

Abstract
Pancreatic adenocarcinoma remains a fatal disease characterized by rapid tumor progression, high metastatic potential and profound chemoresistance. Gemcitabine is the current standard chemotherapy for advanced pancreatic cancer, but it is still far from optimal and novel therapeutic strategies are needed urgently. Mutations in the k‐ras gene have been found in more than 90% of pancreatic cancers and are believed to play a key role in this malignancy. Thus, the goal of this study was to investigate the impact of k‐ras oncogene silencing on pancreatic tumor growth. Additionally, we examined whether combining k‐ras small interfering RNA (siRNA) with gemcitabine has therapeutic potential for pancreatic cancer. The treatment of tumor cell cultures with the corresponding k‐ras siRNA resulted in a significant inhibition of k‐ras endogenous expression and cell proliferation. In vivo, tumor xenografts were significantly reduced with k‐ras siRNAGAT delivered by electroporation. Moreover, combined treatment with pSsik‐rasGAT plus gemcitabine resulted in strong growth inhibition of orthotopic pancreatic tumors. Survival rate was significantly prolonged and the mean tumor volume was dramatically reduced in mice receiving the combined treatment compared with single agents. Collectively, these findings show that targeting mutant k‐ras through specific siRNA might be effective for k‐ras oncogene silencing and tumor growth inhibition. The improvement of gemcitabine‐based chemotherapy suggests that this strategy might be used therapeutically against human pancreatic cancer to potentiate the effects of conventional therapy. (Cancer Sci 2007; 98: 1128–1136)
Worldwide, the incidence of pancreatic carcinoma has increased steadily and it remains one of the major public health problems. Although surgery offers a low cure rate, it is also the only chance for cure. Unfortunately, the diagnosis of ductal pancreatic adenocarcinoma often occurs very late and consequently less than 20% of patients are candidates for tumor resection.( 1 , 2 ) Contraindications for resective surgery are the presence of liver metastases or distant metastases, peritoneal seeding, circular tumor infiltration into mesenteric vessel walls, and locoregional extension of the tumor into the mesentery. Conventional chemotherapy has shown only a minimal survival benefit when combined with surgical resection.( 3 , 4 ) Thus, only 1–3% of all patients diagnosed with pancreatic cancer can expect to survive 5 years.( 2 , 5 ) There is new hope that selective anticancer drugs, with less cytotoxic side effects than conventional cancer chemotherapy, will be developed.
Twenty years ago, the discovery of antisense technology generated considerable enthusiasm in the research and medical community. The principle of this technology is the sequence‐specific binding of an antisense oligonucleotide to target mRNA, resulting in the prevention of gene translation. Because most tumor cells have a different pattern of gene expression in comparison with normal cells, antisense oligonucleotides can theoretically be used to specifically target tumor‐associated genes, or mutated genes, without altering gene expression in normal cells.
Pancreatic carcinoma arises through the accumulation of inherited and acquired genetic alterations.( 6 , 7 ) It is well established that k‐ras mutations are one of the most common types of genetic abnormality in pancreatic adenocarcinomas. Point mutation in the k‐ras gene occurs in 70–90% of cases and the vast majority occur at codon 12 of the oncogene.( 8 , 9 ) Consequently, a considerable research effort has been made to define the function of Ras in normal and neoplastic cells and to target Ras for cancer treatment. Various strategies have been developed to target k‐ras for the treatment of human cancers. These strategies have ranged from inhibiting protein expression via antisense oligonucleotides to blocking post‐translational modification with farnesyltransferase inhibitors (FTI), to inhibiting downstream effectors. In the field of antitumor drug development, antisense oligodeoxynucleotides (ODN) have been studied extensively as a tool to investigate gene function and the identification of potential drug targets.( 10 ) ODN are short (typically 15–17 bases) stretches of synthetic DNA that are complementary to specific regions of cellular mRNA or DNA. Several antisense oligonucleotides have now entered clinical trials in patients with a number of malignancies.( 11 , 12 ) More recently, the discovery of RNA interference (RNAi) started a new era in antisense technology and has offered an additional way to inhibit or knockdown the expression of virtually any gene.( 13 ) RNAi is a process of sequence‐specific, post‐transcriptional gene silencing initiated by double‐stranded RNA (dsRNA) homologous in sequence to the targeted gene. RNAi has been used extensively to determine gene function in a number of organisms, including plants,( 14 ) nematodes( 15 ) and Drosophila.( 16 ) Although the exact mechanism is not fully understood, it was demonstrated that RNAi is a multistep process involving the generation of small interfering RNA (siRNA) through the action of the RNaseIII endonuclease Dicer. The resulting 21–23‐nucleotide (nt) siRNA mediate degradation of their complementary RNA. The attractiveness of RNAi arises from its extremely high inhibitory activity. RNAi has rapidly become a powerful tool for drug target discovery. Consequently, interest is growing in the extension of its application to in vivo systems, such as animal disease models and human therapeutics.
In order to overcome the transient knockdown activity of short oligonucleotides, various vector‐based RNAi have been developed as an alternative. These vector systems use RNA polymerase III promoters, such as U6 and H1, to drive the expression of short hairpin RNA (shRNA), which are very similar to siRNA.( 17 , 18 )
In light of the developments in creating a new anticancer pharmacology based on a k‐ras gene silencing strategy, the primary objective of the present study was to investigate the efficacy of k‐ras siRNA compared with antisense phosphorothioate oligonucleotide (PS‐ODN) specific to the mutated k‐ras oncogene for pancreatic adenocarcinoma treatment.
The cytosine arabinoside analog 2′,2′‐difluorodeoxycytidine (gemcitabine) has been proven to be active in the treatment of this disease,( 19 ) with significant clinical benefit but still with marginal survival advantage.( 20 ) Usually it is well tolerated, although significant toxicities have been reported. Therefore, gemcitabine has become the standard first‐line chemotherapeutic agent for advanced and metastatic pancreatic cancer. Due to the complexity of this disease and the difficulties in treating and curing it, most oncologists are choosing to use combined treatment therapies. In an effort to improve therapeutic efficacy, numerous clinical studies have investigated gemcitabine‐based combination regimens.( 21 , 22 , 23 ) Thus, our secondary objective in the present study was to examine the potential of the combination of k‐ras siRNA together with gemcitabine.
Materials and Methods
Pancreatic tumor cell lines and cell culture conditions. The pancreatic carcinoma cell lines BxPC3 (k‐raswt,Gly), Panc1 (k‐rasasp12) and Capan1 (k‐rasva112) were obtained from American Type Culture Collection (LGC Promochem, France). BxPC3 and Panc‐1/Capan‐1 were cultured in Dulbecco's modified Eagle's medium (DMEM) and RPMI‐1640, respectively, supplemented with 10% (v/v) heat‐inactivated fetal calf serum (FCS), 2 mM glutamine, 100 units/mL penicillin and 100 µg/mL streptomycin. The cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C. All cell culture media and transfection reagent (lipofectamine 2000) were purchased from Invitrogen (Cergy‐Pontoise, France). The bis(guanidinium)‐tren‐cholesterol/dioleoyl phosphatidylethanolamine (BGTC/DOPE) cationic liposomes were obtained from Professor Jean Marie Lehn (Laboratoire de Chimie Supramoléculaire ISIS, Strasbourg, France).
Antisense ODN, synthetic siRNA and siRNA vectors. The antisense (AS) PS‐ODN and the synthetic short dsRNA oligonucleotides for siRNA targeting the different k‐ras mutation at codon 12 (named sik‐ras) corresponded to the region 196–213 (adjacent to the start codon). The k‐ras siRNA were designed based on studies by Elbashir et al.,( 24 ) and their lack of significant interfering homology was validated using BLAST analysis. They were obtained as ready‐annealed, purified duplexes from MWG‐Biotech (Courtaboeuf, France). The control ‘scrambled’ sequences, and the AS PS‐ODN and siRNA k‐ras‐targeted sequences are shown in Table 1.
Table 1.
Construction of small interfering RNA expression plasmids
| K‐ras‐annealed siRNA | K‐ras antisense phosphorothioate oligonucleotides | |
|---|---|---|
| BxPc3 (krasGGT) | 5′‐GUUGGAGCUGGUGGCGUAG‐3′ TT 5′‐CUACGCCACCAGCUCCAAC‐3′ TT | 5′‐CTACGCCACCAG CTCCA‐3′ |
| Panc1 (krasGAT) | 5′‐GUUGGAGCUGAUGGCGUAG‐3′ TT 5′‐CUACGCCAUCAGCUCCAAC‐3′ TT | 5′‐CTACGCCATCAGCTCCA‐3′ |
| Capan1 (krasGTT) | 5′‐GUUGGAGCUGUUGGCGUAG‐3′ TT 5′‐CUACGCCAACAGCUCCAAC‐3′ TT | 5′‐CTACGCCAACAGCTCCA‐3′ |
| Scrambled sequence | 5′‐CGAAGUGUGUGUGUGUGGC‐3′ TT 5′‐GCCACACACACACACUUCG‐3′ TT | 5′‐ACTAGCTATACTAGCTAT‐3′ |
k‐ras mutation sites are indicated by bold type.
Downregulation of gene expression mediated by synthetic siRNA is transient, and usually lasts for only 2–4 days in cell culture. Viral vectors or plasmids can be used for more prolonged and stable expression of RNAi effector molecules. In the present study, pSUPER expression vectors were designed to generate active siRNA from the polymerase III H1 RNA gene promoter.( 17 ) The DNA oligonucleotide sense and antisense strands corresponding to the selected siRNA target site were annealed to form a short duplex with complementary ends to the linearized pSUPER vector. Briefly, in a preliminary annealing reaction (final volume 50 µL), 1 µL of complementary sense and antisense sequences (1 mg/mL) targeting the k‐ras mutation at codon 12 were incubated in the annealing buffer (100 mM NaCl, 50 mM HEPES pH 7.4) at 94°C for 10 min, and then at 70°C for 10 min. Thereafter, the annealed oligos were slowly cooled to 10°C and stored at 4°C (or at –20°C). In parallel, the pSUPER backbone vector was linearized with HindIII/BglII restriction enzymes and used for directional ligation of annealed oligos. To verify the successful insertion of annealed sequences, the ligation reaction products were transformed into DH5α competent bacteria. Several positive colonies were obtained, amplified in Luria Bertani (LB)‐ampicillin‐containing medium and recombinant plasmids were isolated using a mini‐prep kit (Qiagen, Courtaboeuf, France). After verification, the recombinant plasmid DNA expressing k‐ras siRNA (pSsiK‐ras) were prepared using the EndoFree Plasmid Maxi Kit (Qiagen) according to the manufacturer's instructions. An additional plasmid containing an irrelevant hairpin was similarly constructed to generate the pSsiSC control vector.
Transient transfection and gene silencing assays. The day before transfection, cells were transferred into 24‐well plates in appropriate culture medium containing 10% FCS, so that they would be at 70–80% confluence on the day of transfection. Transfection of PS‐ODN and siRNA for targeting endogenous k‐ras was carried out using lipofectamine 2000 according to the manufacturer's guidelines. Briefly, the growth medium was replaced with 200 µL of serum‐free minimum essential medium (MEM) containing 1 µM of AS PS‐ODN, 200 nM of synthetic siRNA duplex or 1 µg of pSsiK‐ras mixed with lipofectamine 2000, and incubated for 4 h at 37°C. Thereafter, 800 µL of fresh medium containing 10% FCS was added and incubation was continued for a further 2–3 days before harvesting the cells for MTT or [3H]thymidine incorporation tests.
For protein and RNA analysis, exponentially growing cells were transfected in 60‐mm Petri dishes with AS PS‐ODN (1 µM), sik‐ras duplex (200 nM) or pSsik‐ras (4 µg/dish) with oligofectamine 2000 according to the manufacturer's instructions. As a mock transfection, cells were exposed to lipofectamine 2000 alone in the absence of ODN or siRNA.
Semiquantitative reverse transcription–polymerase chain reaction analyses for k‐ras mRNA expression evaluation. Total RNA was isolated using TRIzol reagent (Invitrogen), and was subsequently treated with RNase‐free DNaseI to avoid contamination with plasmid or genomic DNA. For cDNA synthesis, 5 µg of purified total RNA was reverse transcribed for 1 h at 42°C using 200 U of SuperScript reverse transcriptase II (Invitrogen) and random hexamer primers (Promega, Charbonnières, France). The reverse transcription (RT) product (1–5 µL) was subjected to polymerase chain reaction (PCR) amplification. Each amplification was carried out twice using independent RT reactions to confirm the results and reproducibility. The primers used to amplify the k‐ras gene around codon 12 were: sense 5′‐GGCCTGCTGAAAATGACTGA‐3′ and antisense 5′‐TGATTCTGAATTAGCTGTAT‐3′. RT‐PCR analysis of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA levels was carried out as a control. GAPDH‐specific primers (sense, 5′‐ACCACAGTCCATGCCAT CAC‐3′; antisense, 5′‐TCCACCACCCTGTTGCTGTA‐3′) were used to PCR amplify a 450‐bp fragment. All of the primers were purchased from Invitrogen. PCR was carried out in a PT100 apparatus (MJ Research‐BioRad, Marnes‐la‐coquette, France) using 5 µL of RT reaction, 0.8 mM of each sense and antisense primer, 0.8 mM dNTP, 2 mM MgCl2 and 2 U of Taq DNA polymerase (final volume 50 µL). After an initial denaturation step of 4 min at 94°C, each PCR mixture was subjected to 35 cycles of amplification (denaturation, 30 s at 94°C; annealing, 30 s at 54°C; and 1 min at 72°C) followed by a final elongation step for 10 min at 72°C. Equal volumes of PCR products were analyzed on a 1.5–2% agarose gel containing ethidium bromide.
Western blot analysis. For protein expression analysis, cells were plated in six‐well plates or in 35‐mm dishes and treated as indicated above. At 48 h after transfection, cells were rinsed twice with ice‐cold phosphate‐buffered saline (PBS) to remove the medium, harvested and lysed with RIPA buffer (0.15 M NaCl, 1% Nonidet‐P40 (NP‐40), 0.01 M deoxycholate, 0.1% sodium dodecylsulfate (SDS), 0.05 M Tris‐HCl pH 8.0, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 10 µg/mL each of aprotinin, pepstatin and leupeptin). Subsequently, the whole‐cell extracts were cleared by centrifugation at 16 831 g , 4°C, and the protein concentration was determined using Bio‐Rad protein assay kit (Bio‐Rad, Marnes‐la‐Coquette, France).
Equal amounts of each protein sample (50 µg) were electrophoresed on 10% SDS–polyacrylamide gels for 90 min at 150 V and transferred onto polyvinylidene fluoride (PVDF) membranes (Amersham Biosciences, Saclay‐Orsay, France) for 60 min at 100 V in 10% methanol transfer buffer. The membranes were blocked with 5% non‐fat dried milk in Tris‐buffered saline pH 7.4 containing 0.1% Tween‐20 for 2 h at room temperature. The membranes were then probed with monoclonal primary antibody overnight at 4°C with shaking. Thereafter, the washed membranes were incubated for 1 h with peroxidase‐conjugated antimouse IgG monoclonal seconday antibody (Amersham‐Biotech, Courtaboeuf, France) at room temperature. Immunoreactive proteins were visualized using an enhanced chemiluminescence detection system (ECL+; Amersham‐Biotech). Monoclonal antibody to k‐ras (clone F234) was purchased from Tébu/Santa‐Cruz (Le Perray‐en‐Yveline, France) and monoclonal antibody to β‐actin (AC15) was from Sigma (Saint Quentin Fallavier, France).
Cell growth assay
[3H]Thymidine incorporation assay. Proliferation of pancreatic tumor cells or DNA synthesis was assessed by the measurement of [methyl‐3H]thymidine incorporation. For the last 24 h of cell culture, 1.85 × 104 Bq of [methyl‐3H]thymidine (37.01 × 107 Bq) was added. Cells were washed twice with PBS and were solubilized by the addition of 0.4 mL of 0.35 M NaOH to each well. The incorporation of [3H]thymidine into DNA was measured by scintillation counting. [3H]Thymidine incorporation was expressed in count per minute (c.p.m.) per well. Assays were carried out in triplicate and the experiment was repeated at least twice.
MTT test. The sensitivity of transfected cells after k‐ras gene silencing was determined using a tetrazolium‐based colorimetric assay (MTT). This test is based on the cellular reduction of MTT by the mitochondrial dehydrogenase of viable cells to a blue formazan product that can be measured with a spectrophotometer. After the desired period of treatment, cell cultures were washed twice with PBS and MTT was added to each well (final concentration 0.5 mg/mL). The plates were incubated at 37°C for 3 h, the medium was removed, 150 µL of dimethylsulfoxide was added and the plates were shaken gently for 15 min to solubilize the formazan blue crystals. Absorbance at 570 nm was measured with a microplate reader (Bio‐Rad). Cell viability was expressed as the percentage of growth of transfected cells with lipofectamine alone. Each experiment was undertaken using three wells per variable and was carried out at least twice.
Colony‐forming cell assay and ex vivo tumor inhibition. Pancreatic tumor cells (Panc1) were transfected with pSsik‐ras or pSsi‐green fluorescent protein (GFP) at 100 nM (2 µg/mL) for 3 days. Then, the transfected and parental cells (500 cells/well) were each plated in three wells of a six‐well plate (in triplicate 60‐mm Petri dishes). At day 12 the plates were fixed in 70% methanol, stained with 0.1% crystal violet (or treated with Giemsa stain), and the numbers of colonies greater than 100 µm in diameter were counted.
For ex vivo experiments, 4 days after transfection, 107 transfected or parental pancreatic tumor cells were injected subcutaneously into athymic nude mice (5 mice/group). The tumors were monitored with a caliper two times weekly over a 5‐week period after initial cells were injected.
Tumor models. Six‐ to seven‐week‐old female athymic nude mice NMRI‐nu (nu/nµ) (Elevage Janvier, Le Genest‐St‐Isle, France) were maintained under specific pathogen‐free conditions. The mice were allowed to acclimate to the facility for at least 2 weeks prior to manipulation. Mice had free access to water and mouse chow at all times.
Subcutaneous tumors. Tumor xenografts were established by subcutaneous bilateral injection of 107 viable cells (in 150 µL) in log‐phase growth into the flanks of athymic mice. When tumors measured an average volume of 80 mm3, mice were randomized into five groups (n = 6), and the electroporation experiments were initiated. Each of the bilateral tumors on a given mouse were treated in the same fashion so as to provide duplicate results. The control group received intratumor vehicle injections of 50 µL of PBS, while the other groups received pSsi GFP (25 µg), pSsik‐ras (25 µg), sik‐ras (25 µg) or AS PS‐ODN (50 µg). Plasmid DNA and oligonucleotides, either siRNA or antisense ODN, were diluted in 50 µL of PBS and slowly injected percutaneously into the tumors using a 27.5‐gauge needle. After 3 min of mechanical massage, a pair of stainless steel electrodes of 10 mm in length was inserted into the tumor to encompass the DNA injection sites. The distance between the electrode needles was adjusted as a function of the tumor diameter (0.5–1 cm). The tumors were pulsed using a BTX‐ECM 830 pulse generator (Q. BIOgene, Strasbourg‐Illkirch, France) and the voltage was set at 150 V/cm, unless otherwise stated. Eight square‐wave pulses were delivered at 150 v/cm with pulse duration of 50 ms and pulse interval of 1 µs. One week thereafter, electroporation was repeated (with the same parameters as the first one). These electroporation parameters were selected based on previous studies and our own preliminary experiments.
The tumor size was measured by caliper twice weekly and the tumor volume was calculated according to the following formula: V = (largest diameter × small diameter × depth) × π/6.
Intrapancreatic orthotopic tumors. In this experiment dedicated to the combined treatment, only one pancreatic tumor cell model (Panc1) was used. We and other authors observed that this cell line is more chemoresistant than other pancreatic cell lines.
The nude mice were anesthetized by isoflurane inhalation, their abdomens were sterilized with alcohol (70%) and were positioned laterally. A small, left abdominal flank incision was made, and the pancreas tail with the spleen was carefully exposed under strict aseptic conditions. The tumor cells (107 cells/50 µL) were injected into the exteriorized pancreatic corpus using a 27‐gauge tuberculin syringe. After replacement of the pancreas into the abdominal cavity, the incision was closed in two layers using an absorbable surgical 6‐0 vicryl suture for the peritoneum and 4‐0 vicryl suture for the skin. After transplantation, mice were inspected daily for the first five postoperative days.
Two weeks after tumor cell implantation, mice were divided randomly into five groups of 15 animals. Control groups included untreated tumor‐bearing mice (group 1). Treated groups included tumor‐bearing mice receiving gemcitabine (group 2), or pSsik‐ras lipoplexes alone (group 3) or in combination with gemcitabine (group 4). The pSsik‐ras lipoplexes were prepared using 100 µg of plasmid diluted in 100 µL of PBS and mixed with 200 µL of BGTC transfection reagent as described previously.( 25 ) The lipoplexes were administered intraperitoneally at 4‐day intervals and gemcitabine was injected 24 h later. Gemcitabine was used at the dose 25 µg/kg in the combination protocol and 45 µg/kg in the single protocol.
Tumor growth was estimated by abdominal palpation. Animals were killed when signs of severe tumor disease became apparent. A palpable abdominal tumor mass with a diameter larger than 1.5 cm, presence of clinical relevant ascites, or severe cachexia were indications. In each group, five mice were killed at day 30 after tumor cell inoculation for the measurement of tumor volume. The remaining 10 mice in each group were maintained for observation of survival time and establishment of Kaplan–Meier survival curves.
Statistical analysis. All data were subjected to statistical analysis using the Excel software package (Microsoft, Courtaboeuf, France). A two‐tailed Student's t‐test was used to determine the difference between the groups. Differences were considered significant at P < 0.05. Data are given as mean ± SEM.
Results
K‐ras siRNA vector construction. It was demonstrated by several groups that siRNA can be transcribed by small nuclear RNA polymerase III promoters (H1 and U6) in human cells, and elicit target‐specific mRNA degradation.( 17 , 18 ) Here, we have designed different pSUPER vector derivatives using the H1 promoter to drive expression of siRNA targeting specific luciferase, GFP or mutated k‐ras gene sequences. Figure 1 shows a schematic diagram of the recombinant H1/siRNA pSUPER expression vector, which produces a hairpin type of the siRNA transcript corresponding to the three k‐ras mutant forms in codon12 (pSUPERsik‐raswt(GGT), pSUPERsik‐rasg1y(GTT), pSUPERsik‐rasva1(GAT)). These plasmids were named pSsik‐rasGGT, pSsik‐rasGTT and pSsik‐rasGAT, respectively.
Figure 1.
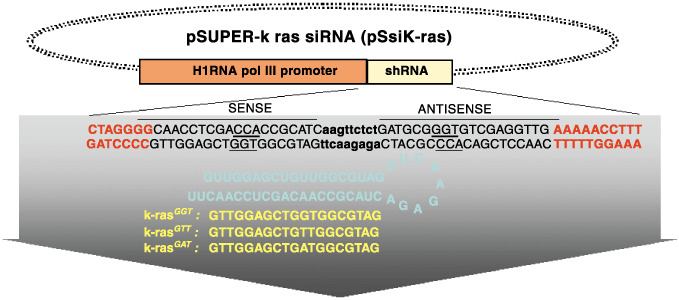
Schematic diagram showing the structure of the short hairpin RNA pSsik‐ras vector (pSsik‐ras). The different sequences, corresponding to wild type and that containing mutated codon 12, were annealed and ligated into the pSUPER vector.
Determination of optimal experimental conditions for gene silencing. In order to establish favorable transfection and electroporation conditions for the optimal delivery of As ODN and siRNA, preliminary experiments were carried out on pancreatic tumor cells (Panc1) using the GFP (pEGFP‐N1) and luciferase (pGL3‐Luc) reporter genes. As shown in Fig. 2a, the transient transfection of Panc1 cells with EGFP‐N1 is rationally efficient. Visualization of cells cotransfected with pEGFP‐N1 and specific GFP siRNA with a phase contrast fluorescence microscope showed significant knockdown of expression. In a second experiment testing the optimal gene silencing conditions, we used cell culture and nude mice bearing a subcutaneous carcinoma xenograft panc1 model to test the functionality of pGL3‐H1Luc expressing siRNA targeting the luciferase gene, on either the target pGL3‐Luc basic vector or the unrelated pEGFP‐N1 plasmid. Results showed that only the cotransfection and coelectroporation of pGL3‐Luc with the siRNA‐Luc‐based vector (pGL3‐H1Luc) significantly reduced the activity of luciferase (Fig. 2b).
Figure 2.
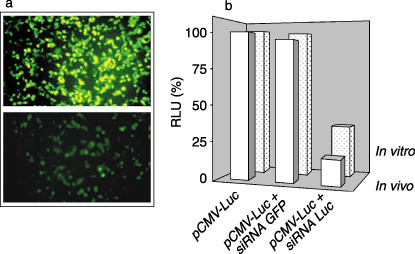
Determination of the optimal conditions for small interfering RNA (siRNA) transfection. (a) After 24 h of cell culture, specific green fluorescent protein (GFP) siRNA was cotransfected with the EGFP‐N1 plasmid using different concentrations of GFP siRNA and the plasmid expressing GFP. (b) In vivo, subcutaneous tumors were coelectroporated with siRNA‐Luc and pGL3‐Luc. After 48 h, tumors were excised and homogenized for bioluminescence measurement.
Silencing of the endogenous k‐ras oncogene: siRNA and As PS‐ODN specificity. The next step was to assess the silencing of k‐ras oncogene expression and to explore the specificity of the designed k‐ras siRNA and AS PS‐ODN. We therefore studied the specificity of k‐ras siRNA and AS PS‐ODN targeting to the wild type (GGT) and two mutant forms (GAT and GTT) of k‐ras in the BxPc3 (wild type, k‐rasGGT), Panc1 (k‐rasGAT) and Capan1 (k‐rasGTT) cell lines. At 48 h after transfection, tumor cells were harvested and k‐ras expression was analyzed by semiquantitative RT‐PCR. At first, as indicated in Fig. 3a, we observed that tumor cells transfected with control scrambled siRNA or PS‐ODN showed no inhibitory effect. Next, Fig. 3a shows that AS PS‐ODN as well as the synthetic and siRNA vectors directed against the GAT mutant form was unable to significantly reduce the endogenous expression of k‐ras in BxPC3 (k‐rasGGT) cells. In contrast, in the same experiment, both synthetic siRNA and pSsik‐ras, and to a lesser extent, AS PS‐ODN targeting the k‐ras GAT and GTT mutations, significantly reduced k‐ras mRNA expression in Panc1 (k‐rasGAT) and Capan1 (k‐rasGTT) tumor cells. Indeed, sik‐ras (100 nM), pSsik‐ras (1 µg/mL) and AS PS‐ODN (1 µM) significantly inhibited the endogenous mRNA levels of k‐ras by between 78 and 88% (P < 0.001). Expression of the GAPDH internal control remained unchanged after these treatments.
Figure 3.
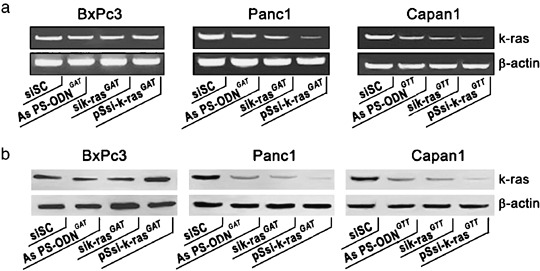
Inhibition of endogenous k‐ras expression after small interfering RNA (siRNA) treatment. (a) Two days after transfection, equal amounts of cell lysate were subjected to electrophoresis and western blot analysis. (b) Total RNA (1 µg) was subjected to semiquantitative reverse transcription–polymerase chain reaction with specific primers for either k‐ras or glyceraldehyde‐3‐phosphate dehydrogenase.
To validate the data from the RT‐PCR experiments, western blot analysis was carried out under the same experimental conditions. The results seen in Fig. 3b reproduce loyally the data mentioned above. Significant k‐ras protein knockdown was observed after tumor cell treatment with the corresponding oligonucleotide targeting the specific k‐ras mutation.
It should be noted that once again the control scrambled siRNA had no effect on endogenous k‐ras protein expression. In addition, β‐actin expression was unaffected by either siRNA or AS PS‐ODN. Together, these data demonstrate that k‐ras siRNA and AS ODN are potent inhibitors of their respective k‐ras targets. Moreover, k‐ras siRNA‐based vectors are more efficient.
Silencing of the endogenous k‐ras oncogene reduces tumor cell growth.
Cell proliferation and cytotoxicity . To evaluate the functionality and efficacy of the designed k‐ras siRNA, we assayed cell proliferation and viability using [3H]thymidine incorporation and MTT tests. In a preliminary experiment, we determined the conditions for optimal tumor cell growth inhibition. We observed that [3H]thymidine incorporation and cytotoxicity are time‐ and oligonucleotide concentration‐dependent. The optimal inhibitory effects were observed after 72 h for [3H]thymidine incorporation, and after 96 h for MTT tests (data not shown). Thus, the cytotoxic effect illustrated by the MTT test seems to be delayed by 24 h compared with the [3H]thymidine incorporation (data not shown). Concerning the comparative effectiveness study of k‐ras siRNA, the pancreatic tumor cell lines were treated over 72 h. As shown in Fig. 4a, the greatest efficacy was seen with pSsik‐ras, which induced a 70 and 62% reduction in thymidine incorporation in Panc1 and Capan1, respectively. Synthetic sik‐ras and AS PS‐ODN showed an inhibitory effect of 58 and 45%, respectively, for Panc1, and 53 and 41%, respectively, for Capan1. Similarly, cell viability assessed by the MTT assay for mitochondrial activity was affected by k‐ras siRNA and AS PS‐ODN treatment (Fig. 4b). The proliferative and cytotoxic results highlight the superiority of k‐ras siRNA. These functional data are thus entirely in good accordance with the efficient endogenous k‐ras expression reduction after mutated oligonucleotide treatment.
Figure 4.
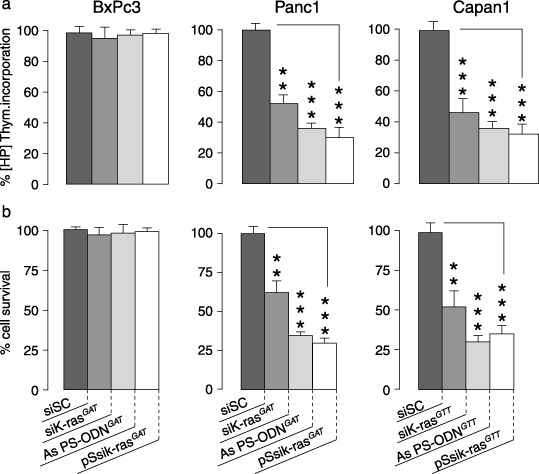
Comparison of the antiproliferative effects induced by specific k‐ras oligodeoxynucleotide (ODN) antisense and k‐ras small interfering RNA (siRNA) on pancreatic tumor cell lines. The different tumor cell lines were untreated or treated with k‐ras antisense ODN, synthetic k‐ras siRNA or pSsik‐ras. Two days after transfection, cell proliferation assays were carried out by [3H]thymidine incorporation measurement (a) and the cytotoxic effects were evaluated using MTT cell viability tests (b). The assays were carried out in triplicate, and each result is representative of at least three independent experiments.
Colony‐forming assay and ex vivo tumorigenicity. We wanted to further assess the cytotoxic effects of stable k‐ras siRNA expression and to verify that the silencing activity of the mutant k‐ras oncogene prevents tumor cell recurrence. To this end, the colony‐forming assay and ex vivo experiments were carried out using the pSsik‐ras and pSsi‐GFP plasmids. As shown in Fig. 5a, the results of the colony‐forming assay were in agreement with those for cell proliferation and cytotoxicity. Notably, a significant decrease in the number of colonies in Panc1 (by 78%; P < 0.001) and Capan1 (by 73%; P < 0.001) was apparent when the cells were transfected with pSsik‐rasGAT and pSsik‐rasGTT, respectively. In contrast, cells in the control group (untreated or pSsi‐GFP‐treated cells) showed little decrease in the number of colonies (Fig. 5b). These results suggest that transfection of tumor cells with pSsik‐ras caused dramatic growth inhibition of the tumor cells. In order to examine the tumor cell recurrence power in vivo, we xenografted the treated tumor cells interscapulary into nude mice. A few weeks later when we examined the mice (results of the ex vivo experiments) there were no detectable (palpable) tumors within 3 weeks, but we observed a small solid residual particle at the site of injection. Thereafter, tumors start to grow in all of the transplanted mice (Fig. 5c). A trivial explanation for our results is that tumor cells escaping the transient transfection were able to induce recurrence and tumor development, or that the transient silencing of oncogene‐induced tumorigenesis is reversible. In contrast, Panc1 and Capan1 cells transfected with pSsi‐GFP showed no delay in tumor growth (Fig. 5c).
Figure 5.
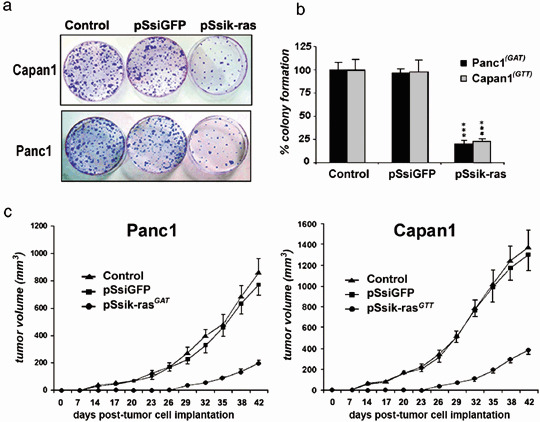
K‐ras downregulation inhibited cell viability and colony formation. (a) Panc1 tumor cells were treated over 48 h. Thereafter, cells were harvested and seeded in Petri dishes (60 mm) at different densities without any treatment. Approximately 10 days later, the plates were fixed with methanol and stained with crystal violet. (b) For each experiment, the colonies visualized by crystal violet staining were counted and the number was plotted in the graph as a percentage. Results are the means of three experiments ± SEM (***P < 0.001). (c) Ex vivo tumorigenesis assay. In vitro cell transfection was done as described in Materials and Methods. After 48 h, parental and small interfering RNA (siRNA)‐transfected Panc1 tumor cells were harvested and injected subcutaneously into nude mice, and the tumor volume was assessed twice weekly during 6 weeks. The left curve represents the tumor volume graph obtained after inoculation of untransfeted Panc1 (control) and transfected with pSsiGFP or pSsik‐rasGAT. The right curve represents the results obtained with Capan1 tranfected in vitro with pSsiGFP or pSsik‐rasGTT.
In vivo k‐ras oncogene silencing induces tumor growth inhibition. To evaluate the therapeutic potential of k‐ras oncogene silencing, athymic nude mice bearing s.c. BxPc3 (k‐raswt) or Panc1 (k‐rasmut) pancreatic carcinomas were assigned randomly to treatment groups when the tumors reached a volume of 75–100 mm3 (on average). Control tumor‐bearing mice were given an intratumoral electroporation of scrambled siRNA and their responses were compared to mice receiving an intratumoral electroporation of AS PS‐ODN, sik‐ras or pSsik‐ras. As shown in Fig. 6a, the BxPc3 tumors injected with the irrelevant AS PS‐ODN (k‐rasGAT), synthetic sik‐rasGAT or pSsik‐rasGAT, as well as the untreated control tumors, resulted in the rapid development of progressive tumors in athymic mice. In contrast to BxPC3 tumors, Panc1 tumors electroporated with k‐ras antisense oligos showed mild suppression of tumor growth (Fig. 6b). In the other treatment groups of Panc1 tumors, synthetic k‐rasGAT siRNA and pSsik‐rasGAT siRNA electroporation dramatically reduced the tumor volumes; this inhibitory effect was significantly greater with pSsik‐rasGAT (Fig. 6b).
Figure 6.
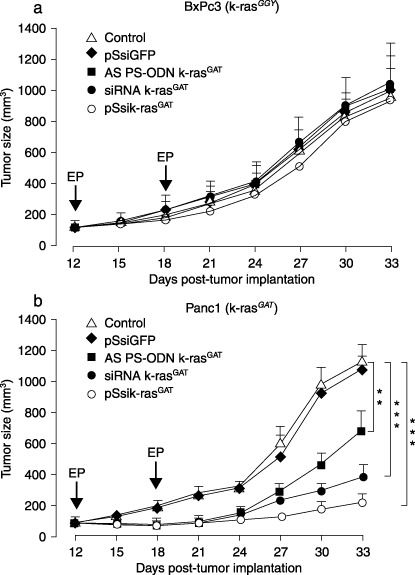
Silencing of K‐ras expression inhibited the growth of tumor xenografts in nude mice. Mice with subcutaneous BxPC3 (k‐rasGGT) and Panc1 (k‐rasGAT) tumors were randomized into groups of six animals and k‐ras antisense oligonucleoides and small interfeing RNA were delivered intratumorally by electropration two times. Individual mice were monitored for tumor growth over a period of 33 days. The volume for individual tumors was calculated and each curve represents the mean tumor volume calculated for each group (n = 6). Standard errors of the mean are represented by error bars.
Enhanced inhibition of tumor growth and prolongation of survival in mice bearing orthotopic pancreatic tumors by combined k‐ras siRNA and gemcitabine treatment. To improve and extend the results of the s.c. tumor xenograft experiments, we established an orthotopic pancreatic cancer model that mimics the clinical features. The antitumor effect of gemcitabine and pSsik‐rasGAT on k‐ras oncogene silencing was determined on the Panc1 pancreatic tumors. This model was chosen because it is considered relatively resistant to many chemotherapeutic regimens. As shown in Fig. 7a, a significant growth‐inhibitory effect was observed in the treated groups compared with the control groups. Indeed, after 30 days of treatment with gemcitabine, pSsik‐rasGAT or gemcitabine plus pSsik‐rasGAT, the mean tumor volume was reduced by 43% (P < 0.01), 40% (P < 0.01) and 68% (P < 0.01), respectively.
Figure 7.
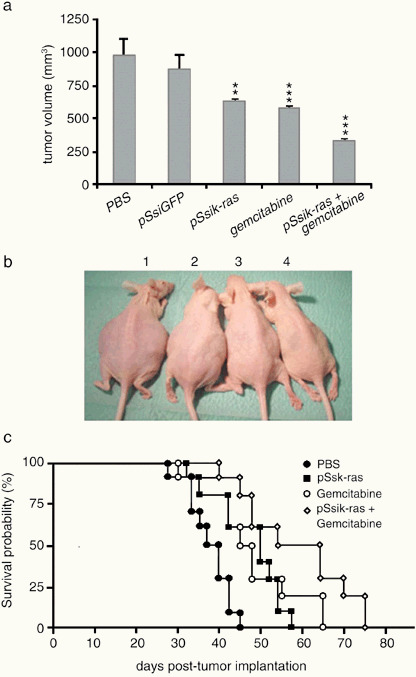
Enhancement of gemcitabine antitumor effects by k‐ras small interfering RNA (siRNA). Panc1 pancreatic tumor cells (k‐rasGAT) were implanted orthotopically. After 10 days, mice were assigned randomly into four groups. The siRNA k‐rasGAT were delivered by intraperitoneal lipofection alone or in combination with gemcitabine (25 µg/kg). Gemcitabine in the single‐treatment protocol was administrated intraperitoneally at the dose 45 µg/kg. (a) Total tumor volume determination after 30 days showed a significant inhibition of tumor growth, particularly in pancreatic tumor‐bearing mice receiving combined treatment. (b) The general appearance of representative mice bearing orthotopic pancreatic tumor xenografts after 45 days. The control mouse (1) had a swelling abdomen and was larger than the mouse treated with pSsik‐rasGAT (2), gemcitabine (3) or pSsik‐rasGAT + gemcitabine (4). (c) Cumulative survival rate of athymic nude mice implanted orthotopically with Panc1 tumor cells. The survival curve was plotted according to the method of Kaplan–Meier (n = 10). Note the high prolongation of mouse survival by combined gemcitabine and k‐ras siRNA (pSsik‐rasGAT) treatment compared with the control mice or with single agent‐treated mice.
External observation after 45 days of the study revealed that untreated control mice had significant abdominal swelling, whereas swelling was much less marked in mice treated with gemcitabine and pSsik‐rasGTT (data not shown). At postmortem examination, we observed that this swelling was due to the formation and accumulation of ascites in the abdomen. In control mice, in addition to the large tumor size, tumor nodules were found on the surface of the peritoneum, intestines and diaphragm, and on the hilus of the liver.
Two and half months after treatment, the Kaplan–Meier plot assessment showed a significant prolongation of surviving mice bearing orthotopic pancreatic tumors after combined treatment (gemcitabine + pSsik‐ras). As shown in Fig. 7b, the median survival times of untreated mice (PBS) was 37 days. In contrast, the median survival time of gemcitabine + pSsik‐rasGTT‐treated mice was significantly longer (64 days) than that of mice treated with gemcitabine or pSsik‐rasGTT (52 and 45 days, respectively).
Discussion
The dismal prognosis associated with pancreatic cancer is no better today than it was 20 years ago. Lack of efficient therapy has prompted intensive investigation into different therapeutic strategies. Ductal pancreatic adenocarcinoma is associated with activation of the k‐ras oncogene in approximately 90% of cases. The present study set out to evaluate the k‐ras oncogene silencing strategy for pancreatic adenocarcinoma. The k‐ras point mutation is described as an early event in pancreatic tumorigenesis.( 26 , 27 ) Even if the exact molecular mechanism of k‐ras involvement in tumor cell proliferation is not fully known, there is no doubt that the Ras oncogene is tightly involved in the carcinogenic and tumorigenic processes. The transforming properties of oncogenic Ras are based on continuous activation of its downstream effectors. Furthermore, experimental evidence has shown that oncogenic Ras plays a key role in human cell transformation. Thus, k‐ras has emerged as an attractive target for cancer therapy. The big challenge is to selectively inhibit oncogenic k‐ras but not the wild‐type k‐ras, which is required for viability and functionality of normal cells. The selective molecular inhibition of k‐ras oncogene signaling is under active investigation and represents a promising strategy for fighting pancreatic cancer. In the present study, we demonstrated that inhibition of k‐ras gene expression can significantly reduce pancreatic tumor growth in vitro and in vivo. We examined the potency and specificity of k‐ras oncogene inhibition achieved by siRNA compared with relevant k‐ras phosphorothioate AS ODN in a variety of human pancreatic cancer cells.
During the last decade, several approaches have been developed to disrupt k‐ras signaling in cancer cells. The first strategy to inhibit Ras activity in tumor cells was the development of FTI. Early in vitro and in vivo studies indicated that FTI selectively inhibit the anchorage‐independent growth of Ras‐transformed cells in soft agar and the growth of Ras‐induced tumors in mouse xenograft models.( 28 , 29 ) However, the antitumor effects of FTI are not accomplished by specific inhibition of oncogenic Ras, and these effects were partially mediated by inhibition of targets other than Ras.( 30 )
A second strategy used to interfere with Ras signaling in cancer cells is the inhibition of Ras protein expression by antisense oligonucleotides. Although the phosphorothioate forms are resistant to endogenous nuclease, therapeutic use of antisense oligodeoxynucleotide technology was limited by its inconsistent non‐specific antitumor activity and induced toxic sequence‐independent effects.( 31 ) After a long period of ups and downs, oligonucleotide technologies have again gained increasing attention in recent years with the discovery of RNAi. RNAi is a sequence‐specific post‐transcriptional gene silencing mechanism induced by dsRNA molecules. This has opened the road for novel, sequence‐specific anticancer therapies.
In agreement with previous reports,( 32 , 33 ) our results demonstrate that k‐ras AS PS‐ODN reduced cell proliferation by 30–40%. The effects of k‐ras AS ODN expression on tumor cell growth and k‐ras endogenous expression were restricted to the relevant k‐ras mutated cells. It is noteworthy that under the same in vitro experimental conditions, k‐ras siRNA induced a more pronounced antiproliferative effect of 50–65%. In the transient k‐ras siRNA‐transfected cells, the endogenous expression of k‐ras was specifically abolished. In the nude mouse xenograft model, intratumoral electroporation of k‐ras‐specific siRNA induced significant inhibition of tumor growth. Intraperitoneal delivery of k‐ras siRNA resulted in a significant improvement in the survival of orthotopic xenograft‐bearing mice, without any apparent signs of toxicity. A key observation of this study is the significant superiority of k‐ras siRNA, which is not due to more efficient uptake of siRNA but is probably the result of a high degree of specificity. The theoretical benefit of such a target‐specific therapy is a lower risk of systemic side effects. Thus, the major advantage of RNAi‐mediated anti‐Ras cancer therapy, in contrast to other Ras‐based therapies, is the specific targeting of oncogenic Ras. However, the potency of siRNA remains limited by intratumoral bioavailability and the transient nature of gene silencing. Even if repeated administration of synthetic siRNA could overcome this limitation, the use of RNAi in cancer therapy requires the development of more stable and suitable delivery systems. Currently, we demonstrated that the k‐ras siRNA expression plasmid led to the most marked inhibition of endogenous k‐ras expression and tumor cell proliferation.
To validate the results of in vitro experiments, s.c. and orthotopic xenograft pancreatic cancer models were initiated in athymic mice which were treated with k‐ras AS PS‐ODN or siRNA reagents. After 5 weeks, the inhibitory effect of k‐ras AS PS‐ODN on pancreatic tumor growth did not exceed 25%. At the same time, the mean tumor volumes of mice treated with synthetic k‐ras siRNA and vector expressing siRNA were reduced by 38 and 62%, respectively. Despite the finding that k‐ras siRNA did not completely prevent xenograft tumor growth, tumors in animals treated with k‐ras siRNA expression vectors grow more slowly than did tumors in synthetic k‐ras siRNA‐treated animals. Furthermore, survival of mice with orthotopic pancreatic tumors was increased after k‐ras siRNA vector intraperitoneal lipofection compared with mice treated with k‐ras AS PS‐ODN or k‐ras synthetic siRNA. However, no mice were cured regardless of the treatment applied in these experiments. Although the electroporation and lipofection were not the most efficient siRNA delivery methods, our study raises the question of whether inhibition of mutated k‐ras expression is potent enough to eradicate or prevent tumor growth. Agami et al. developed a retroviral siRNA vector directed against the region that encodes the point mutation in codon 12 of oncogenic k‐ras.( 34 ) Transduction of this siRNA vector into the human pancreatic cancer cells strongly inhibited the expression of k‐rasV12, whereas wild‐type k‐ras was unaffected. Moreover, retroviral delivery of siRNA targeting k‐rasV12 resulted in the highly selective and stable suppression of mutated k‐ras. More importantly, this led to loss of tumorigenicity. However, from a clinical point of view, the situation is different. In fact, the objective is to destroy an established or unresectable pancreatic tumor, or to treat metastatic tumor cells. Further experimental investigations are clearly needed to more accurately determine if the k‐ras inhibition or k‐ras gene silencing induce direct cytotoxic or cytostatic effects on tumor cells. K‐ras has been shown to affect the expression of a wide range of cancer‐related genes, including growth factors and genes involved in the control of cell cycle and apoptosis.
Our present data on the k‐ras AS‐ODN were nearly the same as those reported by the Watanabe and Aoki groups.( 32 , 33 ) Thus, we confirm and support that the k‐ras antisense strategy reduces pancreatic tumor growth. More importantly, the present study provides substantial evidence that siRNA molecules offer several potential advantages over conventional antisense oligonucleotides.
Taken together, our results and others reported recently in the literature provide evidence that k‐ras siRNA may be used therapeutically against human pancreatic cancer. As with gene therapy, the delivery of RNAi remains the main obstruction for successful application in humans, and further investigations are required to identify the most effective delivery strategies for pancreatic adenocarcinoma therapy using siRNA gene‐silencing approaches. It is hoped that this endeavor will be fruitful and will provide a new weapon in the fight against pancreatic cancer.
To our knowledge, no combination studies have been carried out with currently available chemotherapeutic drugs and k‐ras siRNA in order to test a hypothetical additive or synergistic effect on their anticancer activity.
In recent years, the systemic administration of gemcitabine has been accepted as a standard first‐line treatment for patients with advanced pancreatic cancer. Although treatment with gemcitabine has been shown to result in both clinical benefit and prolongation of survival, objective tumor responses following therapy with gemcitabine are relatively uncommon and median survival times remain short. Efforts are currently being made to increase the therapeutic efficacy of the drug in clinical settings on various types of cancer by combining it with other cytotoxic agents, including 5‐fluorouracil, cisplatin, oxaliplatin, capecitabine, docetaxel or irinotecan.( 23 , 35 )
In the present study, using a human orthotopic pancreatic tumor model (Panc1), we analyzed the antitumor effects of the combination of shRNA plasmid, pSsik‐rasGAT and gemcitabine. Our results indicate a remarkable increase in antitumor response and survival prolongation in mice receiving both therapies compared with those treated with gemcitabine alone. These data are the first to demonstrate that gemcitabine‐based chemotherapy can be improved by mutated k‐ras oncogene silencing for pancreatic cancer treatment. These findings have strong implications for cancer therapeutic strategies. Thus, further experiments with other tumor cell systems need to be carried out to corroborate these results.
The mechanism by which blocking k‐ras signaling enhances or facilitates gemcitabine‐induced killing of mutated k‐ras pancreatic cancer cells is not currently known. For the explanation of this additive or synergistic effect, the k‐ras signal transduction pathway and the activating metabolism of gemcitabine must be elucidated. To determine whether k‐ras oncogene silencing and downregulation of k‐ras activity in pancreatic cancer cells containing mutated k‐ras sensitizes these cells to gemcitabine‐induced killing, experiments are underway in our laboratory. We hypothesized that specific k‐ras siRNA acts by inhibiting downstream oncogenic signaling pathways, such as phosphatidylinositol 3′‐kinase/Akt, mitogen‐activated protein kinase (extracellular signal‐regulated kinase), and IκB/nuclear factor κB, that control cell proliferation, angiogenesis and apoptosis.( 36 , 37 , 38 ) The direct consequence of the downregulation or inhibition of these effectors can abrogate or decrease the resistance to apoptosis induced by gemcitabine treatment, which might happen frequently in growing pancreatic cancer tumors in vivo.
In summary, our study provides a huge window of opportunity for the development of new and more specific therapeutic modalities. We strongly believe that the k‐ras siRNA approach offers great potential as an effective adjuvant treatment modality, for example with chemotherapy regimens and apoptogene therapy, in the clinical management of pancreatic adenocarcinoma. Ultimately, we are persuaded that the rational strategy will be first to determine the genetic tumor status, and second to establish predictive markers that can determine which tumors are responsive to a specific drug in order to adapt or optimize the adequate and appropriate treatment modality. This may lead to less empiric and aggressive therapy for patients in the future.
References
- 1. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet 2004; 363: 1049–57. [DOI] [PubMed] [Google Scholar]
- 2. Bray F, Sankila R, Ferlay J, Parkin DM. Estimates of cancer incidence and mortality in Europe in 1995. Eur J Cancer 2002; 38: 99–166. [DOI] [PubMed] [Google Scholar]
- 3. Wray CJ, Ahmad SA, Matthews JB, Lowy AM. Surgery for pancreatic cancer. Recent Controversies Current Prac Gastroenterol 2005; 128: 1626–41. [DOI] [PubMed] [Google Scholar]
- 4. Berlin JD, Rothenberg M. Chemotherapy for resectable and advanced pancreatic cancer. Oncology 2001; 15: 1241–9. [PubMed] [Google Scholar]
- 5. Beger HG, Rau B, Gansauge F, Poch B, Link KH. Treatment of pancreatic cancer. Challenge Facts World J Surg 2003; 27: 1075–84. [DOI] [PubMed] [Google Scholar]
- 6. Cowgill SM, Muscarella P. The genetics of pancreatic cancer. Am J Surg 2003; 186: 279–86. [DOI] [PubMed] [Google Scholar]
- 7. Rozenblum E, Schutte M, Goggins M et al . Tumor‐suppressive pathways in pancreatic carcinoma. Cancer Res 1997; 57: 1731–4. [PubMed] [Google Scholar]
- 8. Sun C, Yamato T, Furukawa T, Ohnishi Y, Kijima H, Horii A. Characterization of the mutations of the K‐ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol Rep 2001; 8: 89–92. [DOI] [PubMed] [Google Scholar]
- 9. Villanueva A, Reyes G, Cuatrecasas M et al . Diagnostic utility of K‐ras mutations in fine‐needle aspirates of pancreatic masses. Gastroenterology 1996; 110: 1587–94. [DOI] [PubMed] [Google Scholar]
- 10. Scanlon KJ. Anti‐genes: siRNA, ribozymes and antisense. Curr Pham Biotechnol 2004; 5: 415–20. [DOI] [PubMed] [Google Scholar]
- 11. Aboul‐Fadl T. Antisense oligonucleotides: the state of the art. Curr Med Chem 2005; 12: 2193–214. [DOI] [PubMed] [Google Scholar]
- 12. Borkhardt A. Blocking oncogenes in malignant cells by RNA interference – new hope for a highly specific cancer treatment? Cancer Cell 2002; 2: 167–8. [DOI] [PubMed] [Google Scholar]
- 13. Heidenreich O. Oncogene suppression by small interfering RNAs. Curr Pharm Biotechnol 2004; 5: 349–54. [DOI] [PubMed] [Google Scholar]
- 14. Herr AJ. Pathways through the small RNA world of plants. FEBS Lett 2005; 579: 5879–88. [DOI] [PubMed] [Google Scholar]
- 15. Baulcombe D. RNA silencing in plants. Nature 2004; 431: 356–63. [DOI] [PubMed] [Google Scholar]
- 16. Wilhelm JE, Smibert CA. Mechanisms of translational regulation in Drosophila . Biol Cell 2005; 97: 235–52. [DOI] [PubMed] [Google Scholar]
- 17. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296: 550–3. [DOI] [PubMed] [Google Scholar]
- 18. Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 1443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pino SM, Xiong HQ, McConkey D, Abbruzzese JL. Novel therapies for pancreatic adenocarcinoma. Curr Gastroenterol Rep 2004; 6: 119–25. [DOI] [PubMed] [Google Scholar]
- 20. El‐Rayes BF, Philipp PA. A review of systemic therapy for advanced pancreatic cancer. Clin Adv Hematol Oncol 2003; 1: 430–4. [PubMed] [Google Scholar]
- 21. Reni M, Cordio S, Milandri C et al . Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005; 6: 369–76. [DOI] [PubMed] [Google Scholar]
- 22. Richards DA. Chemotherapeutic gemcitabine doublets in pancreatic carcinoma. Semin Oncol 2005; 32 (4 Suppl 6): S9–13. [DOI] [PubMed] [Google Scholar]
- 23. Jacobs AD. Gemcitabine‐based therapy in pancreas cancer: gemcitabine–docetaxel and other novel combinations. Cancer 2002; 95: 923–7. [DOI] [PubMed] [Google Scholar]
- 24. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschel T. Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001; 411: 494–8. [DOI] [PubMed] [Google Scholar]
- 25. Hajri A, Wack S, Lehn P et al . Combined suicide gene therapy for pancreatic peritoneal carcinomatosis using BGTC liposomes. Cancer Gene Ther 2004; 11: 16–27. [DOI] [PubMed] [Google Scholar]
- 26. Deramaudt T, Rustgi AK. Mutant K‐RAS in the initiation of pancreatic cancer. Biochimica Biophysica Acta 2005; 1756: 97–101. [DOI] [PubMed] [Google Scholar]
- 27. Sugio K, Gazdar AF, Albores‐Saavedra J, Kokkinakis DM. High yields of K‐ras mutations in intraductal papillary mucinous tumors and invasive adenocarcinomas induced by N‐nitroso (2‐hydroxypropyl) (2‐oxopropyl) amine in the pancreas of female Syrian hamsters. Carcinogenesis 1996; 17: 303–9. [DOI] [PubMed] [Google Scholar]
- 28. Sun J, Qian Y, Hamilton AD, Sebti SM. Both farnesyltransferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K‐Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene 1998; 16: 1467–73. [DOI] [PubMed] [Google Scholar]
- 29. End DW, Smets G, Todd AV et al . Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro . Cancer Res 2001; 61: 131–7. [PubMed] [Google Scholar]
- 30. Appels NM, Beijnen JH, Schellens JH. Development of farnesyltransferase inhibitors: a review. Oncologist 2005; 10: 565–78. [DOI] [PubMed] [Google Scholar]
- 31. Lebedeva I, Stein CA. Antisense oligonucleotides: promise and reality. Annu Rev Pharmacol Toxicol 2001; 41: 403–19. [DOI] [PubMed] [Google Scholar]
- 32. Morioka CY, Machado MC, Saito S et al . Suppression of invasion of a hamster pancreatic cancer cell line by antisense oligonucleotides mutation‐matched to K‐ras gene. In Vivo 2005; 19: 535–8. [PubMed] [Google Scholar]
- 33. Aoki K, Ohnami S, Yoshida T. Suppression of pancreatic and colon cancer cells by antisense K‐ras RNA expression vectors. Methods Mol Med 2005; 106: 193–204. [DOI] [PubMed] [Google Scholar]
- 34. Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus‐mediated RNA interference. Cancer Cell 2002; 2: 243–7. [DOI] [PubMed] [Google Scholar]
- 35. Heinemann V. Gemcitabine in the treatment of advanced pancreatic cancer: a comparative analysis of randomized trials. Semin Oncol 2002; 29: 9–16. [DOI] [PubMed] [Google Scholar]
- 36. Perugini RA, McDade TP, Vittimberga FJ, Callery MP. Pancreatic cancer cell proliferation is phosphatidylinositol 3‐kinase dependent. J Surg Res 2002; 90: 29–44. [DOI] [PubMed] [Google Scholar]
- 37. De Bono JS, Rowinsky EK. Therapeutics targeting signal transduction for patients with colorectal carcinoma. Br Med Bull 2000; 64: 227–54. [DOI] [PubMed] [Google Scholar]
- 38. Reed JC. Apoptosis‐targeted therapies for cancer. Cancer Cell 2003; 3: 17–22. [DOI] [PubMed] [Google Scholar]