

Abstract
The aim of the present work was to compare the effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives (EPAD) on tumor growth and metastasis to the lung in tumor‐bearing mice. Because EPA is very unstable during long‐term storage, the EPAD were subjected to accelerated testing under storage conditions of 60 ± 5% relative humidity at 37°C for 30 days. EPAD are composed of a mixture of a newly identified EPA ethylester dimer and EPA hydroxyethylester, and known EPA and EPA ethylesters. The inhibitory effects of EPAD, the EPA ethylester dimer and EPA hydroxyethylester on Matrigel‐induced capillary‐like network formation were stronger than the effect of EPA ethylester. The oral administration of EPAD (300 or 1000 mg/kg) inhibited angiogenesis in gels containing Matrigel supplemented with vascular endothelial growth factor (VEGF) and heparin in an in vivo model, but EPA ethylester had no effect. EPA ethylester (300 or 1000 mg/kg) or EPAD (1000 mg/kg) inhibited tumor growth in mice with subcutaneously implanted LLC. Furthermore, EPAD inhibited metastasis to the lung in mice implanted with LLC subcutaneously, but EPA had no effect. EPAD increased the CD8+ T‐cell population in the spleen compared with mice with subcutaneously implanted LLC. EPA ethylester increased the natural killer cell population in the spleen. Thus, it is suggested that the mechanisms of the antitumor and/or antimetastatic actions of EPAD and EPA ethylester involve different immune functions, and that the EPA ethylester dimer and the EPA hydroxyethylester of EPAD may contribute to these actions. (Cancer Sci 2005; 96: 441–450)
There is a large amount of literature regarding the effects of fish oil, including n‐3 polyunsaturated fatty acids (PUFA) such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), on tumor growth and metastasis to the lung in experimental animals.(
1
,
2
,
3
,
4
,
5
) Most of those reports suggest that fish oil has antitumor and antimetastatic effects. Epidemiological studies indicate that the consumption of fish and fish oil correlates with a reduced risk of colorectal cancer.(
6
,
7
,
8
,
9
) However, EPA and DHA are very unstable during long‐term storage. In the present study, we performed stability testing on EPA ethylester under storage conditions of 37°C and 60 ± 5% relative humidity for 30 days. Because n‐3 PUFA are very unstable during long‐term storage, it seems likely that the by‐products (EPA derivatives; EPAD) formed from EPA ethylester during the accelerated stability test might have lower antitumor action compared with EPA ethylester. Therefore, we compared the antitumor and antimetastatic actions of EPA ethylester and the by‐products (EPAD) formed during accelerated stability testing in several experimental models (in vitro and in vivo). We found that the by‐products formed from EPA ethylester in the accelerated stability test were a mixture of four EPAD. In addition, to clarify the antitumor and antimetastatic mechanisms of EPA ethylester and EPAD, we examined their effects on lipid peroxidation,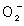 production and DNA synthesis in cultured Lewis lung carcinoma (LLC) cells, Matrigel‐induced angiogenesis, and apoptosis, neovascularization and immune function of tumors by immunohistochemistry. Tumor angiogenesis involves the directional sprouting of new vessels toward a solid tumor. It should be noted that angiogenesis is no less important to the growth of secondary tumor colonies than it is to the growth of primary solid tumors. Secondary tumors often give rise to ternary tumors in distant organs by a process termed the metastatic cascade.(
10
,
11
,
12
,
13
)
production and DNA synthesis in cultured Lewis lung carcinoma (LLC) cells, Matrigel‐induced angiogenesis, and apoptosis, neovascularization and immune function of tumors by immunohistochemistry. Tumor angiogenesis involves the directional sprouting of new vessels toward a solid tumor. It should be noted that angiogenesis is no less important to the growth of secondary tumor colonies than it is to the growth of primary solid tumors. Secondary tumors often give rise to ternary tumors in distant organs by a process termed the metastatic cascade.(
10
,
11
,
12
,
13
)
Materials and Methods
General experimental procedures. Ultraviolet and mass spectrometry (MS) spectra were measured using a Jasco ORD/UV‐5 spectrometer (Tokyo, Japan) and a Hitachi M‐4000H spectrometer (Tokyo, Japan), respectively. The 1H‐nuclear magnetic resonance (NMR) (499.83 Hz) and 13C‐NMR (125.68 Hz) spectra were recorded in CDCl3 using a Varian (Palo Alto, CA, USA) Unity Inova 500 spectrometer.
Materials. Eicosapentaenoic acid ethylester was supplied by Mochida Pharmaceutical Co. (Osaka, Japan). Matrigel basement membrane (with or without growth factor) was obtained from Becton Dickinson Labware (Bedford, MA, USA). [Methyl‐3H]‐thymidine (specific activity; 740 GBq/mmoL) was purchased from NEN Life Science Products (Boston, MA, USA). Dulbecco's modified Eagle's medium (DMEM) and microvascular endothelial base medium (HMEBM) were obtained from Nissui Pharmaceuticals (Tokyo, Japan), and Clonetics (San Diego, CA, USA), respectively, and used as culture media. Medium kits were purchased from Cell Systems (Kirkland, WA, USA). These medium kits consisted of mixtures of DMEM and Ham F12 medium (1 : 1), supplemented with 10% fetal bovine serum (FBS), 15 mmol/L N‐2‐hydroxyethylpiperazine‐N′‐2‐ethanesulfonic acid (HEPES), and CS‐C growth factor. Antibiotic and antimycotic solutions (100×) containing 104 U/mL penicillin, 10 mg/mL streptomycin, and 25 µg/mL amphotericin B in 0.9% NaCl were purchased from Sigma Chemical Co. (St Louis, MO, USA). FBS was purchased from Gibco BRL (Auckland, New Zealand). Six‐, 12‐, 24‐, and 48‐well plates were purchased from Corning Glass Works (NY, USA). Collagen (type I)‐coated 6‐ and 24‐well plates were purchased from Sumitomo Bakelite (Tokyo, Japan). Vascular endothelial growth factor (VEGF) was purchased from Wako Pure Chemical Co. (Osaka, Japan). Mouse lymphocyte separation medium (Lympholytes‐Mouse) was purchased from Dainippon Pharmacy Co. (Osaka, Japan). Fluorescein isothiocyanate (FITC)‐labeled antimouse CD4 and CD8 antibodies, phycoerythrin (PE)‐labeled antimouse natural killer (NK) antibody, and FITC‐labeled antimouse IgG2a and PE‐labeled antimouse IgG2a (used as negative controls) were purchased from Serotec (Oxford, UK). Rabbit polyclonal antiasialo GM1 antibody, mouse monoclonal antihuman CD 8‐α antibody (D‐9), and rat monoclonal antimouse CD34 antibody (MEC 14.7)( 14 ) were purchased from Wako Pure Chemical Co. (Osaka, Japan), Santa Cruz Biotechnology (Santa Cruz, CA, USA), and HyCult Biotechnology (Uden, The Netherlands), respectively. Peroxidase goat antirat IgG was purchased from Oncogene Science (NY, USA), and used as a second‐step reagent for antimouse CD34 antibody. EnVision+, Mouse/horseradish peroxidase (HRP) and EnVision +, Rabbit/HRP were purchased from DAKO Japan (Kyoto, Japan), and used as second step reagents for anti‐CD8‐α antibody and antiasialo GM1 antibody. An apoptosis in situ detection kit was purchased from Wako Pure Chemical Co. (Osaka, Japan), and tumor apoptosis was measured by the in situ terminal deoxyribonucleotidyl transferase‐mediated dUTP–digoxigenin nick end‐labeling (TUNEL) method. Corn starch, α‐starch, casein, cellulose powder, corn oil, a mineral mixture and a vitamin mixture were purchased from Oriental Co. (Osaka, Japan). Other chemicals were of reagent grade.
Formation of by‐products during accelerated testing of EPA ethylester. EPA ethylester was incubated at 60 ± 5% relative humidity at 37°C for 30 days to obtain EPAD according to International Conference on Harmonization (ICH) guidelines.( 15 ) The purification of four EPA derivatives (E‐1, E‐2, E‐3 and E‐4) was performed by chromatography on silica gel thin‐layer plates (Whatman PLK5F silica gel 150 A, 1000 µm, Tokyo, Japan) developed with the solvent n‐hexane‐EtOAc (10 : 3, v/v). Each band was scraped and extracted with CH2Cl2‐MeOH (10 : 19, v/v), and then the extracts were concentrated under a vacuum. Each residue was analyzed by determining the 1H‐ and 13C‐NMR and MS spectra. The EPAD consisted of EPA ethylester (E‐1, 22.5%), the EPA ethylester dimer (E‐2, 6.8%), EPA hydroxyethylester (E‐3, 47.0%), EPA (E‐4, 21.8%) and unknown compounds (1.0%). We isolated two new EPA derivatives (E‐2 and E‐3), as well as EPA ethylester (E‐1) and EPA (E‐4) from EPAD formed during accelerated testing. In in vitro experiments, EPA ethylester (E‐1) and by‐products of EPAD were dissolved in 50% ethanol at a concentration of 33 mg/mL, and the final ethanol concentrations in cell cultures were less than 0.5%. Because E‐2 and E‐3 are very unstable, E‐2 and E‐3 could not be obtained in large quantities during the purification process. Therefore, E‐2 and E‐3 were evaluated in in vitro assays alone. In in vivo experiments, EPA ethylester and EPAD were suspended in phosphate buffered saline (PBS, pH 7.0) containing 10% bovine serum albumin (BSA).
Cells. Lewis lung carcinoma cells were derived from a C57BL/6 mouse and from highly metastatic and drug‐resistant tumors. LLC cells were obtained from the Riken Gene Bank (Tsukuba, Japan) and maintained in DMEM supplemented with 10% FBS, penicillin (100 U/mL), streptomycin (100 µg/mL), and amphotericin B (0.25 µg/mL). Human adult dermal microvascular endothelial cells (HMVEC) were purchased from Cell Systems (Kirkland, WA, USA), seeded onto collagen (type I)‐coated 24‐ or 6‐well plates, and maintained in Clonetics microvascular endothelial basal medium supplemented with growth factor (MEGM), or in CS‐C medium.
Measurement of lipid peroxidation in cultured LLC cells. Lewis lung carcinoma cells were placed in DMEM supplemented with 10% FBS at 5 × 104 cells/well in 12‐well culture plates. After the cells were cultured overnight, the medium was changed to fresh DMEM supplemented with 10% FBS and the indicated amounts of EPA ethylester or EPAD for 24 h, and then the LLC cells were assayed for the amount of thiobarbituric acid (TBA)‐reactive substances as a conventional index for lipid peroxidation according to the method of Ohkawa et al.( 16 ) with some modifications. Briefly, the LLC cells were disrupted by the addition of 1 mL 20% trichloroacetic acid (TCA) to the medium. Cells and medium were transferred to a glass tube, supplemented with 2 mL 0.5% TBA solution, and heated in boiling water for 30 min. After cooling and centrifugation at 1500 g for 5 min, the absorbance of the supernatant was measured at 532 nm. The lipid peroxide value was expressed as nmol of malondialdehyde (MDA) using a standard curve generated with 1,1,3,3‐tetraethoxypropane.
Measurement of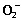 production in cultured LLC cells. Lewis lung carcinoma cells were placed in DMEM (phenol red free) supplemented with 10% FBS at 1 × 104 cells per well in 96‐well culture plates.released into the reaction medium by cultured LLC cells treated with the indicated amounts of EPA ethylester and EPAD was measured in an assay using cytochrome c. Briefly, LLC cells (1 × 104 cells/well) were added to 120 µmol/L cytochrome c with the indicated amounts of EPA ethylester or EPAD, and incubated for 15, 30, 45 or 60 min at 37°C. After the incubation period, the plate was immediately (within 1 min) assayed in a microplate reader at 540 nm. released into the reaction mixture by cultured LLC cells treated with EPA ethylester or EPAD was determined by measuring the rate of reduction of cytochrome c. The data were expressed as µmol/L per h.
production in cultured LLC cells. Lewis lung carcinoma cells were placed in DMEM (phenol red free) supplemented with 10% FBS at 1 × 104 cells per well in 96‐well culture plates.released into the reaction medium by cultured LLC cells treated with the indicated amounts of EPA ethylester and EPAD was measured in an assay using cytochrome c. Briefly, LLC cells (1 × 104 cells/well) were added to 120 µmol/L cytochrome c with the indicated amounts of EPA ethylester or EPAD, and incubated for 15, 30, 45 or 60 min at 37°C. After the incubation period, the plate was immediately (within 1 min) assayed in a microplate reader at 540 nm. released into the reaction mixture by cultured LLC cells treated with EPA ethylester or EPAD was determined by measuring the rate of reduction of cytochrome c. The data were expressed as µmol/L per h.
Animals. Female C57BL/6 strain mice (5 weeks old) were obtained from Clea Japan (Osaka, Japan). They were housed for 1 week in a room maintained at 25 ± 1°C with 60% relative humidity, and given free access to a standard laboratory diet (per 100 g of diet: corn starch 38 g, casein 25 g, α‐starch 10 g, cellulose powder 8 g, corn oil 6 g, sugar 5 g, mineral mixture [AIN‐76] 6 g and vitamin mixture [AIN‐76] 2 g; 332.8 kcal/100 g; Oriental Yeast Co. [Osaka, Japan]) and water. The room was illuminated for 12 h per day starting at 07.00 hours. Animals were treated according to the ethical guidelines of the Animal Center, School of Medicine, Ehime University. The Animal Studies Committee of Ehime University approved the experimental protocol.
Measurement of antitumor and antimetastatic activities in subcutaneous LLC‐implanted mice. Cultured LLC cells were harvested by trypsinization, washed, and suspended at 7 × 105 cells/mL in DMEM supplemented with 10% FBS. Solid‐type LLC was prepared by subcutaneous transplantation of 3.5 × 105 cells (0.5 mL) into the backs of C57BL/6 female mice on day 0. EPA ethylester or EPAD was sonicated in PBS (pH 7.0) containing 10% BSA (without free fatty acid), and the prepared emulsions were used in this study. EPA ethylester (300 or 1000 mg/kg) or EPAD (300 or 1000 mg/kg) was administered orally once (at 07.00 hours) daily for 28 consecutive days, starting 12 h after implantation of the tumor cells. Untreated mice (normal) and LLC‐bearing mice (control) were given PBS containing 10% BSA on the same schedule. The tumor volume was determined every 2–3 days by direct measurement with calipers and calculated using the formula (width2[mm2] × length [mm]/2). On day 29, blood was obtained from the mice via venipuncture with pentobarbital anesthesia, and then the spleen, thymus, lungs, and liver were removed and weighed for evaluation of the antitumor and antimetastatic activities and side‐effects. The blood samples were chilled in test tubes containing heparin, and the number of red cells and leukocytes and the hemoglobin content were measured using a Coulter counter (Japan Scientific Instruments, Tokyo, Japan). Metastases to the lung were counted using a stereoscopic microscope.
Measurement of CD4+ and CD8+ T‐cell and natural killer cell populations in the spleens of mice with subcutaneously implanted LLC. The spleen tissues were gently teased with dissecting forceps to release cells into cold PBS (pH 7.4). The cell suspension (5 mL) was layered on Lympholytes‐Mouse (5 mL) and centrifuged at 1500 g for 30 min to isolate the lymphocytes. The number of lymphocytes was measured using a Coulter counter. The cell concentration was adjusted to 2 × 107 cells/mL, and 10 µL of FITC‐labeled antimouse CD4 and CD8 antibodies, PE‐labeled antimouse NK, or FITC‐ and PE‐labeled negative controls were added to 100 µL of the cell suspension. After incubation for 30 min at 4°C, the lymphocytes were rinsed three times with 1 mL of PBS and centrifuged at 700 g for 5 min. Subsequently, the CD4+ and CD8+ T‐cell and NK cell populations were immediately analyzed by flow cytometry using a FACS Calibur (Becton Dickinson, Mountain View, CA, USA) without cell fixation. The percentages of negative FITC‐labeled lymphocytes (negative control for CD4+ and CD8+ T cells) or negative PE‐labeled lymphocytes (negative control for NK cells) were 0.13 ± 0.01% (n = 92) or 1.70 ± 0.01% (n = 46), respectively.
Histological examination. All tumors were fixed in 10% buffered formalin for at least 24 h, progressively dehydrated in solutions containing an increasing percentage of ethyl alcohol (70, 80, 95 and 100%), cleared in histoclear (FUME HOOD, AS‐ONE, Tokyo, Japan), embedded in paraffin under a vacuum, sectioned into 5‐µm‐thick sections, de‐paraffinized, and stained with Harris hematoxylin and eosin. After the paraffin‐embedded tumor sections were de‐paraffinized, the apoptotic cells in the tumors were quantitated by the TUNEL method using an Apoptosis In Situ Detection Kit (Wako). Four different microscopic fields (×400 magnification) per plate were photographed, and the TUNEL‐positive apoptotic cells in the tumors were counted. Expression of endothelium (tumor‐induced neovascularization) and the invasion of CD8+ T and NK cells into the tumors were also determined by the immunoperoxidase technique using antimouse CD34 monoclonal antibody (clone MEC14.7), antiasialo GM1 antibody, and anti‐CD8‐α antibody (D‐9), respectively.
Measurement of DNA synthesis in LLC cells. Lewis lung carcinoma cells were placed in DMEM supplemented with 10% FBS at 1 × 104 cells/well in 24‐well culture plates. After the cells were cultured overnight, the medium was changed to fresh DMEM supplemented with 10% FBS and the indicated amounts of EPA ethylester, EPAD, E‐2 or E‐3 for 20 h; then the medium was replaced with [3H]thymidine (18.5 kBq = 0.5 µCi/well) in DMEM supplemented with 10% FBS. After further incubation for 4 h, the cells were washed twice with cold phosphate buffered saline (PBS), immersed in 1 mL of 5% trichloroacetic acid (TCA) for 1 h at 4°C, washed twice with 5% TCA, and solubilized with 100 µL of 0.2 mmol/L NaOH containing 0.25% Triton X‐100. Thymidine incorporation into the cells was determined by liquid scintillation counting.
Measurement of DNA synthesis in HMVEC. Human adult dermal microvascular endothelial cells (1 × 104 cells/well) were seeded onto collagen (type I)‐coated 24‐well culture plates in CS‐C medium. After the cells were cultured overnight, the medium was replaced with fresh medium, and the cells were exposed to the indicated amounts of EPA ethylester, EPAD, E‐2 or E‐3 for 20 h. Thymidine incorporation was measured by the method described earlier.
Measurement of Matrigel‐induced neovascularization (in vivo). For in vivo experiments, Matrigel (without growth factor)‐induced neovascularization was assayed according to the method described by Passaniti et al.( 17 ) with some modifications. Briefly, female C57BL/6 mice were injected subcutaneously with 0.5 mL of Matrigel containing 20 ng of VEGF and 32 U of heparin per mL on day 0. EPA (300 or 1000 mg/kg) or EPAD (300 or 1000 mg/kg) was orally administered from day 1 to day 5 to mice implanted with Matrigel containing VEGF and heparin. The mice were killed on day 6 with an overdose of pentobarbital, and the gels were removed and weighed. The hemoglobin contents in the gels were determined using Hemoglobin Test kits (Wako).
Measurement of Matrigel‐induced tube‐like network formation of HMVEC (in vitro). Matrigel‐induced tube‐like formation of HMVEC was assayed according to the methods described in previous reports.( 18 , 19 , 20 ) Briefly, Matrigel supplemented with growth factor (150 µL) was placed into each well of a 48‐well culture plate at 4°C and allowed to polymerize by incubation for 1 h at 37°C. HMVEC (second passage, 2 × 104 cells) were seeded onto the Matrigel in 270 µL of DMEM supplemented with 20% FBS, and incubated with the indicated amounts of EPA ethylester, EPAD, E‐2 or E‐3 at 37°C for 24 h in a humidified 5% CO2/95% air atmosphere. Three different microscopic fields (×40 and ×100 magnification) per well were photographed, and the light micrograph images were stored in a computer. The total lengths of the tube structures in each photograph (×40 magnification) were measured using Adobe Photoshop software (Adobe, Tokyo, Japan).
Statistical analysis. All values were expressed as mean ± SE. Data were analyzed by one‐way anova, and then the differences among the means were analyzed using Fisher's protected least significant difference (LSD) multiple comparison test. Differences were considered significant at P < 0.05.
Results
Structures of E‐1, E‐2, E‐3 and E‐4 isolated from EPAD formed during accelerated testing. Based on the analysis of thin layer chromatography (TLC) and 1H‐NMR data, the by‐products of EPA ethylester formed during accelerated testing were a mixture of EPAD. We attempted to isolate pure compounds from the EPAD by repeated preparative TLC, and thereby obtained E‐1, E‐2, E‐3 and E‐4. E‐1 and E‐4 were identified as EPA ethylester (non‐reactive substance) and EPA (de‐esterified form of EPA ethylester), respectively, by direct comparison of the 1H‐ and 13C‐NMR spectra and MS spectrum with the respective spectra of each authentic sample. E‐2 and E‐3 were determined to be a EPA ethylester dimer and EPA hydroxyethylester, respectively, based on the analysis of 1H‐ and 13C‐NMR and MS spectra. We isolated two new EPA derivatives: the EPA ethylester dimer (E‐2) and EPA hydroxyethylester (E‐3). Our structural analysis of E‐1, E‐2, E‐3 and E‐4 is currently under submission for publication in journals that specialize in the chemical structures of natural products. Therefore, in this study, we compared the antitumor and antimetastatic activities of EPA ethylester and EPAD, including EPA ethylester (E‐1), the EPA ethylester dimer (E‐2), EPA hydroxyethylester (E‐3) and EPA (Fig. 1).
Figure 1.
Structures of E‐1, E‐2, E‐3 and E‐4 isolated from eicosapentaenoic acid derivatives formed during accelerated testing at 60 ± 5% relative humidity at 37°C for 30 days.
Lipid peroxidation and O2 − production in cultured LLC cells. We found that EPA ethylester (E‐1), EPAD (a mixture of E‐1, E‐2, E‐3 and E‐4), E‐2 and E‐3 increased the lipid peroxide levels in a dose‐dependent manner in the absence of LLC cells. The lipid peroxide levels produced in the presence of EPAD, E‐2 and E‐3 were higher than those produced in the presence of EPA ethylester without LLC cells. However, EPA ethylester increased the lipid peroxide levels to a concentration of 330 µg/mL in the presence of LLC cells, whereas EPA had no effect at concentrations of 0.033–33 µg/mL. EPAD and E‐3 increased the lipid peroxide levels at concentrations of 33 µg/mL and 330 µg/mL in the presence of LLC cells. E‐2 isolated from EPAD significantly increased the lipid peroxide levels at concentrations of 0.033–330 µg/mL in the presence of LLC cells, and the effect of E‐2 was stronger than that of EPA ethylester. Furthermore, EPA, EPAD, E‐2 and E‐3 increased the O2 − production by LLC cells at concentrations of 0.033–3.3 µg/mL, and the effect of E‐2 was stronger than that of EPA ethylester. There were no significant differences in O2 − production by LLC cells depending on whether they were treated with EPA ethylester, EPAD, or E‐3 (Table 1).
Table 1.
Lipid peroxidation and O2 − production in cultured Lewis lung carcinoma (LLC) cells treated with eicosapentaenoic acid (EPA) ethylester, EPA derivatives (EPAD), the EPA ethylester dimer (E‐2) and EPA hydroxyethylester (E‐3)
| Concentration (µg/mL) | Lipid peroxide levels (malondialdehyde nmol/well) | O2 − Production (µmol/h per well) (n = 4) | |
|---|---|---|---|
| Without LLC cells (n = 2) | With LLC cells (n = 4) | ||
| EPA ethylester | |||
| None | – | 0.427 ± 0.093 | 0.960 ± 0.320 |
| 0.033 | 0.182 | 0.610 ± 0.085 | 9.81 ± 1.14* |
| 0.33 | 0.273 | 0.547 ± 0.148 | 8.85 ± 2.97* |
| 3.3 | 0.546 | 0.564 ± 0.194 | 11.63 ± 1.86* |
| 33 | 1.694 | 0.416 ± 0.141 | ND |
| 330 | 20.63 | 3.230 ± 0.341* | ND |
| EPAD | |||
| None | – | 0.436 ± 0.071 | 0.959 ± 0.439 |
| 0.033 | 0.251 | 0.592 ± 0.081 | 12.90 ± 0.561* |
| 0.33 | 0.309 | 0.592 ± 0.049 | 13.44 ± 1.22* |
| 3.3 | 1.197 | 0.450 ± 0.025 | 13.55 ± 1.38* |
| 33 | 2.915 | 1.116 ± 0.245*, † | ND |
| 330 | 29.03 | 5.086 ± 0.635* | ND |
| E‐2 | |||
| None | – | 0.453 ± 0.073 | 0.946 ± 0.299 |
| 0.033 | 0.523 | 1.00 ± 0.133*, † | 16.00 ± 0.444*, † |
| 0.33 | 0.733 | 1.38 ± 0.221*, † | 19.83 ± 0.441*, † |
| 3.3 | 4.671 | 1.77 ± 0.238*, † | 21.37 ± 3.09*, † |
| 33 | 15.03 | 2.15 ± 0.171*, † | ND |
| 330 | 66.39 | 3.91 ± 0.293* | ND |
| E‐3 | |||
| None | – | 0.421 ± 0.064 | 0.939 ± 0.337 |
| 0.033 | 0.313 | 0.443 ± 0.066 | 10.31 ± 1.99* |
| 0.33 | 0.422 | 0.509 ± 0.038 | 14.88 ± 3.17* |
| 3.3 | 1.781 | 0.592 ± 0.061 | 15.09 ± 2.11* |
| 33 | 3.222 | 2.03 ± 0.172*, † | ND |
| 330 | 35.18 | 4.99 ± 0.551* | ND |
Significantly different from none (P < 0.05);
Significantly different from EPA ethylester (33 µg/mL; P < 0.05). ND, not determined because of turbidity caused by adding EPA ethylester, EPAD, E‐2 and E‐3 at the concentrations of 33 and 330 µg/mL.
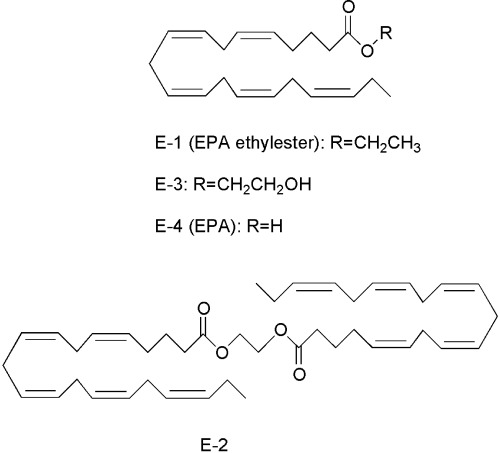
Inhibition of DNA synthesis in LLC cells and HMVEC, and of Matrigel (with growth factor)‐induced capillary‐like network formation by HMVEC. Eicosapentaenoic acid ethylester (E‐1), EPAD (a mixture of E‐1, E‐2, E‐3 and E‐4), E‐2 and E‐3 inhibited DNA synthesis in LLC cells at concentrations of 0.033–330 µg/mL; their IC50 values were 4.46 µg/mL, 5.45 µg/mL, 0.076 µg/mL and 0.060 µg/mL, respectively (Fig. 2a). Thus, the inhibitory effect of EPA ethylester on DNA synthesis in LLC cells was similar to that of EPAD, but the inhibitory effects of E‐2 and E‐3 isolated from EPAD on DNA synthesis in LLC cells were stronger than those of EPA ethylester. EPA ethylester, EPAD, E‐2 and E‐3 also inhibited DNA synthesis in HMVEC at concentrations of 0.33–330 µg/mL; their IC50 values were 39.6 µg/mL, 1.78 µg/mL, 0.39 µg/mL and 1.40 µg/mL, respectively. At concentrations of 3.30–33.0 µg/mL, EPAD, E‐2 and E‐3 significantly inhibited DNA synthesis in HMVEC, compared with the inhibition by EPA ethylester (Fig. 2b). Among these compounds, E‐2 inhibited the DNA synthesis in HMVEC most strongly. HMVEC formed capillary‐like networks on Matrigel 8 h after seeding. EPA ethylester and EPAD inhibited the angiogenesis of HMVEC at 0.33–330 µg/mL in a dose‐dependent manner (Fig. 3). The inhibitory effect of EPAD at a concentration of 33.0 µg/mL on Matrigel‐induced capillary‐like network formation was significantly stronger than that of EPA at the same concentration. Furthermore, E‐2 and E‐3 isolated from EPAD also inhibited the capillary‐like tube formation of HMVEC more potently than EPA ethylester.
Figure 2.
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives E‐2 and E‐3 on 3H‐thymidine incorporation into the DNA of (a) Lewis lung carcinoma cells or (b) human adult dermal microvascular endothelial cells. Values are mean ± SE; n = 4. *Significantly different from medium alone (P < 0.05); #Significantly different from EPA at 0.33, 3.3 and 33 µg/mL (P < 0.05).
Figure 3.
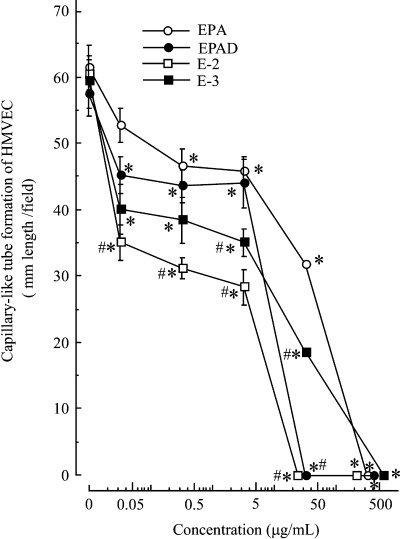
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives E‐2 and E‐3 on Matrigel‐induced capillary‐like network formation by human adult dermal microvascular endothelial cells (HMVEC). Values are mean ± SE; n = 4. *Significantly different from medium alone (P < 0.05); #Significantly (P < 0.05) different from EPA‐treated HMVEC at 0.33, 3.3 and 33 µg/mL.
Matrigel‐induced angiogenesis (in vivo). Based on these results in vitro, we examined the effects of EPA ethylester and EPAD on Matrigel‐induced angiogenesis using an in vivo model. Mice injected with Matrigel supplemented with VEGF (20 ng/mL) and heparin (32 U/mL) showed significantly increased gel weight, and increased hemoglobin content in the gels 6 days after implantation, compared with mice injected with Matrigel (without growth factor) alone (Table 2). Oral administration of EPAD (300 or 1000 mg/kg) significantly inhibited the increase in the weight and hemoglobin content of the gels, but EPA ethylester (300 or 1000 mg/kg) had no effect (Table 2). Moreover, at a dose of 300 mg/kg, oral administration of EPAD inhibited Matrigel‐induced angiogenesis significantly more potently than did EPA ethylester.
Table 2.
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives (EPAD) on Matrigel‐induced angiogenesis (in vivo)
| Treatment | n | Matrigel weight (mg) | Hemoglobin content (mg/Matrigel) |
|---|---|---|---|
| Matrigel alone | 10 | 94.1 ± 9.56* | 2.53 ± 0.22* |
| + VEGF (20 ng/mL) + heparin (32 U/mL) (control) | 11 | 331.8 ± 41.3 | 22.87 ± 2.77 |
| + VEGF, heparin + EPA ethylester (300 mg/kg) | 5 | 323.3 ± 73.3 | 15.31 ± 5.58 |
| + VEGF, heparin + EPA ethylester (1000 mg/kg) | 5 | 220.3 ± 93.1 | 16.27 ± 10.25 |
| + VEGF, heparin + EPAD (300 mg/kg) | 5 | 162.0 ± 32.6*, † | 9.72 ± 3.27* |
| + VEGF, heparin + EPAD (1000 mg/kg) | 5 | 182.0 ± 50.0*, † | 9.60 ± 2.09* |
VEGF, vascular endothelial growth factor. C57BL/6 mice were injected subcutaneously with 0.5 mL of Matrigel alone, or Matrigel supplemented with 20 ng VEGF/mL and 32 U heparin/L on day 0. EPA ethylester (300 or 1000 mg/kg) or EPAD (300 or 1000 mg/kg) was administered orally from day 1 to day 5. The mice were killed on day 6, and the gels were removed and weighed, and the hemoglobin content in the gels was measured. Values are mean ± SE. *Significantly different from Matrigel/VEGF/heparin‐treated groups (P < 0.05); †Significantly different from Matrigel/VEGF/heparin plus EPAD (300 mg/kg)‐treated groups (P < 0.05).
Antitumor and antimetastatic activities, body weight, various tissue weights, blood cell numbers, and hemoglobin content in the blood of mice with subcutaneously implanted LLC. At a dose of 1000 mg/kg, EPAD inhibited tumor growth in mice with subcutaneously implanted LLC, but EPA ethylester (300 or 1000 mg/kg) and EPAD (300 mg/kg) did not inhibit tumor growth (Fig. 4). However, there were no significant differences in the effects caused by EPAD and EPA ethylester. The final tumor weight on day 29 was significantly reduced by the oral administration of EPA ethylester (300 or 1000 mg/kg) and EPAD (1000 mg/kg) compared with that in untreated LLC‐bearing mice (controls; Table 3). The mice with subcutaneously implanted LLC cells showed tumor metastasis to the lungs. EPAD (1000 mg/kg) reduced the number of metastatic tumor colonies in the lungs compared with that in the untreated LLC‐bearing mice, but EPA ethylester (300 or 1000 mg/kg) and EPAD (300 mg/kg) had no effect (Table 3). The final body, liver, and lung weights did not vary among the normal mice, untreated LLC‐bearing mice, EPA ethylester‐, and EPAD‐treated LLC‐bearing mice (data not shown). In the present study, steatosis (such as fatty liver) was not caused by the oral administration of EPA ethylester or EPAD in LLC‐bearing mice. The spleen weight in the LLC‐bearing mice was significantly increased to 195.3 ± 30.4 mg (n = 8), compared with 60.41 ± 5.28 mg (n = 5) in the normal mice (P < 0.05), and the thymus weight in the untreated LLC‐bearing mice was significantly reduced to 44.38 ± 5.77 mg (n = 8), compared to 60.01 ± 3.39 mg (n = 5) in the normal mice (P < 0.05). The administration of EPA or EPAD had no effect on the spleen or thymus weight compared with that in the untreated LLC‐bearing mice (data not shown). The number of leukocytes in the LLC‐bearing mice (4.81 ± 0.710 × 103 cells/µL, n = 8) was significantly greater than that in the normal mice (2.15 ± 0.11 × 103 cells/µL, n = 5; P < 0.05). In contrast, the number of red cells (560.6 ± 69.7 × 104 cells/µL, n = 8) and the hemoglobin concentration (8.48 ± 1.07 g/100 mL) in the untreated LLC‐bearing mice were significantly lower than those in the normal mice (790.4 ± 10.3 × 104 red cells/µL and 11.1 ± 0.10 g hemoglobin/100 mL). EPA ethylester and EPAD did not affect the number of leukocytes or red cells, or the hemoglobin concentration (in comparison with the values from untreated LLC‐bearing mice; data not shown; Fig. 4 and Table 3).
Figure 4.
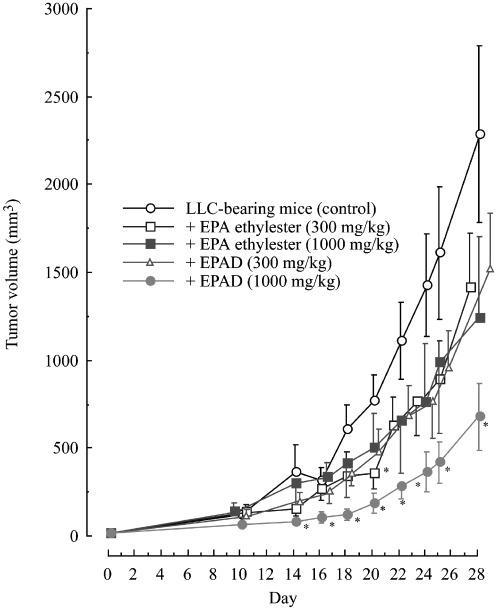
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives (EPAD) on primary solid tumor growth in mice with subcutaneously implanted Lewis lung carcinoma (LLC). Solid‐type LLC were prepared by the subcutaneous implantation of 3.5 × 105 cells (0.5 mL) into the backs of C57BL/6 female mice on day 0. Values are mean ± SE. The untreated group (normal) consisted of five mice; the LLC‐bearing group (control), and EPA ethylester (300 or 1000 mg/kg)‐ or EPAD (300 mg/kg)‐treated groups consisted of eight mice; the EPAD (1000 mg/kg)‐treated groups consisted of nine mice per group. *Significantly different from LLC‐bearing mice (control; P < 0.05).
Table 3.
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives (EPAD) on tumor weight and lung metastasis on day 29 in mice with subcutaneously implanted Lewis lung carcinoma (LLC)
| Treatment | n | Tumor weight (mg) | Metastasis to the lung | |
|---|---|---|---|---|
| (no. tumor colonies) | (no. mice with metastasis) | |||
| LLC‐bearing mice (control) | 8 | 1576.0 ± 480.0 (100%) | 8 ± 3 | 4/8 (50%) |
| + EPA ethylester (300 mg/kg) | 8 | 709.8 ± 194.1* (45.0%) | 3 ± 2 | 3/8 (37.5%) |
| + EPA ethylester (1000 mg/kg) | 8 | 722.4 ± 250.8* (45.8%) | 3 ± 2 | 3/8 (37.5%) |
| + EPAD (300 mg/kg) | 8 | 1093.1 ± 297.0 † (69.4%) | 3 ± 1 | 3/8 (37.5%) |
| + EPAD (1000 mg/kg) | 9 | 485.1 ± 159.5* (30.8%) | 1 ± 1 | 2/9 (22.2%) |
Values are mean ± SE. *Significantly different from untreated LLC‐bearing mice (control; P < 0.05); †Significantly different from LLC‐bearing mice treated with EPA ethylester at a dose of 300 mg/kg (P < 0.05).
2Immune function in the spleen of mice with subcutaneously implanted LLC. The fractions of splenic CD4+ and CD8+ T cells in the LLC‐bearing mice were significantly reduced to 12.8 ± 1.31 and 9.66 ± 1.00% from 20.5 ± 0.27 and 38.8 ± 1.50% in the normal mice, respectively. In contrast, the fraction of splenic NK cells (21.6 ± 3.56%) in the untreated LLC‐bearing mice was significantly greater than that (5.04 ± 0.22%) in the normal mice. The fraction of CD4+ T cells in the untreated LLC‐bearing mice was further reduced by the oral administration of EPA ethylester or EPAD at a dose of 300 or 1000 mg/kg (Table 4). However, the reduction of CD8+ T cells in the untreated LLC‐bearing mice was significantly inhibited by the oral administration of EPA ethylester (1000 mg/kg) or EPAD (300 or 1000 mg/kg), compared with that in the untreated LLC‐bearing mice. The inhibitory effect of EPAD on the reduction of CD8+ T cells in LLC‐bearing mice was significantly stronger than that of EPA. The increase in NK cells in the untreated LLC‐bearing mice was further augmented by the oral administration of EPA ethylester at a dose of 300 or 1000 mg/kg, and the enhancing effect of EPA ethylester (1000 mg/kg) on the increase of NK cells in LLC‐bearing mice was significantly stronger than that of EPAD at the same dose (Table 4). Because the antitumor and antimetastatic actions of E‐2 and E‐3 of EPAD were not evaluated in LLC‐bearing mice, the effects of E‐2 and E‐3 on immune function in LLC‐bearing mice are unknown. The relationship between the antitumor actions and immune functions of E‐2 and/or E‐3 should be clarified in future studies.
Table 4.
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives (EPAD) on splenic CD4+ and CD8+ T cell and natural killer (NK+) cell populations on day 29 in mice with subcutaneously implanted Lewis lung carcinoma (LLC)
| Treatment | n | Lymphocytes (×106 cells per spleen) | CD4+ T cells (%) | CD8+ T cells (%) | NK+ cells (%) |
|---|---|---|---|---|---|
| Normal | 5 | 30.5 ± 0.46 | 20.5 ± 0.27 | 38.8 ± 1.50 | 5.04 ± 0.22 |
| LLC‐bearing mice (control) | 8 | 12.2 ± 0.45* | 12.8 ± 1.31* | 9.66 ± 1.00* | 21.6 ± 3.56* |
| + EPA ethylester (300 mg/kg) | 8 | 11.9 ± 0.76* | 6.82 ± 0.71*, † | 12.8 ± 0.97* | 36.2 ± 2.25*, † |
| + EPA ethylester (1000 mg/kg) | 8 | 11.2 ± 0.62* | 7.87 ± 1.06*, † | 16.0 ± 1.25*, † | 33.4 ± 2.65*, † |
| + EPAD (300 mg/kg) | 8 | 14.5 ± 1.44* | 9.36 ± 1.48*, † | 17.4 ± 1.89*, † , ‡ | 29.3 ± 4.19* |
| + EPAD (1000 mg/kg) | 9 | 11.9 ± 0.96* | 8.31 ± 1.04*, † | 20.2 ± 1.64*, † , § | 23.6 ± 3.68*, § |
Values are mean ± SE. *Significantly different from normal mice (P < 0.05); †Significantly different from LLC‐bearing mice (control; P < 0.05);
Significantly (P < 0.05) different from EPA (300 mg/kg)‐treated LLC‐bearing mice;
Significantly (P < 0.05) different from EPA (1000 mg/kg)‐treated LLC‐bearing mice.
Immunohistochemistry of tumors in mice with subcutaneously implanted LLC. Figure 5a shows that EPA ethylester (1000 mg/kg) and EPAD (300 or 1000 mg/kg) significantly increased the number of apoptotic cells in the tumors of mice with subcutaneously implanted LLC. However, the increases of apoptotic cells in the tumors did not differ between LLC‐bearing mice treated with EPAD and EPA ethylester. In the untreated LLC‐bearing mice, CD 34 (a marker of small vessels and neoformed capillaries) expression was increased in the tumors (Fig. 5b). This finding showed that neovascularization was induced together with the tumor growth. Angiogenesis in the tumors was inhibited by the oral administration of EPA ethylester (1000 mg/kg) or EPAD (300 or 1000 mg/kg; Fig. 5b, Table 5). The inhibition of CD34 expression also did not differ significantly between EPAD‐ and EPA ethylester‐treated LLC‐bearing mice. As shown in Figure 5c, CD8+ T cells invaded the central area of the tumors after the oral administration of EPA ethylester (300 or 1000 mg/kg) or EPAD (300 or 1000 mg/kg) and the increase of the CD8+ T‐cell number caused by EPAD (1000 mg/kg) was significantly greater than that caused by EPA ethylester (1000 mg/kg). However, NK cells invaded the central area of the tumors after the oral administration of EPA ethylester (300 or 1000 mg/kg), but EPAD (300 or 1000 mg/kg) did not cause a significant difference from the tumors in the untreated LLC‐bearing mice (control; Fig. 5d; Table 5).
Figure 5.
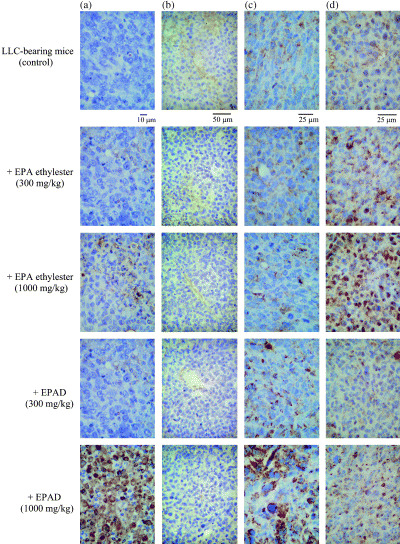
Light micrographs of tumor apoptosis (×400 magnification): (a) stained by the terminal deoxyribonucleotidyl transferase‐mediated dUTP–digoxigenin nick end‐labeling procedure, tumor angiogenesis (×200 magnification); (b) the invasion of CD8+ T cells in tumors (×400 magnification); (c) the invasion of natural killer cells in tumors (×400 magnification); (d) stained by the immunoperoxidase technique using antimouse CD34 rat monoclonal antibody, antihuman CD8α mouse monoclonal antibody and antiasialo GM1 rabbit polyclonal antibody, respectively, and counterstained with hematoxylin on day 29 in mice with subcutaneously implanted Lewis lung carcinoma (LLC). Normal and LLC‐implanted mice (control) were given distilled water alone for 28 days. Other LLC‐implanted mice were administered eicosapentaenoic acid (EPA) ethylester (300 or 1000 mg/kg) or EPA derivatives (300 or 1000 mg/kg) orally once daily for 28 days.
Table 5.
Effects of eicosapentaenoic acid (EPA) ethylester and EPA derivatives (EPAD) on apoptotic cell number, CD34 expression, CD8‐, and natural killer (NK)‐positive cell numbers in tumors of mice with subcutaneously implanted Lewis lung carcinoma (LLC)
| Treatment | n | Apoptotic cells (number/field) | CD34 expression (area µm2/field) | CD8‐positive cells (number/field) | NK‐positive cells (number/field) |
|---|---|---|---|---|---|
| LLC‐bearing mice (control) | 8 | 1 ± 1 | 4246.4 ± 876.9 | 121 ± 11 | 239 ± 61 |
| + EPA ethylester | |||||
| (300 mg/kg) | 8 | 49 ± 22* | 2111.9 ± 656.8* | 287 ± 29* | 436 ± 48* |
| (1000 mg/kg) | 8 | 77 ± 23* | 1480.4 ± 850.1* | 190 ± 32 | 490 ± 47* |
| + EPAD | |||||
| (300 mg/kg) | 8 | 54 ± 13* | 1417.1 ± 761.2* | 289 ± 57* | 320 ± 61 |
| (1000 mg/kg) | 7 | 77 ± 15* | 273.2 ± 147.9* | 525 ± 61*, † | 345 ± 28 |
Values are mean ± SE. Apoptotic cell number was measured using ×400 magnification. CD34 expression area was measured using ×100 magnification. CD8‐ and NK‐positive cell numbers were measured using ×200 magnification. Of the tumors in the nine mice in the EPAD‐treated group, the tumors of two mice had disappeared. *Significantly different from LLC‐bearing mice (control; P < 0.05); †Significantly different from EPA (1000 mg/kg)‐treated LLC‐bearing mice (P < 0.05).
Discussion
Surgery to excise breast carcinoma, colon carcinoma, and osteogenic sarcoma may be followed by the rapid growth of distant metastases in such places as the lungs, liver, and large intestine. It has been reported that subcutaneous LLC implantation in the foot pad or back of C57BL/6 mice resulted in lung metastasis in addition to tumor growth.( 21 , 22 , 23 , 24 ) EPA is orally administered three times daily at doses of 300–900 mg in humans for atherosclerotic obliteration and the treatment of hyperlipidemia. Hirai et al.( 25 ) reported in an epidemiological study in 1980 that the average daily intake of fish in the residents of a fishing village was 260 g per day, which is equivalent to 2.6 g of EPA and 1.8 g of DHA, whereas the equivalent intake in the residents of a farming village was 90 g of fish per day, which is equivalent to 900 mg of EPA and 600 mg of DHA. Therefore, in the present study, excess doses of approximately 10‐fold (300 mg/kg body weight per day) and approximately 30‐fold (1000 mg/kg body weight per day) EPA ethylester or EPAD (formed during accelerated testing) relative to the therapeutic and intake doses of EPA cited here were used to evaluate their pharmacological actions. PUFA (n‐3), like EPA and DHA, have been reported to have antitumor and antimetastatic activities in the lung in tumor‐bearing animals in vivo, and to inhibit tumor cell growth in vitro,( 7 , 8 , 9 , 26 , 27 , 28 , 29 ) and pure EPA and DHA are very unstable during long‐term storage. Therefore, it seems likely that EPAD formed during accelerated testing would have no effect on antitumor and antimetastatic activities. However, we isolated new two EPA derivatives, namely a dimer of EPA ethylester (E‐2) and EPA hydroxyethylester (E‐3), as well as EPA ethylester and EPA from EPAD formed during accelerated testing to evaluate drug stability. Therefore, we attempted to compare the antitumor and antimetastatic effects of EPA and EPAD in this study. In a preliminary study, the lipid peroxide‐inducing activity of EPAD was stronger than that of EPA. It was found that the effects of EPAD on lipid peroxidation and O2 − production in cultured LLC cells tended to be slightly stronger than those of EPA ethylester, although there were no significant differences between EPAD and EPA ethylester. The LLC cytotoxicity of EPAD was similar to that of EPA ethylester, and the antitumor effects of EPAD and EPA ethylester on primary solid tumors in mice with subcutaneously implanted LLC were equal. The same levels of tumor apoptosis were also induced by EPAD and EPA ethylester. These findings suggest that the antitumor actions of EPAD and EPA ethylester may be caused by tumor cell death via tumor apoptosis resulting from lipid peroxidation and O2 − production. Solid tumors cause neovascularization, and the resultant angiogenesis stimulates tumor growth and tumor metastasis.( 12 , 30 , 31 , 32 , 33 ) In vitro, EPA ethylester and EPAD inhibited the Matrigel‐induced capillary‐like tube formation by HMVEC, and the inhibitory effect of EPAD on Matrigel‐induced angiogenesis was stronger than that of EPA ethylester at a concentration of 33.0 µg/mL. Furthermore, in an in vivo study, EPAD (300 and 1000 mg/kg) significantly inhibited the angiogenesis induced by a Matrigel/VEGF/heparin mixture, but EPA ethylester did not at the same doses. The inhibitory effects of EPAD, E‐2 and E‐3 on HMVEC DNA synthesis were stronger that those of EPA ethylester. Therefore, it seems likely that the antiangiogenesis of EPAD and EPA ethylester may be related to the strength of the inhibitory effect on HMVEC DNA synthesis. However, the inhibitory effects of EPAD and EPA ethylester on CD 34 (tumor‐induced neovascularization) expression in the tumors were equally potent at doses of 300 and 1000 mg/kg in an immunohistochemical study. These findings indicate that the inhibition of tumor‐induced neovascularization by EPAD and EPA ethylester might be associated with the inhibition of tumor growth by these fatty acids. However, EPAD (1000 mg/kg) inhibited lung metastasis in LLC‐bearing mice, but EPA did not at the same dose. Therefore, the mechanisms of the antimetastatic actions of EPAD and EPA ethylester could not be sufficiently explained by the antiangiogenic action alone of EPAD or EPA ethylester. This suggests that there are other mechanisms by which these compounds act in addition to direct cytotoxicity against tumors and antiangiogenic actions. To identify other mechanisms of the antitumor and/or antimetastatic actions of EPAD or EPA ethylester, the effects of EPAD and EPA ethylester on immune functions in LLC‐bearing mice were studied. Oral administration of EPAD (300 or 1000 mg/kg) or EPA ethylester (1000 mg/kg) inhibited the reduction of CD8+ T‐cell number in the spleens of untreated LLC‐bearing mice. The inhibitory effect of EPAD on the reduction of CD8+ T cells was significantly stronger than that of EPA ethylester at the same dose. Moreover, the invasion of CD8+ T cells into the tumors was also increased by the oral administration of EPAD (300 and 1000 mg/kg) or EPA ethylester (300 mg/kg) compared to that in untreated LLC‐bearing mice (control). The effect of EPAD on CD8+ T‐cell invasion into the tumors was significantly stronger than that of EPA ethylester at the same dose. However, EPA ethylester (300 and 1000 mg/kg) enhanced the number of NK cells in the spleen compared with that in untreated LLC‐bearing mice (control), but EPAD had no significant effect compared with the control. The invasion of NK cells into the tumors was increased by the oral administration of EPA ethylester (300 and 1000 mg/kg), but not by EPAD at the same doses. It was found that EPA ethylester and EPAD stimulated specific immune functions in the spleen. NK cells have been shown to mediate strong antitumor and antimetastatic activities.( 34 , 35 , 36 ) Both CD4+ and CD8+ T cells have been reported to be important for the induction of tumor regression and development of protective immunity.( 37 , 38 , 39 , 40 ) Recently, it has been reported that interleukin (IL)‐21 exerts potent antitumor effects through the activation of both NK and CD8+ T cells.( 41 ) However, Lo et al. reported that the antitumor and antimetastatic activities of IL‐23 require CD8+ T cells, but not CD4+ T cells or NK cells.( 42 ) Brunda et al. reported that the depletion of CD8+ but not CD4+ T cells significantly reduced the antitumor and antimetastatic activities of IL‐12.( 43 ) It seems likely that the antitumor and antimetastatic activities of EPAD may be partly associated with the activation of CD8+ T cells in immune function. However, EPA ethylester may be partly associated with the activation of NK cells. Further studies will be needed to clarify the differences in the mechanisms of EPAD and EPA ethylester in immune response. The antitumor and antimetastatic activities of EPAD may be due to the combination of inhibition of DNA synthesis in LLC cells, increase of tumor apoptosis, inhibition of tumor‐induced neovascularization, and activation of CD8+ T cells; however, the antitumor activity of EPA ethylester may be due to the combination of inhibition of DNA synthesis in LLC cells, an increase of tumor apoptosis, inhibition of tumor‐induced neovascularization, and activation of NK cells. From our in vitro results, E‐2 (EPA ethylester dimer) inhibited the DNA synthesis in LLC and HMVEC cells most strongly of the four EPAD. Therefore, it seems likely that the EPA ethylester dimer (E‐2) may potently suppress LLC cell growth and metastasis through the inhibition of DNA synthesis in LLC and HMVEC cells, and the enhancement of lipid peroxidation and O2 − production in LLC cells. Further studies are needed to clarify the anitumor and antimetastatic actions of EPA ethylester dimer in in vivo models. Thus, it is suggested that the mechanisms of the antitumor and/or antimetastatic actions of EPAD and EPA ethylester involve different immune functions, and the EPA ethylester dimer and EPA hydroxyethylester may contribute to these actions. Further mechanistic studies will be needed to clarify the interrelations of effects on immune function, angiogenesis, tumor apoptosis, tumor cell proliferation and cytokine production by EPA ethylester, the EPA ethylester dimer and EPA hydroxyethylester from EPAD.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Mochida Pharmaceutical Co. (Osaka, Japan) for providing EPA.
References
- 1. Beck SA, Smith KL, Tisdale MJ. Anticachectic and antitumor effect of eicosapentaenoic acid and its effect on protein turnover. Cancer Res 1991; 51: 6089–93. [PubMed] [Google Scholar]
- 2. Noguchi M, Earashi M, Minami M, Kinoshita K, Miyataki I. Effects of eicosapentaenoic acid and docosahexaenoic acid on cell growth and prostaglandin E and leukotriene B production by a human breast cancer cell line (MDA‐MB‐231). Oncology 1995; 52: 458–64. [DOI] [PubMed] [Google Scholar]
- 3. Pandalai PK, Pilat MJ, Yamazaki K, Naik H, Pienta KJ. The effects of ω‐3 and ω‐6 fatty acids on in vitro prostate cancer growth. Anticancer Res 1996; 16: 815–20. [PubMed] [Google Scholar]
- 4. Suzuki I, Iigo M, Ishikawa C et al. Inhibitory effects of oleic and docosahexaenoic acids on lung metastasis by colon carcinoma 26 cells are associated with reduced matrix metaloproteinase‐2 and – 9 activities. Int J Cancer 1997; 73: 607–12. [DOI] [PubMed] [Google Scholar]
- 5. Yang SP, Morita I, Murota S‐Z. Eicosapentaenoic acid attenuates vascular endothelial growth factor‐induced proliferation via inhibiting Flk‐1 receptor expression in bovine carotid artery endothelial cells. J Cell Physiol 1998; 176: 342–9. [DOI] [PubMed] [Google Scholar]
- 6. Park Y, Albright KJ, Storkson JM, Liu W, Cook ME, Pariza MW. Changes in body composition in mice during feeding and withdrawal of conjugated linoleic acid. Lipids 1999; 34: 243–8. [DOI] [PubMed] [Google Scholar]
- 7. Caygill CPJ, Charlett A, Hill MJ. Fat, fish, fish oil and cancer. Br J Cancer 1996; 74: 159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandez E, Chatenoud L, La Vechia C, Negri E, Franceschi S. Fish consumption and cancer risk. Am J Clin Nutr 1999; 70: 85–90. [DOI] [PubMed] [Google Scholar]
- 9. Kata I, Akhmedkhanov A, Koenig K, Toniola PG, Shore RE, Riboli E. Prospective study of diet and female colorectal cancer: The New York University Women's Health Study. Nutr Cancer 1997; 33: 132–8. [DOI] [PubMed] [Google Scholar]
- 10. Weiss L, Orr FW, Honn KV. Interactions of cancer cells with the microvasculature during metastasis. FASEB J 1998; 2: 12–21. [DOI] [PubMed] [Google Scholar]
- 11. Blood CH, Zetter BR. Tumor interactions with vasculature: angiogenesis and tumor metastasis. Biochim Biopys Acta 1990; 1032: 89–118. [DOI] [PubMed] [Google Scholar]
- 12. Ferrara N. Missing link in angiogenesis. Nature 1995; 376: 467. [DOI] [PubMed] [Google Scholar]
- 13. Kerbel RS. A cancer therapy resistant to resistance. Nature 1997; 390: 335–6. [DOI] [PubMed] [Google Scholar]
- 14. Garlanda C, Berthier R, Garin J et al. Characterization of MEC 14 7, a new monoclonal antibody recognizing mouse CD34: a useful reagent for identifying and characterizing blood vessels and hematopoietic precursors. Eur J Cell Biol 1997; 73: 368–77. [PubMed] [Google Scholar]
- 15. International Conference on Harmonization Steering Committee . Stability Testing of New Drug Substances and Products. ICH Harmonised Tripartite Guideline. Geneva: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2003. [Google Scholar]
- 16. Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979; 95: 351–8. [DOI] [PubMed] [Google Scholar]
- 17. Passaniti A, Taylor RM, Pili R et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin and fibroblast growth factor. Laboratory Invest 1992; 67: 519–28. [PubMed] [Google Scholar]
- 18. Kimura Y, Okuda H. Effects of naturally occurring stilbene glucosides from medicinal plants and wine, on tumor growth and lung metastasis in Lewis lung carcinoma‐bearing mice. J Pharm Pharmacol 2000; 52: 1287–95. [DOI] [PubMed] [Google Scholar]
- 19. Kimura Y, Baba K, Okuda H. Inhibitory effects of active substances isolated from Cassia garrettiana heartwood on tumor growth and lung metastasis in Lewis lung carcinoma‐bearing mice (Part 1). Anticancer Res 2000; 20: 2899–906. [PubMed] [Google Scholar]
- 20. Kimura Y, Okuda H. Resveratrol isolated from Polygonum cuspidateum root prevents tumor growth and metastasis to lung and tumor‐induced neovascularization in Lewis lung carcinoma‐bearing mice. J Nutr 2001; 131: 1844–9. [DOI] [PubMed] [Google Scholar]
- 21. Dewhys WD. Studies correlating the growth rate of a tumor and its metastases and providing evidence for tumor‐related systemic growth‐retarding factor. Cancer Res 1972; 32: 374–9. [PubMed] [Google Scholar]
- 22. Gorelik E, Segel S, Feldman M. Growth of a local tumor exerts a specific inhibitory effect on progression of lung metastases. Int J Cancer 1978; 21: 617–25. [DOI] [PubMed] [Google Scholar]
- 23. Gorelik E, Segal S, Feldman M. Control of lung metastasis progression in mice: role of growth kinetics of 3LL Lewis lung carcinoma and host immune activity. J Natl Cancer Inst 1980; 65: 1257–64. [PubMed] [Google Scholar]
- 24. O'Reilly MS, Homgren L, Shing Y et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994; 79: 315–28. [DOI] [PubMed] [Google Scholar]
- 25. Hirai A, Terano T, Tamura Y, Yoshida S. Eicosapentaenoic acid and adult diseases in Japan: Epidemiological and clinical aspects. J Intern Med 1989; 225 (Suppl. 1): 69–75. [DOI] [PubMed] [Google Scholar]
- 26. Madhavi N, Das UM. Effects of n‐6 and n‐3 fatty acids on the survival of vincristine sensitive and resistant human cervical carcinoma cells in vitro . Cancer Lett 1994; 84: 31–41. [DOI] [PubMed] [Google Scholar]
- 27. Rose DP, Connolly JM, Coleman M. Effect of ω‐3 fatty acids on the progression of metastases after the surgical excision of human breast cancer cell solid tumors growing in nude mice. Clin Cancer Res 1996; 2: 1751–6. [PubMed] [Google Scholar]
- 28. Iwamoto S, Senzaki H, Kiyozuka Y et al. Effects of fatty acids on liver metastasis of ACL‐15 rat colon cancer cells. Nutr Cancer 1998; 31: 143–50. [DOI] [PubMed] [Google Scholar]
- 29. Senzaki H, Iwamoto S, Ogura E et al. Dietary effects of fatty acids on growth and metastasis of KPL‐1 human breast cancer cells in vivo and in vitro . Anticancer Res 1998; 18: 1621–7. [PubMed] [Google Scholar]
- 30. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1: 27–31. [DOI] [PubMed] [Google Scholar]
- 31. Koch AE, Halloran MM, Haskell CJ, Shah MR, Polverini PJ. Angiogenesis mediated by soluble forms of E‐selectin and vascular cell adhesion molecule‐1. Nature 1995; 376: 517–9. [DOI] [PubMed] [Google Scholar]
- 32. O'Reilly MS, Boem T, Shing Y et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997; 88: 277–85. [DOI] [PubMed] [Google Scholar]
- 33. Boem T, Folkman J, Browder TJ, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997; 390: 404–7. [DOI] [PubMed] [Google Scholar]
- 34. Pross HF, Lotzova E. Role of natural killer cells in cancer. Nat. Immun 1993; 12: 279–92. [PubMed] [Google Scholar]
- 35. Lode HN, Xiang R, Dreier T, Varki NM, Gillies SD, Reisfedl RA. Natural killer cell‐mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin‐2 therapy. Blood 1998; 91: 1706–15. [PubMed] [Google Scholar]
- 36. Whiteside TL, Herberman RB. The role of natural killer cells in immune surveillance of cancer. Curr Opin Imuunol 1995; 7: 704–10. [DOI] [PubMed] [Google Scholar]
- 37. Hock H, Dorsch M, Kunzendorf U, Qin Z, Diamanstein T, Blankenstein T. Mechanisms of rejection induced by tumor cell‐targeted gene transfer of interleukin 2, interleukin 4, interleukin 7, tumor necrosis factor, or interferon γ. Proc Natl Acad Sci USA 1993; 90: 2774–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hung K, Hayashi R, Lafond‐Walker A, Lowebstein C, Pardoll D, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med 1998; 188: 2357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Segal BM, Glass DD, Shevach EM. Cutting edge: IL‐10‐producing CD4+ T cells mediate tumor rejection. J Immunol 2002; 168: 1–4. [DOI] [PubMed] [Google Scholar]
- 40. Colombo HP, Forni G. Cytokine gene transfer in tumor inhibition and tumor therapy: where are we now? Imuunol Today 1994; 15: 48–51. [DOI] [PubMed] [Google Scholar]
- 41. Ma H‐L, Whitters MJ, Konz RF et al. IL‐21 activates both innate and adaptive immunity to generate potent antitumor responses that require perforin but are independent of IFN‐γ. J Immunol 2003; 171: 608–15. [DOI] [PubMed] [Google Scholar]
- 42. Lo C‐H, Lee S‐C, Wu P‐Y et al. Antitumor and antimetastatic activity of IL‐23. J Immunol 2003; 171: 600–7. [DOI] [PubMed] [Google Scholar]
- 43. Brunda M, Luistro L, Warrier RR et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 1993; 178: 1223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]