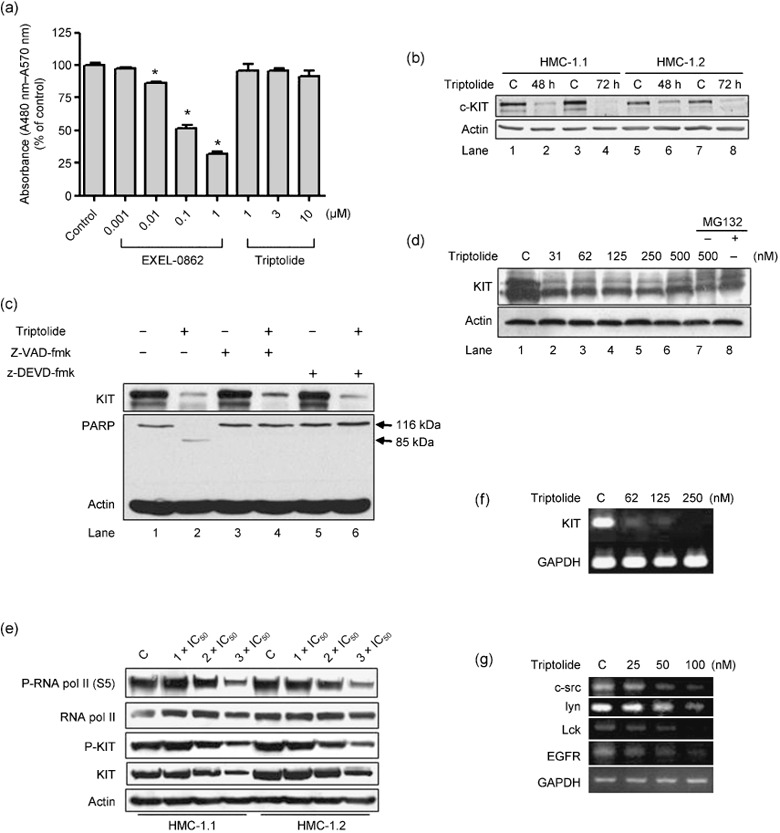
Triptolide inhibits KIT expression. (a) Triptolide did not directly exert inhibitory effect against KIT kinase activity. KIT kinase activity was measured by ELISA assay in the absence or presence of increasing concentrations of EXEL‐0862 (positive control) and triptolide. C denoted control of DMSO containing buffer (DMSO final concentration = 0.1%). Columns, mean; error bars, SE. *P < 0.001, Student's test, n = 4. (b) Triptolide downregulates the level of KIT protein. Western blot analysis of HMC‐1.1 and HMC‐1.2 cells exposed to 250 nM triptolide for 48 or 72 h. Expression was normalized to that of actin. (c) Triptolide downregulates KIT protein level independently of caspase activity. Western blot analysis of HMC‐1.1 cells exposed to 250 nM triptolide in the presence (+) or absence (–) of 20 µM z‐VAD‐fmk (pan‐caspase inhibitor) or 10 µM z‐DEVD‐fmk (caspase‐3 inhibitor) for 24 h. Anti‐KIT, poly(adenosine diphosphate [ADP]‐ribose) polymerase (PARP), and actin levels are shown. (d) Inhibition of proteosome by MG132 does not reverse the inhibitory action of triptolide in HMC‐1.1 cells. Western blot analysis of KIT expression in HMC‐1.1 cells treated with triptolide in the presence or absence of MG132 (1.0 µM) for 24 h (e) Triptolide led to a simultaneous decrease in RNA polymerase activity and KIT protein level. Exposure of HMC‐1.1 and HMC‐1.2 cells to triptolide at concentrations ranged 1‐ to 3‐fold IC50 for 72 h, Western blot analysis was conducted with the specific antibodies indicated. (f) and (g) Triptolide decreases mRNA level of KIT, c‐Src, Lck, Lyn and epidermal growth factor receptor (EGFR). Semi‐quantitative RT‐PCR analysis of mRNA level of KIT in HMC‐1.1 cells (f), and c‐Src, Lyn and EGFR in KBM5 cells (g), which were treated with triptolide for 24 h. Ethidium bromide‐stained gel of the PCR products.
Treatment for 48 or 72 h with triptolide at 250 nM led to a substantial decrease in KIT protein level in HMC‐1.2 as well as HMC‐1.1 cells (Fig. 2b). Interestingly, HMC‐1.1 cells treated with triptolide in the absence or presence of z‐VAD‐fmk (a pan‐caspase inhibitor) or z‐DEVD‐fmk (a caspase‐3 inhibitor) showed no effect of either caspase inhibitor on reversing the triptolide‐mediated decline in KIT level (Fig. 2c). To investigate whether the proteosome pathway was involved in the triptolide‐mediated downregulation of KIT, we pretreated HMC‐1.1 cells with the proteosome inhibitor MG‐132 (1.0 µM) but found no effect on decrease in KIT protein level (Fig. 2d; lane 1 vs. lanes 7 and 8). In contrast, the MG‐132 exhibited an activity to reverse the degradation of Mcl‐1 medicated by small molecule compound EXEL‐0862.( 32 )
Because triptolide was recently reported to induce global transcriptional arrest by inhibiting RNA polymerase in Hela cells,( 27 ) we explored whether triptolide impacted KIT via this mechanism. In deed, 72‐h exposure of HMC‐1.1 and HMC‐1.2 cells to triptolide at concentrations ranged 1‐ to 3‐fold IC50 led to a decline in the phosphorylation of RNA polymerase II in a dose‐dependent manner (Fig. 2e). Concomitantly, a dose‐dependent decline in the levels of KIT and phosphorylated KIT were observed (Fig. 2e). We treated HMC‐1.1 cells with increasing concentrations of triptolide for 24 h; semi‐quantitative RT‐PCR revealed the mRNA level of KIT remarkably diminished (Fig. 2f). The results indicated that triptolide might inhibit KIT at the transcriptional level. Additionally, triptolide decreased the mRNA levels of c‐Src, Lck, Lyn, and epidermal growth factor receptor (EGFR) (Fig. 2g). Taken together, the decrease in expression of KIT, Bcr‐Abl, c‐Src, Lck, lyn, and EGFR was in agreement with inhibition of RNA polymerase II activity by triptolide.( 27 )
Triptolide inhibits activation of KIT downstream targets. Presumably, the loss of total KIT protein would render a reduction in active autophosphorylated KIT leading to subsequent abrogation of downstream signal transduction. Western blot analysis revealed that triptolide decreased the phosphorylation of KIT in human mast cells (Fig. 3a) and murine P815 cells (Fig. 3b). The levels of phosphorylated and total Stat3, Akt, and Erk1/2 were also substantially reduced, whereas that of total JNK was not changed (Fig. 3a). Therefore, in human mast cells, triptolide inhibits the expression of KIT and Stat3, key regulators of cell growth in mast cells.
Figure 3.
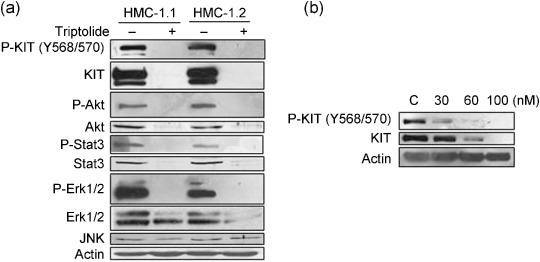
Triptolide inhibits activation of KIT downstream targets. (a) Triptolide reduces the level of phosphorylated and total KIT, Akt, Stat3 and Erk1/2 in human mast cells. Western blot analysis of HMC‐1.1 and HMC‐1.2 cells treated with 250 nM triptolide for 48 h (b) Triptolide reduces phosphorylation of KIT in murine D814Y KIT‐expressing cells. Western blot analysis of P815 cells exposed to triptolide at 30–100 nM for 24 h.
Triptolide induces apoptosis in mast cells harboring D816V KIT. To further define the anti‐tumor activity of triptolide, we evaluated the apoptotic response by annexin V‐PI staining. Flow cytometry of P815 cells exposed to escalating concentrations of triptolide for 24 h revealed a marked concentration‐dependent increase in proportion of annexin V‐positive cells, from 8% to 85% (control vs. 100 nM, Fig. 4a). Further, 100 nM triptolide induced a time‐dependent increase in cleavage of PARP, a specific marker of apoptosis, in P815 cells (Fig. 4b). We next examined whether triptolide induces apoptosis in human mast cells expressing D816V KIT. Triptolide treatment for 24 h induced marked apoptosis in HMC‐1.2 and HMC‐1.1 cells (Fig. 4c), both cell lines exhibiting a plateau in apoptosis at triptolide concentrations ≥62 nM. However, apoptotic cell death was approximately 30% in HMC‐1.2 cells and 60% in HMC‐1.1 cells with 24‐h triptolide treatment (64–1000 nM) (Fig. 4c). Little is known about the molecular basis of this difference because the degree of KIT inhibition by triptolide was similar in these cells (Fig. 2b,e). Taken together, these data suggest that triptolide induces cell death by apoptosis in mast cells harboring imatinib‐resistant D816V KIT mutation.
Figure 4.
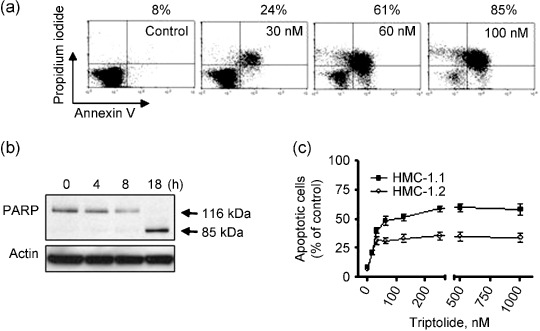
Triptolide induces apoptosis in mast cells expressing D816V KIT. (a) Triptolide induces dose‐dependent apoptosis in P815 cells. P815 cells were exposed to triptolide (30–100 nM) for 24 h, then underwent annexin V–propidium iodide (PI) double staining for flow cytometry. The percentage of annexin V‐positive cells shown above the panels are representative results of three independent experiments. (b) Triptolide induces poly(adenosine diphosphate [ADP]‐ribose) polymerase (PARP) cleavage in a time‐dependent manner. Western blot analysis of PARP level in P815 cells treated with 100 nM triptolide for 4, 8, and 18 h. (c) Apoptotic cell death measured by flow cytometry with annexin V‐PI double staining in human mast cells. HMC‐1.1 and HMC‐1.2 cells were exposed to triptolide at increasing concentrations for 24 h. Points, mean; error bars, SE, n = 3.
Triptolide downregulates Mcl‐1 and XIAP, upregulates Bax and damages mitochondria. We next analyzed the mechanism of the apoptotic response with triptolide exposure. Triptolide was reported to induce apoptosis via the mitochondrial pathway in AML cells.( 36 ) We thus first evaluated mitochondrial transmembrane potential (ΔΨm ) on flow cytometry by CMXRos and MTGreen double staining. As shown in Fig. 5(a) (the first panel), region II represents the cell population losing ΔΨm , whereas region I represents the cell population with normal ΔΨm. P815 cells exposed to escalating concentrations of triptolide showed a substantial concentration‐dependent increase in proportion in region II. In particular, 100 nM triptolide induced 90% of the treated cells to depolarize (region II), as compared with control cells. This result is in agreement with the proportion of annexin V‐positive cells induced by triptolide. 250 nM triptolide significantly increased the proportion of depolarized (region II) human mast HMC‐1.1 and HMC‐1.2 cells (Fig. 5b). Again, HMC‐1.2 cells were less sensitive to triptolide than HMC‐1.1 and P815 cells.
Figure 5.
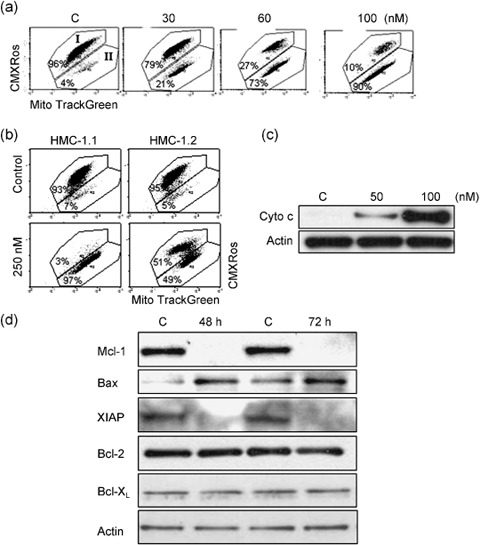
Triptolide induces mitochondrial damage, cytochrome c release, and alteration in level of several apoptosis‐related proteins. (a, b) Mitochondrial damage was elicited by triptolide. (a) P815 cells were exposed to increasing concentrations (30–100 nM) of triptolide and stained with chloromethyl‐X‐rosamine (CMXRos) and MitoTracker Green (MTGreen) for flow cytometry. Region II on the histograms represents the population of cells with mitochondrial depolarization. Representative results from three independent experiments are shown. (b) HMC‐1.1 and HMC‐1.2 cells were exposed to 250 nM triptolide for 72 h, then mitochondrial potential was measured. (c) Triptolide treatment induced cytochrome c release into the cytosol. P815 cells were treated with 50 nM or 100 nM for 24 h. Cytochrome c was detected by immunoblotting, the cytosolic fraction extracted with digitonin buffer. (d) Triptolide decreases level of Mcl‐1 and XIAP while increasing that of Bax, apoptosis‐related proteins. Western blot analysis of HMC‐1.1 cells treated with 250 nM triptolide for 48 or 72 h.
Next, we compared the release of cytochrome c from mitochondria into cytosol in cells treated with or without triptolide. P815 cells were exposed to 50 and 100 nM triptolide for 24 h, cytochrome c in the cytosolic fraction was monitored by Western blot analysis with anti‐cytochrome c antibody. Cytochrome c was undetectable in the cytosol of untreated cells but was greatly increased in level in triptolide‐treated cells (Fig. 5c). Similar results were obtained in HMC‐1.1 and HMC‐1.2 cells (data not shown). Collectively, ΔΨm alteration and release of cytochrome c indicate that triptolide triggers apoptosis through the mitochondrial pathway.
To examine the mediators involved in the mitochondrial apoptosis pathway, we examined apoptosis regulators by Western blot analysis and found a remarkable decline in level of anti‐apoptotic proteins XIAP and Mcl‐1 but increase in level of the pro‐apoptotic protein Bax in triptolide‐treated HMC‐1.1 cells (Fig. 5d), with no alteration in the protein level of Bcl‐2.
Triptolide induces G1 arrest in D816V KIT‐expressing cells. Since HMC‐1.2 cells were less sensitive to triptolide‐mediated apoptosis, we examined the effect of triptolide on the cell cycle of HMC‐1.2 cells. The human mast cells were exposed to triptolide for various times, and the cell cycle was assayed by flow cytometry with PI staining. With triptolide treatment, an increased proportion of HMC‐1.2 cells were in G1 phase accumulation, whereas an increased proportion of HMC‐1.1 cells were in the sub‐G1 phase indicating the occurrence of apoptosis (Fig. 6a). Western blot analysis revealed increased level of p53 and and p27Kip1 in HMC‐1.2 cells but not in HMC‐1.1 cells after treatment with 250 nM triptolide (Fig. 6b). However, levels of cyclin D, cyclin A and CDK4, important G1‐phase progression‐driving forces, were not changed in triptolide‐arrested HMC‐1.2 cells (data not shown). These data suggest that p53 and p27Kip1, two critical negative regulators of cell cycling, may mediate the G1 arrest induced by triptolide.
Figure 6.
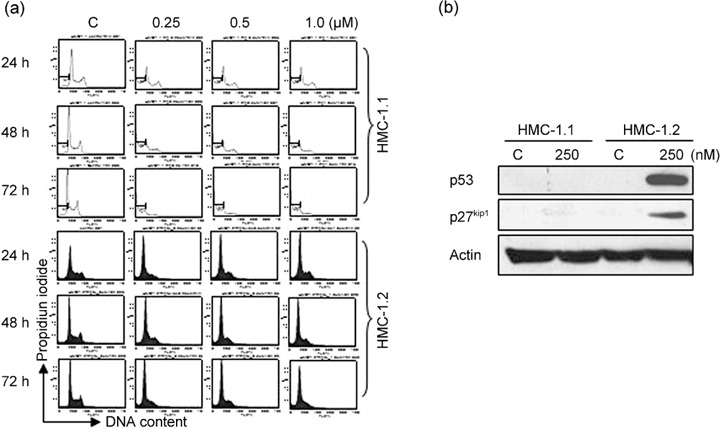
Triptolide induces G1 arrest in human mast cells. (a) HMC‐1.2 cells were exposed to 250 nM triptolide for 24∼72 h, then fixed in ethanol and stained with propidium iodide. Cell cycling was analyzed by flow cytometry. (b) Immunoblot analysis of P27Kip1 and p53 level in HMC‐1.2 cells exposed to 250 nM triptolide for 48 h.
Triptolide potently abrogates the growth of human mast cells carrying D816V KIT in xenografts. We evaluated the effect of triptolide on the growth of tumor xenografts of HMC‐1.2 cells, which are resistant to imatinib, in nude mice. We determined that a dose of 0.15 mg/kg/day i.p. injection of triptolide was tolerable to nude mice in our preliminary studies and from the literature.( 35 ) Five days after mice were inoculated with HMC‐1.2 cells, when the size of tumor reached approximately 50 mm3, mice were randomized to receive treatment with DMSO containing media (control) or triptolide, 0.15 mg/kg/day, for 21 days (10 animals in each group). The growth curves (estimated tumor size calculated from the tumor dimension vs. time) are shown in Fig. 7(a). Triptolide remarkably abrogated the growth of tumors. On day 21, the tumor xenografts were dissected and weighed. The weights of the tumors were significantly decreased in triptolide‐treated mice (Fig. 7c; P < 0.0001, n = 10). The body weight of the mice remained stable, without significant differences between treated and control mice (data not shown). Motor activity and feeding behavior of all mice were normal. Whole blood cell counts did not reveal any significant myelosuppression in treated mice (Table 2). Liver enzyme analysis revealed no increase in aspartate and alanine aminotransferase activity in the treatment group (data not shown). No mice died. Overall, surveillance of morbidity and mortality revealed no significant toxic effects of triptolide at the dosage used.
Figure 7.
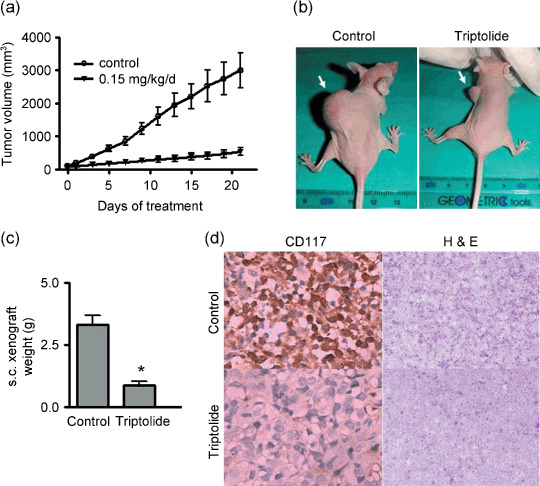
Triptolide potently abrogates the growth of cells and decreases KIT expression in xenografts of human mast cells carrying D816V KIT. BALB/c nu/nu nude mice bearing s.c.‐innoculated HMC‐1.2 xenografts were randomized into two groups (10 animals each) for treatment with DMSO containing medium (control) or triptolide 0.15 mg/kg/day. The tumor growth curves are plotted (a). The vertical axis represents the tumor size, and the horizontal axis represents the number of days since triptolide treatment began. Error bars, SE. Images for one representative mouse from each group are shown (b). On day 21, tumor xenografts in mice were dissected and measured. The bar chart (c) shows the weight of the tumors from each group (n = 10). Error bars, SE. *P < 0.0001 by Student's t‐test. (d) The expression of KIT was greatly inhibited by triptolide. Immnunohistochemical analysis with anti‐CD117 antibody (KIT) and H&E in xenograft tissues from mice on day 21 after triptolide treatment.
Table 2.
Count of whole blood cells with triptolide treatment in nude mice
| Blood cell type | Control treatment (n = 9) | Triptolide treatment (0.15 mg/kg/day, for 21 days) (n = 9) | P |
|---|---|---|---|
| Erythrocytes (× 1012/L) | 9.792 ± 0.2002 | 9.664 ± 0.221 | 0.169 |
| Leukocytes (× 109/L) | 14.16 ± 0.2187 | 14.24 ± 0.216 | 0.732 |
| Platelet (× 109/L) | 193.2 ± 2.687 | 189.1 ± 1.798 | 0.284 |
Values are mean ± SE.
To gain insight into the underlying mechanism by which triptolide inhibits the growth of mast cells in vivo, we examined the level of KIT in HMC‐1.2 xenograft tumor tissues from mice treated with DMSO (control) or triptolide. Immunohistochemistry revealed the expression of KIT much lower in tumor tissue sections of mice treated with triptolide than those mice treated with DMSO (control) (Fig. 7d). The in vivo data strengthened our in vitro findings and suggested that triptolide may exert anti‐SM activity via decreasing KIT level in vivo.
Discussion
This study investigated the activity of triptolide in the mast cell lines bearing D816V KIT in vitro and in xenografts of nude mice in vivo to determine whether this compound may have potential benefit for patients with SM. Triptolide at nanomolar concentrations potently downregulated the expression of KIT, inhibited cell growth and induced apoptosis in mast cells bearing D816V and D816Y KIT mutant isoforms, which are resistant to imatinib.
The expression of KIT was substantially downregulated in both cultured cells and xenograft tissues following treatment with triptolide. The downregulation of KIT by triptolide did not depend on activation of caspase and the proteosome pathway. Triptolide potently inhibited transcription of KIT and related kinases including c‐Src, Lck, lyn, EGFR (Fig. 2g), Bcr‐Abl, 38 the underlying mechanism may be associated with its inhibiting global transcription via inhibiting RNA polymerase, which is supported by our findings (Fig. 2e) and that of others.( 27 )
KIT mutation in SM is one of the typical models of oncogene addiction. In other words, SM is highly dependent on the constitutive activation of KIT kinase. Although triptolide may represent an inhibitor of global transcription, the oncogene addiction of these SM cells to KIT may allow triptolide to provide a therapeutic window. The loss of total KIT protein would result in reduced active autophosphorylated KIT, which leads to subsequent abrogation of downstream signal transduction.( 15 ) We and others have confirmed this notion.( 15 ) A decrease in level of multiple targets may confer growth retardation and apoptosis of mast cells. Both the juxtamembrane V560G and kinase domain D816V mutations give rise to constitutively activated KIT isoforms that preferentially activate several downstream signal pathways, including Stat3‐Stat5, PI3 K‐Akt, and Ras‐Raf‐Erk1/2. These key regulators play important roles in the carcinogenesis caused by constitutively active KIT mutations. We showed that triptolide treatment decreased the level of Stat3, Akt, as well as Erk1/2.
The responses of different mast cell lines to triptolide differed, although the degree of KIT inhibition was similar. With triptolide treatment, HMC‐1.1 and P815 cells underwent massive cell death by apoptosis, while HMC‐1.2 cells exhibited the reduced ability to undergo apoptosis, in which similar response was also seen after treatment with tyrosine kinase inhibitors.( 9 , 13 ) HMC‐1.2 cells underwent an obvious G1 arrest associated with increased p53 and p27 level. The G1 arrest inhibits tumor proliferation but allows the arrested malignant cells to survive chemotherapy. This may partially help explain why HMC‐1.2 cells exhibit a lower apoptotic cell death rate than HMC‐1.1 cells upon triptolide treatment, even though HMC‐1.2 cells had a lower IC50 (MTS assay). Future work should elucidate if additional targets could explain the higher sensitivity of HMC‐1.2 cells against triptolide when measured in terms of cell viability. Nevertheless, triptolide significantly inhibited the growth of HMC‐1.2 cells in xenografts in nude mice (Fig. 7).
Our data clearly demonstrated that triptolide activated the mitochondrial apoptotic pathway, as reflected by the loss of ΔΨm and the release of cytochrome c into the cytoplasm in triptolide‐treated mast cells. However, how inhibition of transcription by triptolide can trigger the intrinsic mitochondrial apoptotic pathway is still unclear. It may be related to the pronounced decline in antiapoptotic proteins such as Mcl‐1 and XIAP and upregulation of a proapoptotic protein Bax. Targeting Mcl‐1 by antisense oligonucleotides or small interfering RNA has been shown to induce apoptosis in D816V KIT cells and increase sensitivity to tyrosine kinase inhibitors, including PKC412, AMN107, and imatinib.( 37 )
In summary, our in vitro data in mast cells and in vivo data in mouse xenografts demonstrate that triptolide can effectively inhibit the growth of cells and induce apoptosis in cells bearing both juxtamembrane and activation loop mutants of KIT, including the imatinib‐resistant D816V KIT, involved in SM. The mechanism of action may involve the inhibition of the transcription of KIT and antiapoptotic proteins such as Mcl‐1 and XIAP. Our data warrant further investigation of triptolide derivatives as potential therapeutic agents against human neoplasms driven by gain‐of‐function KIT mutations. A phase I clinical trial of the effect of a water‐soluble derivative of triptolide on solid tumors is ongoing in Europe.( 36 ) A clinical trial of triptolide derivatives such as PG490–88 in malignant disease involved in imatinib‐resistant D816V KIT may be justified.
Authorship Contribution
Y.J. – designed, performed experiments and wrote the manuscript; Q.C., X.S., Z.L., C.C., Y.L, and Q.Z. – performed experiments; J.P. – designed, performed research, analyzed data and wrote the manuscript.
Acknowledgments
This study was supported by grants from the National High Technology Research and Development Program of China (863 Program grant 2007AA02Z490 to J.P.), the Major Research Plan of the National Natural Science Fund of China (grant 90713036 to J.P.), National Basic Research Program of China (973 Program grant 2009CB825506 to J.P.). The authors thank Dr Sai‐Ching Jim Yeung (The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA) for a critical reading of the manuscript.
References
- 1. Tefferi A, Pardanani A. Systemic mastocytosis: current concepts and treatment advances. Curr Hematol Rep 2004; 3: 197–202. [PubMed] [Google Scholar]
- 2. Corbin AS, Demehri S, Griswold IJ et al . In vitro and in vivo activity of ATP‐based kinase inhibitors AP23464 and AP23848 against activation‐loop mutants of Kit. Blood 2005; 106: 227–34. [DOI] [PubMed] [Google Scholar]
- 3. Quintas‐Cardama A, Aribi A, Cortes J, Giles FJ, Kantarjian H, Verstovsek S. Novel approaches in the treatment of systemic mastocytosis. Cancer 2006; 107: 1429–39. [DOI] [PubMed] [Google Scholar]
- 4. Pardanani A. Systemic mastocytosis: bone marrow pathology, classification, and current therapies. Acta Haematol 2005; 114: 41–51. [DOI] [PubMed] [Google Scholar]
- 5. Valent P, Akin C, Sperr WR et al . Mastocytosis: pathology, genetics, and current options for therapy. Leuk Lymphoma 2005; 46: 35–48. [DOI] [PubMed] [Google Scholar]
- 6. Hirota S, Isozaki K, Moriyama Y et al . Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–80. [DOI] [PubMed] [Google Scholar]
- 7. Akin C, Brockow K, D’Ambrosio C et al . Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild‐type or mutated c‐kit. Exp Hematol 2003; 31: 686–92. [DOI] [PubMed] [Google Scholar]
- 8. Furitsu T, Tsujimura T, Tono T et al . Identification of mutations in the coding sequence of the proto‐oncogene c‐kit in a human mast cell leukemia cell line causing ligand‐independent activation of c‐kit product. J Clin Invest 1993; 92: 1736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gleixner KV, Mayerhofer M, Aichberger KJ et al . PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V‐mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006; 107: 752–9. [DOI] [PubMed] [Google Scholar]
- 10. Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild‐type c‐kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther 2002; 1: 1115–24. [PubMed] [Google Scholar]
- 11. Ma Y, Zeng S, Metcalfe DD et al . The c‐KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild‐type kinases and those with regulatory‐type mutations. Blood 2002; 99: 1741–4. [DOI] [PubMed] [Google Scholar]
- 12. Longley BJ Jr, Metcalfe DD, Tharp M et al . Activating and dominant inactivating c‐KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA 1999; 96: 1609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah NP, Lee FY, Luo R, Jiang Y, Donker M, Akin C. Dasatinib (BMS‐354825) inhibits KITD816V, an imatinib‐resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood 2006; 108: 286–91. [DOI] [PubMed] [Google Scholar]
- 14. Fletcher JA, Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev 2007; 17: 3–7. [DOI] [PubMed] [Google Scholar]
- 15. Sambol EB, Ambrosini G, Geha RC et al . Flavopiridol targets c‐KIT transcription and induces apoptosis in gastrointestinal stromal tumor cells. Cancer Res 2006; 66: 5858–66. [DOI] [PubMed] [Google Scholar]
- 16. Morris DG, Bramwell VH, Turcotte R et al . A phase II study of Flavopiridol in patients with previously untreated advanced soft tissue sarcoma. Sarcoma 2006, 2006: 64374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell 2007; 130: 769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tao XL, Sun Y, Dong Y et al . A prospective, controlled, double‐blind, cross‐over study of tripterygium wilfodii hook F in treatment of rheumatoid arthritis. Chin Med J (Engl) 1989; 102: 327–32. [PubMed] [Google Scholar]
- 19. Jiang X. Clinical observations on the use of the Chinese herb Tripterygium wilfordii Hook for the treatment of nephrotic syndrome. Pediatr Nephrol 1994; 8: 343–4. [DOI] [PubMed] [Google Scholar]
- 20. Kupchan SM, Court WA, Dailey RG Jr, Gilmore CJ, Bryan RF. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc 1972; 94: 7194–5. [DOI] [PubMed] [Google Scholar]
- 21. Tao X, Schulze‐Koops H, Ma L, Cai J, Mao Y, Lipsky PE. Effects of Tripterygium wilfordii hook F extracts on induction of cyclooxygenase 2 activity and prostaglandin E2 production. Arthritis Rheum 1998; 41: 130–8. [DOI] [PubMed] [Google Scholar]
- 22. Jiang XH, Wong BC, Lin MC et al . Functional p53 is required for triptolide‐induced apoptosis and AP‐1 and nuclear factor‐kappaB activation in gastric cancer cells. Oncogene 2001; 20: 8009–18. [DOI] [PubMed] [Google Scholar]
- 23. Fidler JM, Li K, Chung C et al . PG490–88, a derivative of triptolide, causes tumor regression and sensitizes tumors to chemotherapy. Mol Cancer Ther 2003; 2: 855–62. [PubMed] [Google Scholar]
- 24. Efferth T, Li PC, Konkimalla VS, Kaina B. From traditional Chinese medicine to rational cancer therapy. Trends Mol Med 2007; 13: 353–61. [DOI] [PubMed] [Google Scholar]
- 25. Tao X, Lipsky PE. The Chinese anti‐inflammatory and immunosuppressive herbal remedy Tripterygium wilfordii Hook F. Rheum Dis Clin North Am 2000; 26: 29–50. [DOI] [PubMed] [Google Scholar]
- 26. Qiu D, Kao PN. Immunosuppressive and anti‐inflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. F Drugs R D 2003; 4: 1–18. [DOI] [PubMed] [Google Scholar]
- 27. Leuenroth SJ, Crews CM. Triptolide‐induced transcriptional arrest is associated with changes in nuclear substructure. Cancer Res 2008; 68: 5257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res 1988; 12: 345–55. [DOI] [PubMed] [Google Scholar]
- 29. Tsujimura T, Furitsu T, Morimoto M et al . Ligand‐independent activation of c‐kit receptor tyrosine kinase in a murine mastocytoma cell line P‐815 generated by a point mutation. Blood 1994; 83: 2619–26. [PubMed] [Google Scholar]
- 30. Dong F, Guo W, Zhang L et al . Downregulation of XIAP and induction of apoptosis by the synthetic cyclin‐dependent kinase inhibitor GW8510 in non‐small cell lung cancer cells. Cancer Biol Ther 2006; 5: 165–70. [DOI] [PubMed] [Google Scholar]
- 31. Pan J, Quintas‐Cardama A, Kantarjian HM et al . EXEL‐0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. Blood 2007; 109: 315–22. [DOI] [PubMed] [Google Scholar]
- 32. Pan J, Quintas‐Cardama A, Manshouri T et al . The novel tyrosine kinase inhibitor EXEL‐0862 induces apoptosis in human FIP1L1‐PDGFR‐alpha‐expressing cells through caspase‐3‐mediated cleavage of Mcl‐1. Leukemia 2007; 21: 1395–404. [DOI] [PubMed] [Google Scholar]
- 33. Pan J, Xu G, Yeung SC. Cytochrome c release is upstream to activation of caspase‐9, caspase‐8, and caspase‐3 in the enhanced apoptosis of anaplastic thyroid cancer cells induced by manumycin and paclitaxel. J Clin Endocrinol Metab 2001; 86: 4731–40. [DOI] [PubMed] [Google Scholar]
- 34. Dackiw A, Pan J, Xu G, Yeung SC. Modulation of parathyroid hormone‐related protein levels (PTHrP) in anaplastic thyroid cancer. Surgery 2005; 138: 456–63. [DOI] [PubMed] [Google Scholar]
- 35. Yang S, Chen J, Guo Z et al . Triptolide inhibits the growth and metastasis of solid tumors. Mol Cancer Ther 2003; 2: 65–72. [PubMed] [Google Scholar]
- 36. Carter BZ, Mak DH, Schober WD et al . Triptolide induces caspase‐dependent cell death mediated via the mitochondrial pathway in leukemic cells. Blood 2006; 108: 630–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aichberger KJ, Mayerhofer M, Gleixner KV et al . Identification of MCL1 as a novel target in neoplastic mast cells in systemic mastocytosis: inhibition of mast cell survival by MCL1 antisense oligonucleotides and synergism with PKC412. Blood 2007; 109: 3031–41. [DOI] [PubMed] [Google Scholar]
- 38. Shi X, Jin Y, Cheng C et al . Triptolide inhibits Bcr‐AbI transcription and induces apoptosis in STI571‐resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res 2009; 15: 1686–97. [DOI] [PubMed] [Google Scholar]