

Abstract
MicroRNAs (miRNAs) are ∼22 nt long, non‐coding RNAs that guide post‐transcriptional gene silencing of their target genes and regulate diverse biological processes including cancer. miRNAs do not act alone, but require assembly into RNA‐induced silencing complex (RISC). In this review, we summarize how miRNAs are produced, assembled into RISC, and regulate target mRNAs, and discuss how the miRNA pathway is involved in cancer. (Cancer Sci 2010; 101: 2309–2315)
In the last decade, many non‐coding RNAs were found to regulate a wide variety of biological processes. Among these, microRNAs (miRNAs) are the most well‐characterized. miRNAs are small RNAs, ∼22 nt long, that induce post‐transcriptional gene silencing. miRNAs recognize their target mRNAs by base complementarity. However, miRNAs are, on their own, unable to regulate expression of their target mRNAs. Rather, they need to be assembled into a complex termed RNA‐induced silencing complex (RISC). Within RISC, miRNAs act as the specificity determinants while protein components achieve target mRNA silencing. Therefore, considering miRNAs in the context of RISC is crucial to understanding how miRNAs function.
MicroRNAs regulate surprisingly diverse biological processes. Inhibition of the miRNA biogenesis pathway incurs severe developmental defects and lethality in many organisms. To date, more than 900 human miRNA genes are registered in the miRNA repository miRBase (release 15.0), and it is proposed that miRNAs regulate more than one‐third of all human genes. Not surprisingly, it has been suggested that a considerable number of miRNAs are involved in cancer. In this review, we focus on how miRNAs are produced and assembled into RISCs, how miRNA‐containing RISCs silence their target genes, and how the miRNA pathway is involved in cancer or vice‐versa.
Biogenesis of miRNAs
MicroRNA genes encoded in the genome are transcribed into long primary miRNAs (pri‐miRNAs) by RNA polymerase II or, in some cases, by RNA polymerase III (Fig. 1).( 1 , 2 , 3 ) Typically, animal pri‐miRNAs display a ∼33 bp stem and a terminal loop structure with flanking segments.( 4 ) Many pri‐miRNAs originate from clusters; loci of miRNAs positioned closely together. Primary miRNA processing begins in the nucleus where an RNase III enzyme, Drosha, removes the flanking segments and a ∼11 bp stem region, thereby catalyzing conversion of pri‐miRNAs into precursor miRNAs (pre‐miRNAs) (Fig. 1).( 1 , 5 ) Precursor miRNAs are generally 60–70 nt long hairpin RNAs with 2‐nt overhangs at the 3′ end. Drosha typically acts together with the DiGeorge syndrome critical region 8 protein (DGCR8) for efficiency and precision.( 6 , 7 , 8 ) However, a Drosha/DGCR8‐independent processing pathway can also produce pre‐miRNAs (Fig. 1). In this pathway, the nuclear splicing machinery provides pre‐miRNA from introns. miRNAs produced by this pathway are appropriately called mirtrons.( 9 , 10 , 11 ) Mirtrons encompass a small group of miRNAs but are found in many organisms. Precursor miRNAs are exported from the nucleus to the cytoplasm by the exportin‐5/RanGTP heterocomplex (Fig. 1).( 12 , 13 , 14 ) In the cytoplasm, pre‐miRNAs are processed by RNase III enzyme Dicer (Fig. 1).( 15 ) Dicer is thought to act with dsRBDs‐containing partner proteins HIV TAR RNA‐binding protein (TRBP)( 16 , 17 , 18 ) and/or PKR activating protein (PACT)( 18 , 19 ). Dicer cleaves pre‐miRNAs into 21–25 nt long miRNA/miRNA* duplexes, each strand of which bears 5′ monophosphate, 3′ hydroxyl group and a 3′ 2‐nt overhang. Of a miRNA/miRNA* duplex, only one strand, designated the miRNA strand, is selected as the guide of mature RISC, whereas the other strand, the miRNA* strand, is discarded during RISC assembly (Fig. 1). Such biased strand selection depends on the balance of at least three properties of a miRNA/miRNA* duplex: the structure; the 5′ nucleotide identity; and the thermodynamic asymmetry (reviewed in Kawamata and Tomari( 20 )).
Figure 1.
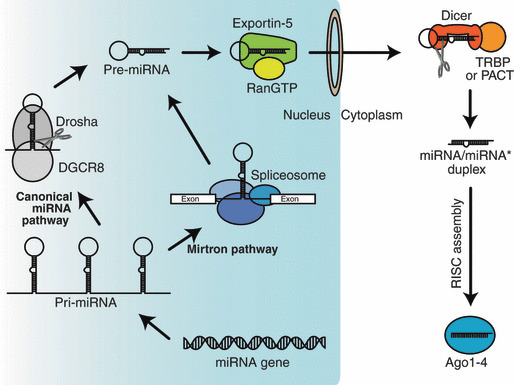
Biogenesis of microRNA (miRNA). miRNAs are encoded in the genome and upon transcription give rise to primary miRNAS (pri‐miRNAs). The Drosha/DiGeorge syndrome critical region 8 protein (DGCR8) complex or the spliceosome catalyzes pri‐ to precursor miRNA (pre‐miRNA) conversion. Exportin‐5/RanGTP complex exports pre‐miRNAs from the nucleus to the cytoplasm. Dicer interacts with TAR RNA‐binding protein (TRBP) or PKR activating protein (PACT) and cleaves pre‐miRNAs into miRNA/miRNA* duplexes. These duplexes are assembled into Argonaute (Ago)1–4, and eventually form mature RNA‐induced silencing complexes (RISCs), which contain only single‐stranded guides.
RISC Assembly
The core component of RISC is a member of Argonaute (Ago) subfamily proteins, of which there are four paralogs (Ago1–4) in humans. RISC assembly follows a multi‐step pathway. The first well‐characterized event of RISC assembly is RISC loading (Fig. 2). During RISC loading, miRNA/miRNA* duplexes are incorporated into Ago proteins. Native gel analyses revealed that central mismatches (guide position 8–11) in miRNA/miRNA* duplexes are preferred for RISC loading into human Ago1–4 (Fig. 2).( 21 ) Ago1–4 disfavor duplexes bearing only non‐central mismatches, but can incorporate siRNA‐like perfectly complementary duplexes (Fig. 2). RISC loading is not a simple binding of miRNA/miRNA* duplexes and Ago proteins, but rather an ATP‐dependent active process.( 21 , 22 ) Structural analyses of Thermus thermophilus Ago protein suggest that a generic miRNA/miRNA* duplex is too bulky to fit directly.( 23 , 24 ) To enable loading, drastic conformational opening of Ago protein appears necessary; an energetic reaction likely powered by ATP hydrolysis.( 20 ) We and others recently reported that Hsc70/Hsp90 chaperone machinery mediates such conformational opening (Fig. 2).( 25 , 26 )
Figure 2.
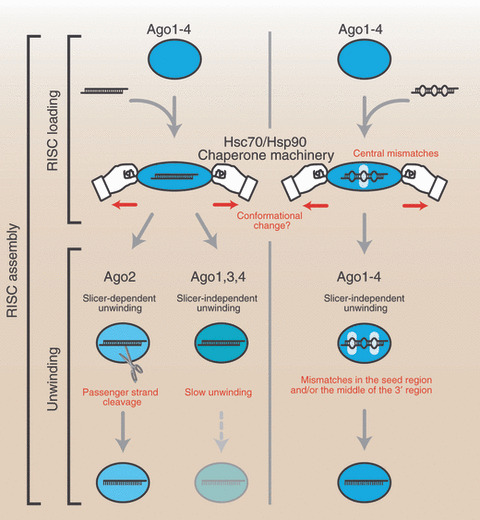
RNA‐induced silencing complex (RISC) assembly can be divided into at least two steps, RISC loading and unwinding. The former prefers central mismatches in microRNA (miRNA)/miRNA* duplexes. Perfectly complementary siRNA duplexes can also be loaded into Argonaute (Ago)1–4, but duplexes without central mismatches are not favored for RISC loading. The RISC loading of small RNA duplexes is ATP‐dependent and facilitated by Hsc70/Hsp90 chaperone machinery. Mismatches in the seed region and/or the middle of the 3′ region enhance the slicer‐independent unwinding efficiency of all four Ago proteins. In contrast, only Ago2 can efficiently unwind perfectly complementary duplexes by passenger strand cleavage.
After RISC loading, the duplex is unwound and the miRNA* strand is discarded from Ago protein (Fig. 2). Surprisingly, unwinding does not require ATP; presumably unwinding is linked to the release of the structural opening incurred upon Ago proteins during RISC loading.( 20 , 21 ) Unwinding can be further classified into slicer‐dependent unwinding and slicer‐independent unwinding. In humans, only Ago2 retains the cleavage activity( 27 ) and thus can facilitate unwinding by slicing the miRNA* strand (Fig. 2).( 21 , 28 ) However, passenger strand cleavage occurs only if the duplex is extensively base‐paired, especially around position 10–11 of the guide. Most miRNA/miRNA* duplexes bear central mismatches and are therefore unwound by a slicer‐independent mechanism. Mismatches in the seed region (guide position 2–8) and/or the middle of the 3′ region (guide position 12–16) greatly enhance the unwinding efficiency in all four Ago proteins (Fig. 2).( 21 )
RISC Functions
MicroRNA target sites often lie in the 3′ untranslated region (UTR) rather than in the 5′ UTR or ORF, probably because translation (i.e. movement of ribosomes) will counteract RISC binding.( 29 , 30 ) Typically, a target mRNA bears multiple binding sites of the same miRNA (e.g. let‐7 and HMGA2( 31 )) and/or several different miRNAs (e.g. miR‐375, miR‐124, and let‐7b and Mtpn( 32 )). Importantly, not all nucleotides of a miRNA contribute equally to RISC target recognition. This target recognition is largely determined by base‐pairing of nucleotides in the seed region and is enhanced by additional base‐pairing in the middle of the 3′ region.( 29 , 32 , 33 , 34 ) How RISC acts upon target mRNA is determined by both the character of the Ago protein in which the miRNA is incorporated and complementarity between the miRNA strand and the target mRNA (Fig. 3). Ago2 is capable of RNA cleavage,( 27 ) but this reaction requires extensive base‐pairing between the miRNA strand and mRNA target. Some miRNAs silence target mRNAs through cleavage, just as siRNAs do (Fig. 3a). For example, mouse miR‐196 induces cleavage of HOXB8 mRNA because of the extensive complementarity between them.( 35 ) In contrast to this example, the complementarity between the miRNA strand and target mRNA is typically limited,( 29 , 32 , 33 , 34 ) which renders RISC incapable of target cleavage. In such a case, Ago protein provides a platform to recruit factors, including GW182 proteins (TNRC6A–C in humans), required for translational repression and mRNA deadenylation/degradation of target mRNAs (Fig. 3b,c). Such slicer‐independent silencing can be reconstituted by artificial tethering of an Ago protein or a GW182 protein to an mRNA, independent of the presence of a miRNA.( 36 , 37 ) This contrasts with target mRNA cleavage, where the presence of a guiding small RNA is a de facto requirement.
Figure 3.
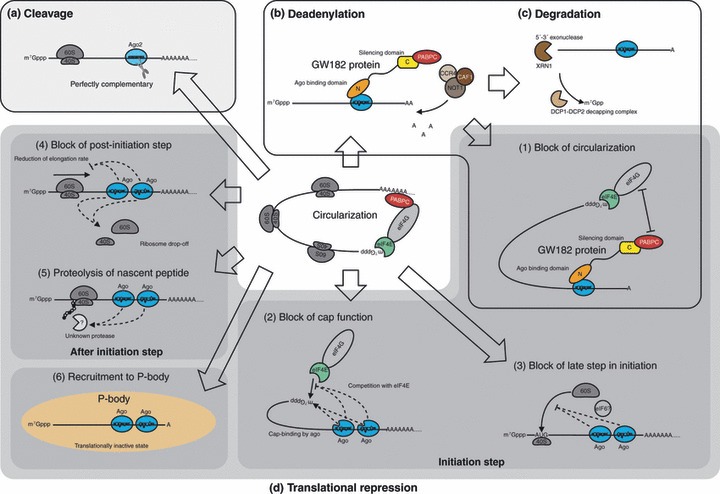
RNA‐induced silencing (RISC) regulates gene expression through various mechanisms: (a) cleavage; (b) deadenylation; (c) degradation; and (d) translational repression of target mRNA. (1–6) Proposed mechanisms of translational repression. Mechanisms 1–3 target the initiation step of translation; mechanisms 4 and 5 target the steps after initiation. AAA..., poly(A) tail; Ago, Argonaute; C, C‐terminal domain of GW182 protein: silencing domain; CAF1, CCR4 associated factor 1; CCR4, carbon catabolite repression 4; N, N‐terminal domain of GW182 protein: Ago binding domain; NOT1, negative on TATA‐less 1; PABPC, cytoplasmic poly(A) binding protein; P‐body, processing body; XRN1, 5′‐exoribonuclease 1.
Proposed Mechanisms of Translational Repression
Despite recent remarkable progress in this field, the exact mechanisms of miRNA‐mediated translational repression are still subjects of ongoing debate. To date, at least six models of translational repression have been proposed (Fig. 3d): (i) RISC induces deadenylation which causes decrease of translational efficiency by blocking target mRNA circularization;( 38 , 39 , 40 ) (ii) RISC blocks cap function by interacting with either the cap or eIF4E;( 39 , 41 , 42 , 43 , 44 , 45 , 46 ) (iii) RISC blocks a late step in initiation of translation such as recruitment of 60S ribosomal subunit;( 47 , 48 , 49 ) (iv) RISC blocks a post‐initiation step such as elongation and/or ribosome dropoff;( 50 , 51 , 52 , 53 , 54 ) (v) RISC induces proteolysis of nascent peptides during translation;( 55 ) or (vi) RISC recruits target mRNAs to processing bodies, in which mRNA is degraded and/or stored in a translationally inactive state.( 56 , 57 , 58 ) These models do not necessarily exclude each other. Further research is warranted to clarify the exact molecular mechanisms of translational repression.
Cancer and miRNA
Cancer is characterized by abnormally proliferative cells that undergo rapid, uncoordinated cell growth. Malignant cancer, in contrast to benign cancer, is further hallmarked by aggressive neoplasms that have the ability to invade and annihilate adjacent tissues and/or metastasize to more distant, and sometimes specific, tissues. Genes involved in cancer, be it inceptionally or during the later invasive or metastasis stages, are generally classified into oncogenes or tumor suppressor genes. During the last decade, a unique set of cancer regulator miRNAs have emerged and these are divided into oncomiRs and anti‐oncomiRs. OncomiRs and anti‐oncomiRs negatively regulate tumor suppressor genes and oncogenes, respectively. Interestingly, let‐7, the first identified conserved miRNA, is an anti‐oncomiR. let‐7 functions as a post‐transcriptional gatekeeper of cell proliferation genes, such as RAS lung cancer oncogenes, which in cancerous cells disturb normal cell cycle progression.( 59 , 60 ) Differential miRNA expression profiles of cancerous and normal tissues have revealed signatures that facilitate identifying and monitoring cancers.( 61 ) Researchers are now trying to use these miRNA signatures therapeutically to support diagnosis, prognosis, or treatment of cancer. We will not detail the whole body of identified oncomiRs and anti‐oncomiRs (the readers should consult other reviews including references 62–64),( 62 , 63 , 64 ) but rather illustrate mechanistically how the miRNA pathway is involved in, or affected by, cancer at the hand of representative examples.
Genomic Abnormalities
Aberrant RNA silencing by miRNAs is often caused by phenomena similar to those of protein‐coding genes involved in cancer. Chromosomal abnormalities can trigger oncogenic actions of miRNAs by modulating miRNA expression in the wrong cell type or at wrong times (Fig. 4a). Lymphoma karyotype analysis identified a chromosomal translocation containing the miR‐17–92 cluster (also designated as oncomiR‐1).( 65 ) This cluster comprises six miRNAs of which most prominently expressed is miR‐19, an oncomiR that protects lymphocytes from apoptosis after depletion of growth factors, thereby facilitating neoplasia. In addition to overexpression of miR‐19, the translocation coincided with a second re‐arrangement that activates Notch1. Expression of both miR19a and Notch1 synergistically induces T‐cell acute lymphoblastic leukemia.( 65 )
Figure 4.

MicroRNA (miRNA) pathway and cancer. (a) Genomic abnormalities, such as chromosomal translocations and point mutations, can attenuate or stimulate miRNA transcription leading to an increase or decrease of primary miRNA (pri‐miRNA). (b) Mutations in a miRNA gene can lead to RNA‐induced silencing (RISC) assembly abnormalities. For example, when nucleotides that establish the thermodynamic asymmetry are substituted, this can lead to “flipped” strand selection, resulting in the miRNA* strand, instead of the miRNA strand, being favored for RISC incorporation. (c) Furthermore, single nucleotide polymorphisms or mutations in either the miRNA or the target RNA can abrogate proper target recognition, especially when located within the seed region. Consequently, genes might escape being regulated by RISCs or perhaps even become differently regulated as a result of erroneous targeting. (d) Genomic abnormalities can also result in aberrant miRNA processing. Some pri‐miRNAs require additional proteins for efficient conversion. p53 mutants affect the interaction between p68/p72 RNA helicases and Drosha and this decreases pri‐ to precursor miRNA (pre‐miRNA) conversion of a subset of miRNAs. Additionally, p68 associates with receptor‐regulated Smads. This interaction has been shown to be important for efficient pri‐ to pre‐miRNA conversion of another subset of miRNAs. In the cytoplasm, TAR RNA‐binding protein (TRBP) is phosphorylated by MAPK/Erk signaling. The phosphorylation of TRBP increases pre‐miRNA to miRNA/miRNA* conversion for general miRNAs, but it decreases conversion for let‐7 family miRNAs. AAA..., poly(A) tail; Ago, Argonaute; DGCR8, DiGeorge syndrome critical region 8 protein.
Point mutations within miRNAs or their flanking regions can lead to altered transcription levels (Fig. 4a). An anti‐oncomiR cluster consisting of miR‐15a/miR‐16‐1 was found to be transcribed less efficiently in some chronic lymphocytic leukemia (CLL) patients.( 66 ) The markedly decreased expression level of these miRNAs was caused by a single nucleotide polymorphism in the 3′‐region flanking sequence. This specific germline mutation has so far only been detected in CLL patients, although similar mutations have been found in a CLL‐prone species of mice.( 67 ) Human HEK293 cells transiently expressing either the wild‐type allele of the miR‐15a–miR‐16‐1 cluster or the mutated allele revealed that the single substitution mutant is also transcribed less efficiently in vitro.( 66 ) In theory, mutations within miRNA genes can also result in severe abnormality in RISC loading, unwinding, and strand selection (Fig. 4b), but such a mutant involved in cancer has not been reported.
When point mutations occur in miRNA binding sites of target mRNAs, these too can compromise the ability of small RNAs to silence target mRNA transcripts (Fig. 4c). miR‐155 and tumor suppressor gene, suppressor of cytokine signaling 1 (SOCS1), portray such a relationship. miR‐155 is an oncomiR overexpressed in breast, lung, colon, pancreatic, and thyroid cancer tumors.( 68 ) SOCS‐1, an evolutionarily conserved target of miR‐155, is part of a feedback loop that modulates cytokine receptor signal transduction.( 69 ) In breast tumors, a point mutation in the 3′‐UTR binding site of SOCS‐1 was found to markedly avoid repression by miR‐155.( 70 ) Knockdown of SOCS‐1 mimics the oncogenic effects of miR‐155, and overexpression of miR‐155 promotes proliferation of breast cancer cells, underlining miR‐155’s role as an oncomiR.( 70 )
Transcriptional Activation
Another oncomiR overexpressed in breast cancer cells, miR‐10b, was identified together with miR‐155 in a miRNA microarray profile of breast carcinoma.( 71 ) This miRNA is specifically highly expressed in cancer cells capable of metastasizing. miR‐10b expression is directly regulated by the transcription factor Twist.( 72 ) Twist regulates cell movement and tissue reorganization during early embryogenesis, but is also known to play a key role in the invasion–metastasis cascade of tumor cells. Further scrutiny of miR‐10b, using antisense oligonucleotides for in vitro and retroviral transduction and immunohistochemical analyses for in vivo assays, elucidated that this oncomiR promotes invasion and induces distant metastasis.( 72 )
Dysregulation of the miRNA Pathway
In addition to the above‐mentioned mechanisms, protein components of the miRNA pathway can be compromised by the derailed genome (Fig. 4d). If one of the components associated with the miRNA pathway is under‐ or overexpressed or qualitatively impaired, it would destabilize the pathway as a whole. In human cells, master watchman p53 interacts with p68 and p72 RNA helicases.( 73 ) p53 is a prominent tumor suppressor; its function as a transcription factor for apoptosis or cell‐cycle arrest is so central that, in order for most cancers to proliferate, the p53 gene needs to be disabled or the p53 pathway needs to be disrupted.( 74 ) p68 and p72 RNA helicases are required for efficient maturation of a subset of miRNAs.( 75 ) Recently it was revealed that their association with the Drosha/DGCR8 complex facilitates the conversion of pri‐ to pre‐miRNAs. p53 mutants affect the interaction between p68/p72 and Drosha.( 75 ) Consequently, this decreases pri‐ to pre‐miRNA conversion, and such a cutback affects the composition and total pool of mature miRNAs (Fig. 4d, upper). As not just one but a whole subset of miRNAs are affected by mutant p53, this causes pleiotropical changes in gene expression.
Additionally, receptor‐regulated Smads (R‐Smads) interact with p68 RNA helicase. R‐Smads are known for their role as signal transducers of transforming growth factor‐β (TGF‐β) and bone morphogenetic protein receptors. Recently, a non‐canonical function of R‐Smads was revealed, showing that TGF‐β and bone morphogenetic protein stimulation of R‐Smads enhances pri‐ to pre‐miRNA conversion of a subset of miRNAs including pri‐miR‐21 and pri‐miR‐199a (Fig. 4d, middle).( 76 ) Receptor‐regulated Smads not only interact with p68 but also associate with Drosha, an affiliation dependent on RNA. What qualitatively defines the miRNAs that are affected by R‐Smad‐p68/Drosha interaction remains unknown. miR‐21 is a well‐researched, potent oncomiR that is upregulated in many tumors, including breast, colon, lung, pancreas, prostate, and stomach tumors.( 77 ) Enhancing pre‐miRNA conversion could be a mechanism by which TGF‐β superfamily signaling contributes to oncogenesis. However, a direct link between R‐Smad‐mediated miR‐21 maturation and its role in oncogenesis is yet to be established.
Further downstream, miRNA biogenesis interacts with another signaling pathway (Fig. 4d, lower). As described, Dicer and TRBP process pre‐miRNAs into miRNA/miRNA* duplexes. A recent study found that TRBP is a phosphoprotein.( 78 ) Strikingly, expressing phospho‐mimic TRBP enhances mature miRNA production, whereas a phospho‐mutant does not.( 78 ) Phosphorylation of TRBP is dependent on serine/threonine‐selective protein kinases belonging to the MAPK/Erk pathway, a mitosis‐inducing signal transduction pathway that relays extracellular‐activated signals to the nucleus and promotes cell division. MAPK‐mediated activation of TRBP enhances pre‐miRNA processing and, as such, alters mature miRNA composition.( 78 ) Stimulation or inhibition of MAPK/Erk signal transduction leads to pro‐growth or anti‐proliferative miRNA profiles, respectively; when MAPK/Erk signaling is stimulated (inhibited), general miRNAs, including pro‐growth miRNAs such as miR‐17, miR‐20a, and miR‐92a, are globally upregulated (downregulated), while anti‐oncomiR let‐7 family is specifically downregulated (upregulated).( 78 ) RAS subfamily oncogenes are under negative regulation by let‐7.( 59 ) RAS protein acts in the same signaling cascade, but upstream of the TRBP kinases, so a feedback loop would be expected, akin to a cumulative causation loop where a signal keeps on amplifying itself. Apart from phosphorylation, TRBP genes are frequently found to contain mutations in colon and gastric cancer tissues or cell lines. Frameshift mutations within TRBP diminish TRBP protein expression and this correlates with Dicer destabilization. Reintroduction of TRBP in deficient cells recovers efficient pre‐miRNA processing and reduces tumor growth in mice.( 79 )
Concluding Remarks
Our current understanding of miRNA expression and miRNA pathway regulation is still in its infancy, but its involvement in destabilizing the balance between oncogenes and tumor suppressor genes is irrefutable. Cancer can influence the miRNA pathway at nearly all steps, so it is of great importance that we clearly discriminate between what is cause and what is effect in miRNA expression and regulation. To this end, it is crucial not only to identify miRNA targets, but also precisely understand the molecular mechanisms of the miRNA pathway, including biogenesis, RISC assembly, and RISC functions.
Acknowledgments
We apologize to our colleagues whose important work is not cited because of space limitations. Research in the Tomari laboratory is supported by a Grant‐in‐Aid for Young Scientists (A) and a Grant‐in‐Aid for Scientific Research on Innovative Areas (“functional machinery for non‐coding RNAs”) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology, and a Carrier Development Award from The International Human Frontier Science Program Organization. S.I. is a recipient of a Japan Society for the Promotion of Science (JSPS) Research Fellowship.
References
- 1. Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 2002; 21: 4663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee Y, Kim M, Han J et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23: 4051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 2006; 13: 1097–101. [DOI] [PubMed] [Google Scholar]
- 4. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97. [DOI] [PubMed] [Google Scholar]
- 5. Lee Y, Ahn C, Han J et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003; 425: 415–9. [DOI] [PubMed] [Google Scholar]
- 6. Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature 2004; 432: 231–5. [DOI] [PubMed] [Google Scholar]
- 7. Gregory RI, Yan KP, Amuthan G et al. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004; 432: 235–40. [DOI] [PubMed] [Google Scholar]
- 8. Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha‐DGCR8 complex in primary microRNA processing. Genes Dev 2004; 18: 3016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol Cell 2007; 28: 328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA‐class regulatory RNAs in Drosophila . Cell 2007; 130: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature 2007; 448: 83–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP‐dependent dsRNA‐binding protein that mediates nuclear export of pre‐miRNAs. RNA 2004; 10: 185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science 2004; 303: 95–8. [DOI] [PubMed] [Google Scholar]
- 14. Yi R, Qin Y, Macara IG, Cullen BR. Exportin‐5 mediates the nuclear export of pre‐microRNAs and short hairpin RNAs. Genes Dev 2003; 17: 3011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA‐interference enzyme Dicer in the maturation of the let‐7 small temporal RNA. Science 2001; 293: 834–8. [DOI] [PubMed] [Google Scholar]
- 16. Chendrimada TP, Gregory RI, Kumaraswamy E et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005; 436: 740–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haase AD, Jaskiewicz L, Zhang H et al. TRBP, a regulator of cellular PKR and HIV‐1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep 2005; 6: 961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kok KH, Ng MH, Ching YP, Jin DY. Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J Biol Chem 2007; 282: 17649–57. [DOI] [PubMed] [Google Scholar]
- 19. Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO J 2006; 25: 522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kawamata T, Tomari Y. Making RISC. Trends Biochem Sci 2010; 35: 368–76. [DOI] [PubMed] [Google Scholar]
- 21. Yoda M, Kawamata T, Paroo Z et al. ATP‐dependent human RISC assembly pathways. Nat Struct Mol Biol 2010; 17: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawamata T, Seitz H, Tomari Y. Structural determinants of miRNAs for RISC loading and slicer‐independent unwinding. Nat Struct Mol Biol 2009; 16: 953–60. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ. Structure of an Argonaute silencing complex with a seed‐containing guide DNA and target RNA duplex. Nature 2008; 456: 921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Juranek S, Li H et al. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 2009; 461: 754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iki T, Yoshikawa M, Nishikiori M et al. In vitro assembly of plant RNA‐induced silencing complexes facilitated by molecular chaperone HSP90. Mol Cell 2010; 39: 282–291. [DOI] [PubMed] [Google Scholar]
- 26. Iwasaki S, Kobayashi M, Yoda M et al. Hsc70/Hsp90 chaperone machinery mediates ATP‐dependent RISC loading of small RNA duplexes. Mol Cell 2010; 39: 292–299. [DOI] [PubMed] [Google Scholar]
- 27. Liu J, Carmell MA, Rivas FV et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004; 305: 1437–41. [DOI] [PubMed] [Google Scholar]
- 28. Leuschner PJ, Ameres SL, Kueng S, Martinez J. Cleavage of the siRNA passenger strand during RISC assembly in human cells. EMBO Rep 2006; 7: 314–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lewis BP, Shih IH, Jones‐Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003; 115: 787–98. [DOI] [PubMed] [Google Scholar]
- 30. Gu S, Jin L, Zhang F, Sarnow P, Kay MA. Biological basis for restriction of microRNA targets to the 3′ untranslated region in mammalian mRNAs. Nat Struct Mol Biol 2009; 16: 144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee YS, Dutta A. The tumor suppressor microRNA let‐7 represses the HMGA2 oncogene. Genes Dev 2007; 21: 1025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krek A, Grun D, Poy MN et al. Combinatorial microRNA target predictions. Nat Genet 2005; 37: 495–500. [DOI] [PubMed] [Google Scholar]
- 33. Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA‐target recognition. PLoS Biol 2005; 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grimson A, Farh KK, Johnston WK, Garrett‐Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 2007; 27: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yekta S, Shih IH, Bartel DP. MicroRNA‐directed cleavage of HOXB8 mRNA. Science 2004; 304: 594–6. [DOI] [PubMed] [Google Scholar]
- 36. Behm‐Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 2006; 20: 1885–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pillai RS, Artus CG, Filipowicz W. Tethering of human Ago proteins to mRNA mimics the miRNA‐mediated repression of protein synthesis. RNA 2004; 10: 1518–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beilharz TH, Humphreys DT, Clancy JL et al. microRNA‐mediated messenger RNA deadenylation contributes to translational repression in mammalian cells. PLoS ONE 2009; 4: e6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwasaki S, Kawamata T, Tomari Y. Drosophila Argonaute1 and Argonaute2 employ distinct mechanisms for translational repression. Mol Cell 2009; 34: 58–67. [DOI] [PubMed] [Google Scholar]
- 40. Wakiyama M, Takimoto K, Ohara O, Yokoyama S. let‐7 microRNA‐mediated mRNA deadenylation and translational repression in a mammalian cell‐free system. Genes Dev 2007; 21: 1857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Humphreys DT, Westman BJ, Martin DI, Preiss T. MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proc Natl Acad Sci U S A 2005; 102: 16961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kiriakidou M, Tan GS, Lamprinaki S, De Planell‐Saguer M, Nelson PT, Mourelatos Z. An mRNA m7G cap binding‐like motif within human Ago2 represses translation. Cell 2007; 129: 1141–51. [DOI] [PubMed] [Google Scholar]
- 43. Mathonnet G, Fabian MR, Svitkin YV et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap‐binding complex eIF4F. Science 2007; 317: 1764–7. [DOI] [PubMed] [Google Scholar]
- 44. Pillai RS, Bhattacharyya SN, Artus CG et al. Inhibition of translational initiation by let‐7 MicroRNA in human cells. Science 2005; 309: 1573–6. [DOI] [PubMed] [Google Scholar]
- 45. Thermann R, Hentze MW. Drosophila miR2 induces pseudo‐polysomes and inhibits translation initiation. Nature 2007; 447: 875–8. [DOI] [PubMed] [Google Scholar]
- 46. Wang B, Love TM, Call ME, Doench JG, Novina CD. Recapitulation of short RNA‐directed translational gene silencing in vitro. Mol Cell 2006; 22: 553–60. [DOI] [PubMed] [Google Scholar]
- 47. Chendrimada TP, Finn KJ, Ji X et al. MicroRNA silencing through RISC recruitment of eIF6. Nature 2007; 447: 823–8. [DOI] [PubMed] [Google Scholar]
- 48. Nissan T, Parker R. Computational analysis of miRNA‐mediated repression of translation: implications for models of translation initiation inhibition. RNA 2008; 14: 1480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang B, Yanez A, Novina CD. MicroRNA‐repressed mRNAs contain 40S but not 60S components. Proc Natl Acad Sci U S A 2008; 105: 5343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA‐binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci U S A 2007; 104: 9667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maroney PA, Yu Y, Fisher J, Nilsen TW. Evidence that microRNAs are associated with translating messenger RNAs in human cells. Nat Struct Mol Biol 2006; 13: 1102–7. [DOI] [PubMed] [Google Scholar]
- 52. Olsen PH, Ambros V. The lin‐4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN‐14 protein synthesis after the initiation of translation. Dev Biol 1999; 216: 671–80. [DOI] [PubMed] [Google Scholar]
- 53. Petersen CP, Bordeleau ME, Pelletier J, Sharp PA. Short RNAs repress translation after initiation in mammalian cells. Mol Cell 2006; 21: 533–42. [DOI] [PubMed] [Google Scholar]
- 54. Seggerson K, Tang L, Moss EG. Two genetic circuits repress the Caenorhabditis elegans heterochronic gene lin‐28 after translation initiation. Dev Biol 2002; 243: 215–25. [DOI] [PubMed] [Google Scholar]
- 55. Nottrott S, Simard MJ, Richter JD. Human let‐7a miRNA blocks protein production on actively translating polyribosomes. Nat Struct Mol Biol 2006; 13: 1108–14. [DOI] [PubMed] [Google Scholar]
- 56. Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA‐mediated translational repression in human cells subjected to stress. Cell 2006; 125: 1111–24. [DOI] [PubMed] [Google Scholar]
- 57. Liu J, Valencia‐Sanchez MA, Hannon GJ, Parker R. MicroRNA‐dependent localization of targeted mRNAs to mammalian P‐bodies. Nat Cell Biol 2005; 7: 719–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sen GL, Blau HM. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol 2005; 7: 633–6. [DOI] [PubMed] [Google Scholar]
- 59. Johnson SM, Grosshans H, Shingara J et al. RAS is regulated by the let‐7 microRNA family. Cell 2005; 120: 635–47. [DOI] [PubMed] [Google Scholar]
- 60. Johnson CD, Esquela‐Kerscher A, Stefani G et al. The let‐7 microRNA represses cell proliferation pathways in human cells. Cancer Res 2007; 67: 7713–22. [DOI] [PubMed] [Google Scholar]
- 61. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–66. [DOI] [PubMed] [Google Scholar]
- 62. Esquela‐Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–69. [DOI] [PubMed] [Google Scholar]
- 63. Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol 2009; 4: 199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med 2009; 60: 167–79. [DOI] [PubMed] [Google Scholar]
- 65. Mavrakis KJ, Wolfe AL, Oricchio E et al. Genome‐wide RNA‐mediated interference screen identifies miR‐19 targets in Notch‐induced T‐cell acute lymphoblastic leukaemia. Nat Cell Biol 2010; 12: 372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Calin GA, Ferracin M, Cimmino A et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005; 353: 1793–801. [DOI] [PubMed] [Google Scholar]
- 67. Raveche ES, Salerno E, Scaglione BJ et al. Abnormal microRNA‐16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 2007; 109: 5079–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR‐155 gene: a typical multifunctional microRNA. Biochim Biophys Acta 2009; 1792: 497–505. [DOI] [PubMed] [Google Scholar]
- 69. Davey GM, Heath WR, Starr R. SOCS1: a potent and multifaceted regulator of cytokines and cell‐mediated inflammation. Tissue Antigens 2006; 67: 1–9. [DOI] [PubMed] [Google Scholar]
- 70. Jiang S, Zhang HW, Lu MH et al. MicroRNA‐155 functions as an oncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res 2010; 70: 3119–27. [DOI] [PubMed] [Google Scholar]
- 71. Iorio MV, Ferracin M, Liu CG et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005; 65: 7065–70. [DOI] [PubMed] [Google Scholar]
- 72. Ma L, Teruya‐Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA‐10b in breast cancer. Nature 2007; 449: 682–8. [DOI] [PubMed] [Google Scholar]
- 73. Bates GJ, Nicol SM, Wilson BJ et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 2005; 24: 543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Junttila MR, Evan GI. p53 – a Jack of all trades but master of none. Nat Rev Cancer 2009; 9: 821–9. [DOI] [PubMed] [Google Scholar]
- 75. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature 2009; 460: 529–33. [DOI] [PubMed] [Google Scholar]
- 76. Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA‐mediated microRNA maturation. Nature 2008; 454: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Krichevsky AM, Gabriely G. miR‐21: a small multi‐faceted RNA. J Cell Mol Med 2009; 13: 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA‐generating complex mediates MAPK/Erk signaling. Cell 2009; 139: 112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Melo SA, Ropero S, Moutinho C et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet 2009; 41: 365–70. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]