

Abstract
In previous studies, we demonstrated that the breast cancer resistance protein (BCRP, ABCG2) forms an S–S homodimer. The BCRP‐C603S mutant substituting Ser for Cys‐603 in the third extracellular domain formed both a 70–75‐kDa monomer and 140–150‐kDa dimer, suggesting that Cys‐603 is an important residue in the covalent bridge. These results also suggested the involvement of other Cys residues in dimer formation. In the present study, we examined the possible involvement of the other extracellular Cys residues, Cys‐592 and Cys‐608, in the dimerization and transporter functions of BCRP using double and triple Cys‐mutant BCRP transfectants. In SDS–PAGE under non‐reducing conditions, BCRP‐C592S·C603S and BCRP‐C592S·C608S were detected as dimers whereas BCRP‐C603S·C608S and BCRP‐C592S·C603S·C608S were found only as monomers. This finding indicated that no Cys residues other than the three extracellular Cys are responsible for the dimer formation. The formation of BCRP‐C592S·C603S dimer suggested the involvement of Cys‐608 in the covalent linkage of this mutant BCRP. PA/C592S·C603S·C608S‐cl.7 cells showed a significant level of multiple drug resistance and low‐level accumulation of mitoxantrone. These results clearly demonstrate that BCRP functions as a drug resistance protein without covalent dimerization. Among drug‐resistant Cys‐mutant BCRP transfectants, PA/C603S, PA/C592S·C608S, and PA/C592S·C603S·C608S were found to be more resistant to the reversal effects of fumitremorgin C than PA/WT, suggesting some alteration in the substrate recognition in Cys‐mutant BCRPs. In conclusion, Cys‐mediated covalent dimerization is not required for BCRP to function as a transporter. In addition to Cys‐603, Cys‐608 may also be involved in BCRP dimer formation. (Cancer Sci 2010)
Cancer cells that acquire resistance to certain chemotherapeutic agents sometimes develop cross‐resistance to other structurally unrelated drugs. This phenomenon is known as multidrug resistance. ATP‐binding cassette (ABC) transporters such as P‐glycoprotein and multidrug resistance‐associated protein 1 (MRP1) have an internally duplicated structure with two transmembrane domains and two ATP‐binding domains. These proteins pump out various structurally unrelated anticancer agents in an energy‐dependent manner. Thus cells expressing these transporters manifest a multidrug‐resistant phenotype.( 1 , 2 , 3 , 4 , 5 ) In contrast, breast cancer resistance protein (BCRP/ABCG2/MXR) is a half transporter, possessing a C‐terminal transmembrane domain and an N‐terminal ATP‐binding domain.( 6 , 7 , 8 , 9 ) Breast cancer resistance protein (BCRP) mediates the resistance of various anticancer drugs such as 7‐ethyl‐10‐hydroxycamptothecin (SN‐38), which is an active metabolite of irinotecan, topotecan, and mitoxantrone, by pumping out them out of cells.( 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 )
We have previously shown that BCRP forms a homodimer bridged by disulfide bonds.( 15 ) To identify Cys residues that are involved in dimerization and explore the role of the covalent binding in protein activity, we performed cysteine‐scanning mutagenesis substituting Ser for Cys. We established 12 transfectants expressing Cys‐mutant BCRPs. Among these, BCRP‐C603S mutant substituting Ser for Cys‐603 was found to migrate both as a 70–75‐kDa monomer and as a 140–150‐kDa dimer in SDS–PAGE under non‐reducing conditions.( 16 ) The PA/C603S transfectant showed the drug resistance phenotype. Cys‐603 is located in the third extracellular domain (between the 5th and 6th transmembrane domains) of BCRP, which comprises 62 amino acids (Fig. 1). This suggested that Cys‐603 is an important residue in the formation of the covalent bridge between BCRP monomers. However, the existence of a small amount of 150‐kDa dimer in the PA/C603S transfectant suggested the involvement of other Cys residues in this process. In addition, it was unclear whether this non‐covalently bound BCRP really functions as a drug efflux pump. Two other cysteine residues (Cys‐592 and Cys‐608) are also present in this same domain (Fig. 1). These are candidates that would participate in BCRP dimer formation.
Figure 1.
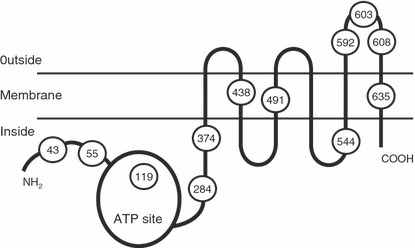
Schematic depiction of the breast cancer resistance protein (BCRP) with 12 cysteine residues indicated. Three of these residues, Cys‐592, Cys‐603, and Cys‐608, are localized in the third extracellular domain.
In the present study, to elucidate the possible involvement of Cys‐592 and Cys‐608 in BCRP dimerization and protein activity, we established double (C592S·C603S, C592S·C608S, and C603S·C608S) and triple (C592S·C603S·C608S) mutant BCRP transfectants, and compared the expression, dimer formation, and function of these BCRP mutants with wild‐type and single (C592S, C603S, and C608S) mutant proteins.
Materials and Methods
Mutant BCRP transfectants. Double (C592S·C603S, C592S·C608S, and C603S·C608S) and triple (C592S·C603S·C608S) mutant BCRP cDNAs were generated using a site‐directed mutagenesis kit (Takara Bio, Otsu, Japan), according to the manufacturer’s instructions. The mutant BCRP cDNAs were cloned into pHa‐IRES‐DHFR bicistronic expression vector harboring a mutant dihydrofolate reductase (DHFR) cDNA under the control of an internal ribosome entry site (IRES). The wild‐type and single (C592S, C603S, and C608S) mutant BCRP cDNAs in the same vector were also used as controls.( 16 ) Murine fibroblast PA317 cells were transfected with the mutant BCRP vectors as described previously.( 16 ) The transfected cells were then selected with 120 ng/mL methotrexate for 8 days. The resulting methotrexate‐resistant cells were mixed and used as mixed populations of transfectants. Clonal cells were obtained from the mixed populations of transfectants by a standard limiting dilution technique. The mutant BCRP cDNAs in the transfectants were amplified by PCR and the nucleotide sequences were confirmed by direct sequencing.
Western blot analysis. Cell lysates from the BCRP transfectants were prepared in the presence or absence of 5% 2‐mercaptoethanol as described previously,( 16 ) resolved using 5–20% SDS–PAGE (5 μg of protein per lane, unless specified), and then electro‐transferred onto nitrocellulose membranes. The membranes were incubated with 1 μg/mL of rabbit anti‐BCRP polyclonal antibody 3488( 15 ) and/or mouse anti‐GAPDH monoclonal antibody (Chemicon, Temecula, CA, USA). This was followed by washing and treatment with peroxidase‐conjugated sheep antirabbit and/or antimouse IgGs (Amersham, Buckinghamshire, UK), respectively. The membrane‐bound antibodies were visualized using an enhanced chemiluminescence detection system (Amersham).
Growth inhibition assay. The sensitivity of the mutant BCRP transfectants to anticancer agents was evaluated using cell growth inhibition assays after incubation of the cells for 4 days at 37°C in the presence or absence of various concentrations of anticancer agents. Cell numbers were determined with a Coulter counter (Beckman Coulter, Brea, CA, USA). The IC50 value (the drug dose causing a 50% inhibition of cell growth) was determined from growth inhibition curves, and the degree of resistance (x‐fold) was calculated by dividing the IC50 values of the BCRP transfectants by those of the parental PA317 cells. The effects of fumitremorgin C (FTC), a BCRP inhibitor, were also examined.( 17 , 18 ) The reversal index (RI) was determined by dividing the degree of resistance in the absence of FTC by the degree of resistance in the presence of FTC.
Intracellular accumulation of mitoxantrone. The intracellular accumulation of mitoxantrone in the mutant BCRP transfectants was determined by flow cytometry. Cells (5 × 105) were incubated in the presence or absence of 1 μM mitoxantrone for 40 min at 37°C and then washed and resuspended in ice‐cold phosphate‐buffered saline (PBS). Fluorescence was determined using a BD LSR ΙΙ flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
Immunofluorescent microscopy. PA317 or BCRP transfectants were seeded onto Lab‐Tek II chamber slides (5 × 104 cells/1.7 cm2‐well; Nalge Nunc International, Naperville, IL, USA) and cultured overnight. The cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed with PBS three times, and then permeabilized with 0.5% Triton X‐100 in PBS for 15 min at room temperature. After washing the cells with PBS three times, non‐specific binding sites were blocked with 3% bovine serum albumin (BSA)/PBS for 15 min at room temperature. Cells were then incubated with 2.5 μg/mL of mouse anti‐BCRP monoclonal antibody BXP‐21 (Chemicon) for 60 min at room temperature. After washing cells with PBS three times, the cells were incubated with 1 μg/mL of an Alexa Fluor 488 goat antimouse antibody (Molecular Probes, Eugene, OR, USA) for 30 min at room temperature, washed with PBS three times and mounted in a Prolong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA, USA). Acquisition of images was performed using FV1000‐D confocal microscopy (Olympus, Tokyo, Japan).
Results
Establishment of mutant BCRP transfectants. PA317 cells were transfected with the wild‐type, single (C592S, C603S, and C608S), double (C592S·C603S, C592S·C608S, and C603S·C608S), and triple (C592S·C603S·C608S) mutant BCRP cDNAs in pHa‐IRES‐DHFR bicistronic expression vector as described previously.( 16 ) In cells transfected with pHa‐BCRP‐IRES‐DHFR, a single mRNA is transcribed under the control of a retroviral LTR promoter, and two gene products are translated independently from a bicistronic mRNA. Cells expressing mutant DHFR show a high‐level resistance to methotrexate. The transfected cells were therefore selected with 120 ng/mL methotrexate for 8 days. The resulting mixed populations of the drug‐selected cells were designated as PA/WT‐mix, PA/C592S‐mix, PA/C603S‐mix, PA/C608S‐mix, PA/C592S·C603S‐mix, PA/C592S·C608S‐mix, PA/C603S·C608S‐mix, and PA/C592S·C603S·C608S‐mix. Nine clonal cells were established from each of these mixed populations by limiting dilution.
Expression of mutant BCRP in mixed populations of transfected cells. The BCRP expression levels in the mixed populations of methotrexate‐selected cells were evaluated by western blot analysis under reducing conditions (Fig. 2a). Wild‐type BCRP and BCRP‐C603S both migrated as a 75‐kDa molecule. BCRP‐C592S·C608S migrated as 75‐kDa and 70‐kDa species. Other BCRP mutants migrated mostly as a 70‐kDa molecule. A high level of BCRP expression was observed in PA/WT‐mix, PA/C603S‐mix, and PA/C592S·C608S‐mix cells (Fig. 2a).
Figure 2.
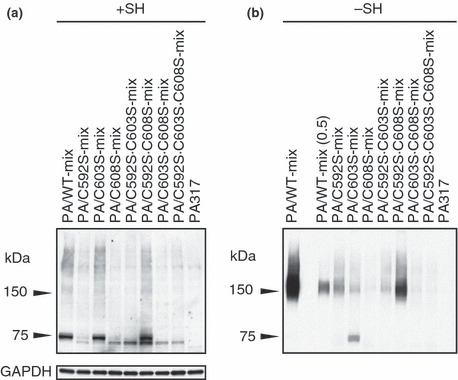
Expression of mutant breast cancer resistance protein (BCRP) in mixed populations of transfected cells. (a) Western blot analysis was performed under reducing (+SH) conditions. GAPDH expression was analyzed as an internal control of protein loading in reducing conditions. (b) Western blot analysis was performed under non‐reducing (−SH) conditions. In lane PA/WT‐mix(0.5), 0.5 μg protein was loaded on SDS–PAGE.
Breast cancer resistance protein (BCRP) dimerization in each of the PA317 transfectants was evaluated by western blotting under non‐reducing conditions (Fig. 2b). As reported previously, BCRP‐C603S migrated as both a 150‐kDa dimer and a 70–75‐kDa monomer. Breast cancer resistance protein (BCRP) dimer formation was observed in PA/WT‐mix, PA/C592S‐mix, PA/C592S·C603S‐mix, and PA/C592S·C608S‐mix cells. The BCRP expression levels in PA/C608S‐mix, PA/C603S·C608S‐mix, and PA/C592S·C603S·C608S‐mix cells were too low to evaluate dimer/monomer formation (Fig. 2b).
Expression of mutant BCRP in mutant BCRP transfectant clones. Since BCRP expression of PA/C592S·C603S·C608S‐mix cells was low, we next isolated the mutant BCRP transfectant clones. Western blot analyses of all of the clones are shown in Figure S1. Figure 3 shows the BCRP expression profiles in the selected clones, together with PA/WT and the corresponding mixed populations. The BCRP expression levels and monomer/dimer formation in PA/WT and single mutant BCRP transfectants (PA/C592S, PA/C603S, and PA/C608S) were essentially the same as those in our previous report (Fig. 3a–d).( 16 ) Among double mutant BCRP transfectants, BCRP dimer formation was prominent in PA/C592S·C603S and PA/C592S·C608S cells (Fig. 3e,f). In contrast, monomer formation in these transfectants was marginal, although single mutation in Cys‐603 or Cys‐608 resulted in monomer formation. It should be noted that BCRP‐C592S·C603S formed a BCRP dimer in the absence of Cys‐603. Another double mutant BCRP transfectant, PA/C603S·C608S, expressed a small amount of BCRP monomer (Fig. 3g). As shown in Figure 3(h), a triple mutant BCRP transfectant, PA/C592S·C603S·C608S‐cl.7, expressed a fairly high level of the BCRP monomer. No dimer formation was evident in this cell line. This finding indicated that the three extracellular Cys residues, Cys‐592, Cys‐603, and/or Cys‐608, are responsible for BCRP dimer formation. It also suggested that dimer formation of BCRP‐C592S·C603S might be due to the involvement of Cys‐608 in the covalent linkage of this mutant BCRP.
Figure 3.
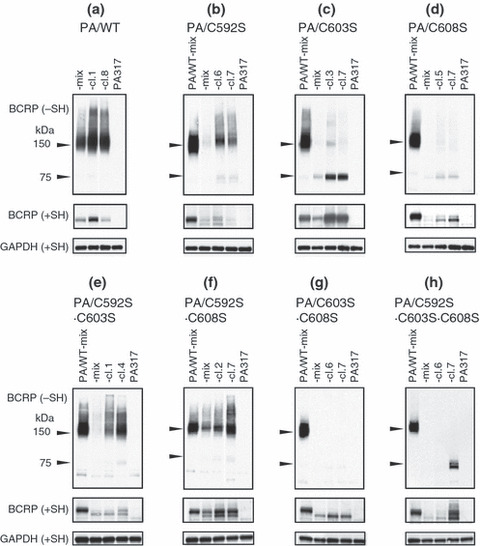
Expression of mutant breast cancer resistance protein (BCRP) in selected transfectant clones. Breast cancer resistance protein (BCRP) expression in a mixed population of cells (‐mix) and in two transfectant clones with relatively high BCRP expression (‐cl.X) was examined. PA/WT‐mix was used as a control cell line. Western blot analysis was performed under reducing (+SH) and non‐reducing (−SH) conditions. GAPDH expression was analyzed as an internal control of protein loading in reducing conditions. (a) PA/WT cells; (b) PA/C592S cells; (c) PA/C603S cells; (d) PA/C608S cells; (e) PA/C592S·C603S cells; (f) PA/C592S·C608S cells; (g) PA/C603S·C608S cells; (h) PA/C592S·C603S·C608S cells.
Two species of BCRP, 75‐kDa and 70‐kDa, were detected in western blot analyses under reducing conditions (Fig. 3). The relative expression levels of 75‐kDa and 70‐kDa BCRP in transfectants to the PA/WT‐mix cells were calculated from each BCRP band under reducing conditions (Fig. 3) and are presented in Figure 4. PA/WT cells expressed 75‐kDa BCRP only, and PA/C603S cells mostly expressed the 75‐kDa molecule. The ratio of 75‐kDa BCRP in PA/C592S, PA/C592S·C608S, and PA/C592S·C603S·C608S cells was two‐third to half of the total BCRP. The expression levels of 70‐kDa BCRP were higher than those of 75‐kDa BCRP in other transfectants, especially PA/C603S·C608S cells.
Figure 4.
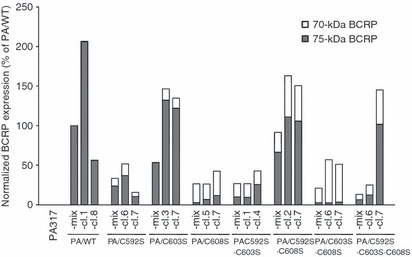
Normalized breast cancer resistance protein (BCRP) expression in transfectant clones. The relative expression levels of total and 75‐kDa BCRP in transfectants as compared with PA/WT‐mix cells were calculated from each BCRP band under reducing conditions (Fig. 3).
Drug resistance of mutant BCRP transfectants. The effects of substituting Ser for Cys in BCRP on cellular drug resistance were evaluated using a cell growth inhibition assay. The SN‐38 resistance levels of the mutant BCRP transfectant sublines are listed in Table 1. PA/WT, PA/C603S‐cl.3, and PA/C603S‐cl.7 transfectants showed high levels of resistance to SN‐38. PA/C592S transfectants exhibited intermediate levels of resistance to SN‐38 whereas PA/C608S transfectants showed only marginal levels of resistance to this drug. The growth inhibition curves of double‐ and triple‐mutant BCRP transfectants are described in Figure 5. Among double mutant BCRP transfectants, PA/C592S·C608S transfectants showed intermediate levels of resistance to SN‐38 whereas PA/C592S·C603S transfectants showed only marginal levels of resistance to this drug. PA/C603S·C608S transfectants did not show SN‐38 resistance. It is noteworthy that PA/C592S·C603S·C608S‐cl.7 cells exhibited a 3‐fold higher level of resistance to SN‐38 as compared with parental PA317 cells, indicating that this triple‐mutant BCRP can also confer drug resistance.
Table 1.
SN‐38 resistance of mutant BCRP transfectants
| Cell line | IC50 to SN‐38† (nM) | Degree of resistance‡ (x‐fold) | Relative expression level | |
|---|---|---|---|---|
| Total BCRP§ | 75‐kDa BCRP¶ | |||
| PA317 | 3.7 ± 0.2 | 1 | – | – |
| PA/WT‐mix | 90 ± 4.4 | 24 | 100 | 100 |
| PA/WT‐cl.1 | 172 ± 6.1 | 47 | 207 | 206 |
| PA/WT‐cl.8 | 77 ± 10.3 | 21 | 56 | 56 |
| PA/C592S‐mix | 12.4 ± 0.8 | 3.4 | 34 | 24 |
| PA/C592S‐cl.6 | 28.5 ± 1.4 | 7.7 | 52 | 37 |
| PA/C592S‐cl.7 | 13.7 ± 0.5 | 3.7 | 16 | 11 |
| PA/C603S‐mix | 9.7 ± 0.5 | 2.6 | 54 | 51 |
| PA/C603S‐cl.3 | 409 ± 15.9 | 110 | 146 | 132 |
| PA/C603S‐cl.7 | 158 ± 17.7 | 43 | 135 | 122 |
| PA/C608S‐mix | 4.8 ± 0.3 | 1.3 | 27 | 3 |
| PA/C608S‐cl.5 | 5.6 ± 0.2 | 1.5 | 26 | 7 |
| PA/C608S‐cl.7 | 6.3 ± 0.6 | 1.7 | 43 | 12 |
| PA/C592S·C603S‐mix | 3.6 ± 0.2 | 1.0 | 27 | 10 |
| PA/C592S·C603S‐cl.1 | 5.1 ± 1.1 | 1.4 | 27 | 10 |
| PA/C592S·C603S‐cl.4 | 6.2 ± 0.8 | 1.7 | 43 | 26 |
| PA/C592S·C608S‐mix | 16.7 ± 1.8 | 4.6 | 92 | 67 |
| PA/C592S·C608S‐cl.2 | 22.8 ± 2.0 | 6.2 | 163 | 111 |
| PA/C592S·C608S‐cl.7 | 34.5 ± 4.9 | 9.4 | 151 | 106 |
| PA/C603S·C608S‐mix | 2.9 ± 0.2 | 0.8 | 21 | 3 |
| PA/C603S·C608S‐cl.6 | 4.4 ± 0.4 | 1.2 | 57 | 3 |
| PA/C603S·C608S‐cl.7 | 3.4 ± 0.1 | 0.9 | 51 | 4 |
| PA/C592S·C603S·C608S‐mix | 4.1 ± 0.2 | 1.1 | 13 | 7 |
| PA/C592S·C603S·C608S‐cl.6 | 4.0 ± 0.4 | 1.1 | 25 | 13 |
| PA/C592S·C603S·C608S‐cl.7 | 11.1 ± 1.5 | 3.0 | 145 | 102 |
†The cells were incubated for 4 days at 37°C in the presence or absence of various concentrations of 7‐ethyl‐10‐hydroxycamptothecin (SN‐38). The IC50 value (the drug dose causing a 50% inhibition of cell growth) was determined from growth inhibition curves. Data are the mean ± SD from triplicate determinations. ‡The degree of resistance (x‐fold) was calculated by dividing the IC50 values of the breast cancer resistance protein (BCRP) transfectants by those of the parental PA317 cells. §Each total BCRP (75‐ and 70‐kDa) band intensity was measured (Fig. 3), and the relative expression levels were calculated by dividing BCRP expression values in each cell line as compared with PA/WT‐mix cells. ¶Each 75‐kDa BCRP band intensity (not including 70‐kDa band) was measured (Fig. 3), and the relative expression levels were calculated by dividing BCRP expression values in each cell line as compared with PA/WT‐mix cells.
Figure 5.

SN‐38 resistance of PA/C592S·C603S, PA/C592S·C608S, PA/C603S·C608S, and PA/C592S·C603S·C608S transfectants. The cells were incubated for 4 days at 37°C in the presence or absence of various concentrations of 7‐ethyl‐10‐hydroxy‐camptothecin (SN‐38). Cell numbers were determined with a Coulter counter. Data are the mean ± SD of triplicate determinations. PA317 and PA/WT‐mix were used as control cell lines. (a) PA/C592S·C603S cells; (b) PA/C592S·C608S cells; (c) PA/C603S·C608S cells; (d) PA/C592S·C603S·C608S cells.
We next examined the drug resistance of the triple‐mutant BCRP transfectant, PA/C592S·C603S·C608S, to mitoxantrone, topotecan, and doxorubicin. As shown in Figure 6, PA/C592S·C603S·C608S‐cl.7 cells showed resistance to mitoxantrone and topotecan, but not to doxorubicin. This cross‐resistance pattern was very similar to that of PA/WT‐mix cells. This result also supported our finding that BCRP‐C592S·C603S·C608S can confer multiple drug resistance similar to that of the wild‐type BCRP.
Figure 6.
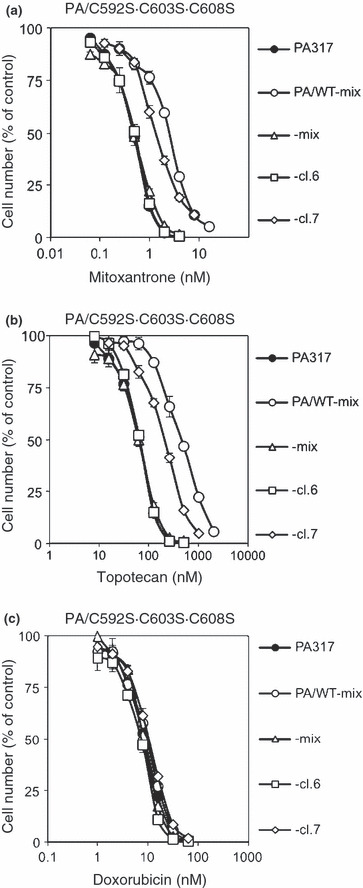
Cross‐resistance pattern of triple‐mutant breast cancer resistance protein (BCRP) transfectants. The cells were incubated for 4 days at 37°C in the presence or absence of various concentrations of mitoxantrone, topotecan, or doxorubicin. Cell numbers were determined with a Coulter counter. Data are the mean ± SD of triplicate determinations. PA317 and PA/WT‐mix were used as control cell lines. (a) Mitoxantrone resistance in PA/C592S·C603S·C608S cells. (b) Topotecan resistance in PA/C592S·C603S·C608S cells. (c) Doxorubicin resistance in PA/C592S·C603S·C608S cells.
Mitoxantrone accumulation in mutant BCRP transfec‐tants. Based on the results of drug sensitivity assay, we selected PA/WT‐mix, PA/C592S‐cl.6, PA/C603S‐cl.3, PA/C592S·C608S‐cl.7, and PA/C592S·C603S·C608S‐cl.7 cells as representative drug‐resistant transfectants for further study. To confirm that the drug resistance of the mutant BCRP transfectants was attributable to a lowered accumulation of the drug, a mitoxantrone uptake experiment was performed. As shown in Figure 7(a), intracellular mitoxantrone was at low levels in PA/WT‐mix and PA/C603S‐cl.3 cells and at intermediate levels in PA/C592S‐cl.6, PA/C592S·C608S‐cl.7, and PA/C592S·C603S·C608S‐cl.7 cells. These data were in good agreement with the results of the drug sensitivity assay. Fluorescence‐activated cell sorter (FACS) fluorograms of mitoxantrone accumulation experiments are shown in S2.
Figure 7.

Mitoxantrone accumulation in mutant breast cancer resistance protein (BCRP) transfectants. (a) Cells (5 × 105) were incubated in the presence or absence of 1 μM mitoxantrone for 40 min at 37°C and then washed and resuspended in ice‐cold PBS. Fluorescence was determined by flow cytometry. Bars indicate median channels of fluorescence in the transfectants. (1) PA317 cells; (2) PA/WT‐mix cells; (3) PA/C592S‐cl.6 cells; (4) PA/C603S‐cl.3 cells; (5) PA/C592S·C608S‐cl.7 cells; (6) PA/C592S·C603S·C608S‐cl.7 cells. (b) Cells were seeded onto 60 mm dishes (2 × 105 cells/dish) and cultured overnight. Cells were transfected with non‐silencing control or BCRP siRNA using Lipofectamine 2000. Western blot analysis was performed under non‐reducing conditions (−SH). GAPDH expression was analyzed as an internal control of protein loading. (c) The mitoxantrone accumulation assay was performed in siRNA‐transfected cells as described in (a). (1) PA317 cells transfected with control siRNA. (2) PA317 cells transfected with BCRP siRNA. (3) PA/WT‐mix cells transfected with control siRNA. (4) PA/WT‐mix cells transfected with BCRP siRNA. (5) PA/C592S·C603S·C608S‐cl.7 cells transfected with control siRNA. (6) PA/C592S·C603S·C608S‐cl.7 cells transfected with BCRP siRNA.
To confirm that BCRP‐C592S·C603S·C608S expression caused lowered accumulation of mitoxantrone, we then performed a mitoxantrone accumulation assay in BCRP‐knocked down PA/C592S·C603S·C608S‐cl.7 cells (Fig. 7c). PA317 and PA/WT‐mix cells were also used in this experiment as negative and positive controls, respectively. Breast cancer resistance protein (BCRP) small‐interfering RNA (siRNA) was found to suppress both wild‐type BCRP and BCRP‐C592S·C603S·C608S expression (Fig. 7b). In PA317 cells, BCRP siRNA hardly affected intracellular mitoxantrone levels. Again, intracellular mitoxantrone levels in PA/WT‐mix and PA/C592S·C603S·C608S‐cl.7 cells transfected with control siRNA was much lower than that in PA317 cells. Transfection with BCRP siRNA restored intracellular mitoxantrone levels in both PA/WT‐mix and PA/C592S·C603S·C608S‐cl.7 cells. In particular, intracellular mitoxantrone in PA/C592S·C603S·C608S‐cl.7 cells transfected with BCRP siRNA was present at almost the same level as that in PA317 cells. These results suggest that lower level occurrence of intracellular mitoxantrone depends on BCRP expression in both PA/WT‐mix and PA/C592S·C603S·C608S‐cl.7 cells. Fluorescence‐activated cell sorter (FACS) fluorograms of mitoxantrone accumulation experiments are shown in Figure S3.
Localization of mutant BCRP in mutant BCRP transfec‐tants. Immunofluorescent microscopy analysis using an anti‐BCRP antibody BXP‐21 was performed to examine whether BCRP‐C592S·C603S·C608S is expressed on the cell surface. As shown in Figure 8, wild‐type BCRP expression was detected predominantly on the cell surface. In contrast, only a small amount of BCRP‐C592S·C603S·C608S was expressed on the surface of the transfectant, and most of the triple mutant BCRPs was present in the cytoplasm. Subcellular localization of other mutant BCRPs in the transfectants are shown in Figure S4.
Figure 8.
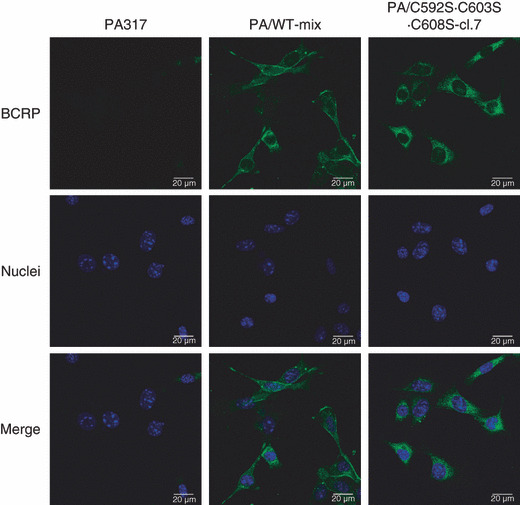
Localization of breast cancer resistance protein (BCRP) protein in PA/WT and PA/C592S·C603S·C608S cells. The cells were stained with anti‐BCRP antibody BXP‐21 following an Alexa Fluor 488 goat antimouse antibody (green). Nuclei were stained with DAPI (blue). PA317 cells were also stained with BXP‐21 and DAPI as a negative control. Breast cancer resistance protein (BCRP) and nuclei were observed using confocal microscopy.
Effect of FTC on SN‐38 and mitoxantrone resistance of the mutant BCRP transfectants. To examine the sensitivity of mutant BCRPs to a BCRP inhibitor, namely FTC, the reversal effect of FTC on the mutant BCRP transfectants was examined. PA/C603S‐cl.7 was added to the panel of drug‐resistant transfectants in this experiment. As shown in Table 2, FTC (1 μM) completely reversed the resistance of PA/WT‐mix and PA/C592S‐cl.6 cells to SN‐38 and mitoxantrone. Fumitremorgin C (FTC) reversed the drug resistance of PA/C603S‐cl.3 and PA/C603S‐cl.7 cells, although these clones still showed 4‐ to 15‐fold higher levels of resistance to SN‐38 and mitoxantrone in the presence of FTC. Fumitremorgin C (FTC) slightly lowered the resistance of PA/C592S·C608S‐cl.7 cells to SN‐38, but did not affect their resistance to mitoxantrone. Fumitremorgin C (FTC) had no effect on the resistance of PA/C592S·C603S·C608S‐cl.7 cells to SN‐38 and mitoxantrone (Table 2). These results demonstrate that substitution of Cys residues in the extracellular domain of BCRP alters the sensitivity of the transporter to FTC.
Table 2.
Effect of FTC on the SN‐38 and mitoxantrone resistance of mutant BCRP transfectants
| Cell line | SN‐38 | Mitoxantrone | Relative expression level of total BCRP§ | ||||
|---|---|---|---|---|---|---|---|
| Degree of resistance† | RI‡ | Degree of resistance† | RI‡ | ||||
| −FTC | +FTC | −FTC | + FTC | ||||
| PA317 | 1.0 ± 0.1 | 0.9 ± 0.1 | – | 1.0 ± 0.1 | 0.9 ± 0.1 | – | – |
| PA/WT‐mix | 19.3 ± 1.7 | 1.4 ± 0.1 | 13.8 | 9.0 ± 0.3 | 1.2 ± 0.1 | 7.5 | 100 |
| PA/C592S‐cl.6 | 4.2 ± 0.3 | 1.2 ± 0.1 | 3.5 | 3.7 ± 0.1 | 1.3 ± 0.1 | 2.8 | 52 |
| PA/C603S‐cl.3 | 106 ± 7.3 | 7.9 ± 0.3 | 13.4 | 57 ± 5.9 | 15 ± 0.7 | 3.8 | 146 |
| PA/C603S‐cl.7 | 18 ± 2.0 | 4.1 ± 0.2 | 4.4 | 28 ± 3.9 | 12 ± 1.0 | 2.3 | 135 |
| PA/C592S·C608S‐cl.7 | 8.4 ± 0.5 | 4.0 ± 0.2 | 2.1 | 3.3 ± 0.2 | 3.3 ± 0.1 | 1.0 | 151 |
| PA/C592S·C603S·C608S‐cl.7 | 4.1 ± 0.3 | 4.6 ± 0.3 | 0.9 | 7.5 ± 0.2 | 6.0 ± 0.8 | 1.3 | 145 |
†The cells were incubated for 4 days at 37°C in the presence or absence of various concentrations of anticancer agents in the presence or absence of 1 μM fumitremorgin C (FTC). The IC50 value (the drug dose causing a 50% inhibition of cell growth) was determined from growth inhibition curves, and the degree of resistance (x‐fold) was calculated by dividing the IC50 values of the breast cancer resistance protein (BCRP) transfectants by those of the parental PA317 cells. ‡The reversal index (RI) was determined by dividing the degree of resistance in the absence of FTC by the degree of resistance in the presence of FTC. Data are the mean ± SD of triplicate determinations. §Each total BCRP (75‐ and 70‐kDa) band intensity was measured (Fig. 3), and the relative expression levels were calculated by dividing BCRP expression values in each cell line as compared with PA/WT‐mix cells.
Discussion
We have previously reported that Cys‐603 is an important residue in the formation of the covalent bridge between BCRP monomers.( 16 ) In the present study, we generated cells expressing double (C592S·C603S, C592S·C608S, and C603S·C608S) and triple (C592S·C603S·C608S) mutant BCRP products to elucidate the possible involvement of the Cys‐592 and Cys‐608 residues in the dimerization and function of this protein. We employed the bicistronic vector pHa‐BCRP‐IRES‐DHFR, which allows for the direct selection of transfected cells with methotrexate.( 19 , 20 , 21 , 22 ) We have shown in a previous study that mixed populations of Cys mutant BCRP‐transfected cells express similar levels of exogenous BCRP mRNA.( 16 ) We used the same strategy in our current study. However, as shown in Figure 2, some of the mixed population of the transfected cells expressed very low amounts of mutant BCRP. We therefore isolated mutant BCRP transfectant clones to obtain cells that highly expressed the exogenous mutant protein to evaluate BCRP dimerization and the resistance levels of the cells to anticancer agents.
Consistent with our previous study, PA/C603S cells expressed both monomeric and dimeric BCRP.( 16 ) PA/C608S cells also expressed a BCRP monomer whereas PA/C592S cells expressed a BCRP dimer. The triple‐mutant BCRP transfectant, PA/C592S·C603S·C608S‐cl.7, expressed a BCRP monomer only. This indicated that the three extracellular Cys residues, Cys‐592, Cys‐603, and/or Cys‐608, are responsible for BCRP dimer formation, and that no other Cys residue is involved. Among double mutant BCRP transfectants, PA/C592S·C603S and PA/C592S·C608S expressed BCRP dimers, but PA/C603S·C608S expressed only a BCRP monomer. It has been discussed by Henriksen et al. ( 23 ) that an intramolecular disulfide bridge exists between Cys‐592 and Cys‐608. From this discussion and the findings of our study, we propose disulfide‐binding modes in wild‐type and mutant BCRPs. Free Cys‐603 is normally allowed to configure a homodimer of BCRP. Single mutation on Cys‐603 loses this homo‐dimerization site, and it can therefore be concluded that BCRP‐C603S protein mainly existed as a monomer. The intramolecular disulfide bridge between Cys‐592 and Cys‐608 is partially disrupted for some reason, and small amount of the BCRP dimer could be detected in PA/C603S. Introduction of the C592S mutation into BCRP‐C603S extinguishes the counter partner against Cys‐608 in the intramolecular disulfide bridge. As a result, free Cys‐608 is produced and dimerization of BCRP may be restored via this site. Double mutant BCRP on Cys‐603 and Cys‐608 has no more dimerization sites, and thus this mutant is thought to exist as a monomer. In PA/C608S cells, Cys‐603 can also construct an intramolecular disulfide bridge with Cys‐592, and therefore a single mutation on Cys‐608 may weaken dimerization of BCRP. The above scheme is illustrated in Figure S5. Altogether, Cys‐608 in addition to Cys‐603, but not Cys‐592, is involved in the dimerization of BCRP.
We successfully demonstrated that the triple‐mutant BCRP, namely BCRP‐C592S·C603S·C608S, functions as an active transporter whose expression confers drug resistance. In a previous study Henriksen et al. ( 23 ) reported a low expression of triple‐mutant BCRP, which prevented an evaluation of its functional activity. We also found that the generation of transfectants expressing high levels of the triple‐mutant BCRP was somewhat difficult and thus performed the selection of multiple clones. PA/C592S·C603S·C608S‐cl.7 cells showed resistance to mitoxantrone and topotecan, but not to doxorubicin (Fig. 5), and a cross‐resistance pattern which was very similar to that of the PA/WT‐mix cells. We carefully confirmed the complete sequence of the mutant BCRP cDNA in PA/C592S·C603S·C608S‐cl.7 cells. The possibility of unexpected additional mutations in this mutant, or contamination of the clonal PA/C592S·C603S·C608S‐cl.7 cell culture by wild‐type or other mutant BCRP‐expressing cells was thus excluded. We concluded that triple‐mutant BCRP (BCRP‐C592S·C603S·C608S) has some potential to function as a transporter protein and maintains a substrate specificity that is similar to wild‐type BCRP with no covalent disulfide bonds. However, our findings do not rule out the possibility that BCRP‐C592S·C603S·C608S functions as a dimer or an oligomer without covalent linkages in cells. In this regard, Xu et al. ( 24 , 25 ) have reported that BCRP can function as a homo‐oligomer via interactions in the third extracellular domain containing the Cys‐592, Cys‐603, and Cys‐608 residues.
PA/WT, PA/C592S, PA/C603S, PA/C592S·C608S, and PA/C592S·C603S·C608S‐cl.7 cells exhibited drug resistance and low accumulation of mitoxantrone. PA/WT and PA/C603S cells expressed a 75‐kDa BCRP product (Fig. 2). Significant expression of a 75‐kDa BCRP was also found in PA/C592S, PA/C592S·C608S, and PA/C592S·C603S·C608S‐cl.7 cells, whereas the PA/C608S cells mainly expressed a 70‐kDa BCRP (Fig. 3). The 70‐kDa BCRP seems to be an under‐molecule. Thus, the expression of a 75‐kDa BCRP in these transfectants is likely to be related to the drug resistance phenotype. However, when correlation of SN‐38 resistance with both total and 75‐kDa BCRP expression levels under reducing conditions was investigated, we found that some mutant BCRP transfectants showed lower drug resistance despite expressing 75‐kDa BCRP. This finding suggests that the degree of drug resistance depends on the quality and function of mutant BCRPs rather than on expression levels.
We examined the reversal effects of FTC on the drug resistance levels of wild‐type and mutant BCRP transfectants. Fumitremorgin C (FTC) strongly reversed the resistance of PA/WT and PA/C592S cells to SN‐38 and mitoxantrone, and partially reversed the drug resistance of PA/C603S cells. However, FTC had no effect on the drug resistance of PA/C592S·C603S·C608S‐cl.7 cells. In addition, FTC slightly lowered the resistance of PA/C592S·C608S‐cl.7 cells to SN‐38, but not to mitoxantrone. These results indicate that a substitution of Ser for Cys in the extracellular domain altered the FTC sensitivity. Similarly, it has been reported that a substitution of Val for Gly in the first intracellular loop of P‐glycoprotein alters its substrate specificity and the effects of inhibitors.( 26 , 27 )
Breast cancer resistance protein (BCRP) expression has been found in the placenta, intestine, kidney, liver, blood–brain barrier, and hematopoietic stem cells, and is assumed to play a role in the protective functions of such normal tissues against toxic substances and metabolites.( 28 , 29 , 30 ) Competitive inhibitors of BCRP might alter the tissue distribution and blood concentration of substrate anticancer agents. Currently, small molecule tyrosine kinase inhibitors (TKIs) such as imatinib, nilotinib, gefitinib, erlotinib, lapatinib, and sunitinib have been used in various clinical settings. Most of these TKIs are competitive inhibitors for ATP, and good substrates of BCRP and other ABC transporters. We and others have shown that these TKIs strongly inhibit BCRP and sensitize cancer cells to substrate anticancer agents.( 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 ) Therefore, co‐administration of such TKIs might alter the tissue distribution and blood concentration of substrate anticancer agents.
Functional single nucleotide polymorphisms (SNPs) also appear to be able to alter the expression and function of BCRP. We previously reported that BCRP 421C>A SNP, substituting Lys‐141 for Gln, is associated with a low expression of the protein.( 40 ) In a phase Ι study of diflomotecan, a camptothecin‐derivative anticancer agent, patients with a BCRP 421C>A allele had a significantly higher blood concentration of the drug.( 41 ) Cusatis et al. ( 42 ) have reported that patients with a BCRP 421C>A allele had a significantly higher risk of developing gefitinib‐induced diarrhea. Rudin et al. ( 43 ) have demonstrated that the diplotype of two polymorphic loci in the BCRP promoter, involving –15622C>T and 1143C>T, conferred lower BCRP levels associated with higher erlotinib pharmacokinetic parameters, including area under the curve and maximum concentration. We have also reported that two germ line mutations of BCRP in our laboratory, namely 623T>C (F208S) and 1291T>C (F431L), also resulted in the loss of function or severely reduced the function of BCRP.( 44 ) Sunitinib clearly reversed the SN‐38 resistance of PA/WT cells but not of BCRP‐F431L transfectants. In addition, sunitinib did not inhibit [125I]iodoarylazidoprazosin‐binding to BCRP‐F431L.( 45 ) Future systematic analyses of the effects of amino acid substitutions on BCRP expression and function will be helpful in furthering our understanding of the structure‐function relationship of this protein.
The present study demonstrated that the extracellular Cys residues of BCRP, Cys‐603, and Cys‐608, are important for the dimer formation of this protein. However, covalent dimerization of BCRP is not necessary for its transporter function. Moreover, substitution of Cys residues in the extracellular domain of BCRP alters the specificity of a specific inhibitor of this transporter.
Supporting information
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank K. Muto, S. Yoshioka, Y. Inoue, Y. Ogi, and M. Kunitake for their expert technical assistance and helpful discussions. This study was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology, and the Ministry of Health, Labour and Welfare, Japan.
References
- 1. Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet 1995; 29: 607–49. [DOI] [PubMed] [Google Scholar]
- 2. Chen CJ, Chin JE, Ueda K et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P‐glycoprotein) gene from multidrug‐resistant human cells. Cell 1986; 47: 381–9. [DOI] [PubMed] [Google Scholar]
- 3. Ueda K, Clark DP, Chen CJ, Roninson IB, Gottesman MM, Pastan I. The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J Biol Chem 1987; 262: 505–8. [PubMed] [Google Scholar]
- 4. Pastan I, Gottesman MM, Ueda K, Lovelace E, Rutherford AV, Willingham MC. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P‐glycoprotein in MDCK cells. Proc Natl Acad Sci USA 1988; 85: 4486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cole SP, Bhardwaj G, Gerlach JH et al. Overexpression of a transporter gene in a multidrug‐resistant human lung cancer cell line. Science 1992; 258: 1650–4. [DOI] [PubMed] [Google Scholar]
- 6. Doyle LA, Yang W, Abruzzo LV et al. A multidrug resistance transporter from human MCF‐7 breast cancer cells. Proc Natl Acad Sci USA 1998; 95: 15665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. The mouse Bcrp1/Mxr/Abcp gene: amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res 1999; 59: 4237–41. [PubMed] [Google Scholar]
- 8. Ross DD, Yang W, Abruzzo LV et al. Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone‐selected cell lines. J Natl Cancer Inst 1999; 91: 429–33. [DOI] [PubMed] [Google Scholar]
- 9. Maliepaard M, Van Gastelen MA, De Jong LA et al. Overexpression of the BCRP/MXR/ABCP gene in a topotecan‐selected ovarian tumor cell line. Cancer Res 1999; 59: 4559–63. [PubMed] [Google Scholar]
- 10. Litman T, Brangi M, Hudson E et al. The multidrug‐resistant phenotype associated with overexpression of the new ABC half‐transporter, MXR (ABCG2). J Cell Sci 2000; 113: 2011–21. [DOI] [PubMed] [Google Scholar]
- 11. Kawabata S, Oka M, Shiozawa K et al. Breast cancer resistance protein directly confers SN‐38 resistance of lung cancer cells. Biochem Biophys Res Commun 2001; 280: 1216–23. [DOI] [PubMed] [Google Scholar]
- 12. Jonker JW, Smit JW, Brinkhuis RF et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 2000; 92: 1651–6. [DOI] [PubMed] [Google Scholar]
- 13. Sugimoto Y, Tsukahara S, Ishikawa E, Mitsuhashi J. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmacokinetics/pharmacodynamics. Cancer Sci 2005; 96: 457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noguchi K, Katayama K, Mitsuhashi J, Sugimoto Y. Functions of the breast cancer resistance protein (BCRP/ABCG2) in chemotherapy. Adv Drug Deliv Rev 2009; 61: 26–33. [DOI] [PubMed] [Google Scholar]
- 15. Kage K, Tsukahara S, Sugiyama T et al. Dominant‐negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S‐S dependent homodimerization. Int J Cancer 2002; 97: 626–30. [DOI] [PubMed] [Google Scholar]
- 16. Kage K, Fujita T, Sugimoto Y. Role of Cys‐603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci 2005; 96: 866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rabindran SK, He H, Singh M et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res 1998; 58: 5850–8. [PubMed] [Google Scholar]
- 18. Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res 2000; 60: 47–50. [PubMed] [Google Scholar]
- 19. Sugimoto Y, Tsukahara S, Sato S et al. Drug‐selected co‐expression of P‐glycoprotein and gp91 in vivo from an MDR1‐bicistronic retrovirus vector Ha‐MDR‐IRES‐gp91. J Gene Med 2003; 5: 366–76. [DOI] [PubMed] [Google Scholar]
- 20. Sugimoto Y, Aksentijevich I, Gottesman MM, Pastan I. Efficient expression of drug‐selectable genes in retroviral vectors under control of an internal ribosome entry site. Biotechnology 1994; 12: 694–8. [DOI] [PubMed] [Google Scholar]
- 21. Sugimoto Y, Hrycyna CA, Aksentijevich I, Pastan I, Gottesman MM. Coexpression of a multidrug‐resistance gene (MDR1) and herpes simplex virus thymidine kinase gene as part of a bicistronic messenger RNA in a retrovirus vector allows selective killing of MDR1‐transduced cells. Clin Cancer Res 1995; 1: 447–57. [PubMed] [Google Scholar]
- 22. Sugimoto Y, Sato S, Tsukahara S et al. Coexpression of a multidrug resistance gene (MDR1) and herpes simplex virus thymidine kinase gene in a bicistronic retroviral vector Ha‐MDR‐IRES‐TK allows selective killing of MDR1‐transduced human tumors transplanted in nude mice. Cancer Gene Ther 1997; 4: 51–8. [PubMed] [Google Scholar]
- 23. Henriksen U, Fog JU, Litman T, Gether U. Identification of intra‐ and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J Biol Chem 2005; 280: 36926–34. [DOI] [PubMed] [Google Scholar]
- 24. Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric human half‐ABC transporter ATP‐binding cassette G2. J Biol Chem 2004; 279: 19781–9. [DOI] [PubMed] [Google Scholar]
- 25. Xu J, Peng H, Chen Q, Liu Y, Dong Z, Zhang JT. Oligomerization domain of the multidrug resistance‐associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res 2007; 67: 4373–81. [DOI] [PubMed] [Google Scholar]
- 26. Choi K, Chen CJ, Kriegler M, Roninson IB. An altered pattern of cross‐resistance in multidrug‐resistant human cells results from spontaneous mutations in the mdr1 (P‐glycoprotein) gene. Cell 1988; 53: 519–29. [DOI] [PubMed] [Google Scholar]
- 27. Cardarelli CO, Aksentijevich I, Pastan I, Gottesman MM. Differential Effects of P‐glycoprotein inhibitors on NIH3T3 cells transfected with wild‐type (G185) or mutant (V185) multidrug transporters. Cancer Res 1995; 55: 1086–91. [PubMed] [Google Scholar]
- 28. Maliepaard M, Scheffer GL, Faneyte IF et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 2001; 61: 3458–64. [PubMed] [Google Scholar]
- 29. Zhou S, Schuetz JD, Bunting KD et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side‐population phenotype. Nat Med 2001; 7: 1028–34. [DOI] [PubMed] [Google Scholar]
- 30. Jonker JW, Merino G, Musters S et al. The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat Med 2005; 11: 127–9. [DOI] [PubMed] [Google Scholar]
- 31. Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y. Gefitinib reverses breast cancer resistance protein–mediated drug resistance. Mol Cancer Ther 2004; 3: 1119–25. [PubMed] [Google Scholar]
- 32. Burger H, Van Tol H, Boersma AW et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004; 104: 2940–2. [DOI] [PubMed] [Google Scholar]
- 33. Elkind NB, Szentpetery Z, Apati A et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res 2005; 65: 1770–7. [DOI] [PubMed] [Google Scholar]
- 34. McDowell HP, Meco D, Riccardi A et al. Imatinib mesylate potentiates topotecan antitumor activity in rhabdomyosarcoma preclinical models. Int J Cancer 2007; 120: 1141–9. [DOI] [PubMed] [Google Scholar]
- 35. Shi Z, Peng XX, Kim IW et al. Erlotinib (Tarceva, OSI‐774) antagonizes ATP‐binding cassette subfamily B member 1 and ATP‐binding cassette subfamily G member 2‐mediated drug resistance. Cancer Res 2007; 67: 11012–20. [DOI] [PubMed] [Google Scholar]
- 36. Brendel C, Scharenberg C, Dohse M et al. Imatinib mesylate and nilotinib (AMN107) exhibit high‐affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007; 21: 1267–75. [DOI] [PubMed] [Google Scholar]
- 37. Polli JW, Humphreys JE, Harmon KA et al. The role of efflux and uptake transporters in N‐{3‐chloro‐4‐[(3‐fluorobenzyl)oxy]phenyl}‐6‐[5‐({[2‐(methy‐lsulfonyl)ethyl]amino} methyl)‐2‐furyl]‐4‐quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos 2008; 36: 695–701. [DOI] [PubMed] [Google Scholar]
- 38. Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib. (Sutent, SU11248), a small‐molecule receptor tyrosine kinase inhibitor, blocks function of the ABC transporters, P‐glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos 2008; 37: 359–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dai CL, Liang YJ, Wang YS et al. Sensitization of ABCG2‐overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett 2009; 279: 74–83. [DOI] [PubMed] [Google Scholar]
- 40. Imai Y, Nakane M, Kage K et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low‐level drug resistance. Mol Cancer Ther 2002; 1: 611–6. [PubMed] [Google Scholar]
- 41. Sparreboom A, Gelderblom H, Marsh S et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther 2004; 76: 38–44. [DOI] [PubMed] [Google Scholar]
- 42. Cusatis G, Gregorc V, Li J et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 2006; 98: 1739–42. [DOI] [PubMed] [Google Scholar]
- 43. Rudin CM, Liu W, Desai A et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol 2008; 26: 1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoshioka S, Katayama K, Okawa C et al. The identification of two germ‐line mutations in the human breast cancer resistance protein gene that result in the expression of a low/non‐functional protein. Pharm Res 2007; 24: 1108–17. [DOI] [PubMed] [Google Scholar]
- 45. Kawahara H, Noguchi K, Katayama K, Mitsuhashi J, Sugimoto Y. Pharmacologicalinteraction with sunitinib is abolished by a germ‐line mutation (1291T>C) of BCRP/ABCG2 gene. Cancer Sci 2010; DOI: 10.1111/j.1349-7006.2010.01539.x.. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item