

Abstract
The platelet aggregation‐inducing factor, Aggrus (also known as podoplanin), is reported to contribute to cancer metastasis by mediating cancer cell–platelet interaction. Aggrus has been shown to be upregulated in many different types of cancers. Thus, not only the functional inhibition of Aggrus, but also its application as a cancer‐specific antigen has therapeutic potential. Among a series of anti‐Aggrus mAb established previously, no mouse anti‐human Aggrus mAb exists that possesses the ability to neutralize platelet aggregation. For precise preclinical examinations of mouse and monkey models, the establishment of Aggrus‐neutralizing mouse mAb and their chimeric Abs is needed. In this study, we established two mouse anti‐human Aggrus mAb, P2‐0 and HAG‐3. A precise analysis of their epitopes revealed that P2‐0 recognized the conformation near the bioactive O‐glycosylation site at the Thr52 residue. In contrast, HAG‐3 recognized the amino‐terminus side at a short distance from the conformation recognized by P2‐0. We observed that only P2‐0 attenuated Aggrus‐induced platelet aggregation and Aggrus binding to its platelet receptor, that is, the C‐type lectin‐like receptor‐2. Consistent with these data, only P2‐0 prevented the experimental metastasis of human Aggrus‐overexpressing CHO cells. Subsequently, we cloned the complementary determining region of P2‐0 and produced the murine/human chimeric P2‐0 antibody. This chimeric antibody maintained its inhibitory activity of Aggrus‐induced platelet aggregation and experimental metastasis. Thus, P2‐0 and its chimeric antibody are expected to aid the development of preclinical and clinical examinations of Aggrus‐targeted cancer therapy. (Cancer Sci 2011; 102: 2051–2057)
Aggrus/podoplanin, a type‐I transmembrane sialoglycoprotein, has been shown to be upregulated in many cancers, such as different types of squamous cell carcinoma, mesothelioma, Kaposi’s sarcoma, testicular germ cell tumors, and brain tumors.( 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 ) In some reports, Aggrus overexpression was correlated with a poorer prognosis,( 3 , 4 ) suggesting the important contribution of Aggrus to cancer progression. We previously discovered that Aggrus was a platelet aggregation‐inducing factor, and its expression promoted both experimental and spontaneous pulmonary metastasis in mice.( 10 , 11 ) The platelet aggregation‐inducing activity of Aggrus is directly linked to metastasis formation, because the introduction of a point mutation that suppresses platelet aggregation attenuates pulmonary metastasis formation.( 10 ) Cancer cell‐induced platelet aggregation is believed to form a large cancer cell–platelet aggregate, resulting in enhanced cancer cell embolization in the microvasculature and protection from immunological assault in the circulation.( 12 , 13 , 14 ) The C‐type lectin‐like receptor‐2 (CLEC‐2) expressed on platelets was recently identified as one of the counter receptors of Aggrus.( 15 , 16 ) When CLEC‐2 binds to Aggrus expressed on tumor cells, it generates activation signals in platelets, with no requirement for plasma components.( 17 ) Thus, the therapeutic potential of this pathological interaction between Aggrus and CLEC‐2 is now ready to be examined.
An mAb can bind firmly and specifically to cell surface antigens and induce an immunological response in the target cell. Thus, many mAb are now used in cancer therapy. The mAb used in cancer therapy show anticancer effects via three representative modes of action: neutralizing, antibody‐dependent cellular cytotoxic (ADCC), and complement‐dependent cytotoxic (CDC) activities.( 18 ) These three modes of action are specified by the isotype and subclass of the antibody, the traits of the antigen, and the recognition site. Aggrus’s critical domains for the interaction to CLEC‐2 have been identified.( 11 , 19 ) Therefore, the antibody against Aggrus that specifically recognizes the interaction site is expected to have all three modes of action. Many mAb against human or mouse Aggrus have been established so far, but most cannot interfere with the Aggrus–CLEC‐2 interaction.( 20 ) Although one anti‐human Aggrus mAb exists that can inhibit the Aggrus–CLEC‐2 interaction and platelet aggregation,( 21 ) it is a rat antibody and cannot be precisely examined in universally‐used mouse cancer models because of the species barrier.
In the present study, we established a novel mouse mAb that inhibited the Aggrus–CLEC‐2 interaction and Aggrus‐induced platelet aggregation and experimental metastasis. Furthermore, we succeeded in the development of murine/human chimeric P2‐0 and showed that the chimeric antibody (hP2‐0) retained the ability to inhibit Aggrus‐induced platelet aggregation and experimental metastasis. Thus, these antibodies are expected to aid the development of preclinical and clinical examinations of Aggrus‐targeted cancer therapy.
Materials and Methods
Cell lines. CHO, H226, and HT1080 cells were cultured in RPMI‐1640 media (Wako, Osaka, Japan), supplemented with 10% FBS (Sigma, St Louis, MO, USA). CHO cells that had been stably transfected with vectors (CHO/mock) and human aggrus (CHO/Aggrus) were previously described.( 10 )
Animals. Female BALB/c mice, BALB/c‐nu/nu mice, and CB‐17/Icr‐scid mice were purchased from Charles River Japan (Kanagawa, Japan). All animal procedures were performed using protocols approved by the Japanese Foundation for Cancer Research Animal Care and Use Committee.
Plasmids and recombinant proteins. A human aggrus cDNA region encoding the TT679 antigen (112–153 bp) was cloned( 11 ) and connected eight times repeatedly on a pGEX‐6P‐3 vector (TT679‐repeat; GE Healthcare, Buckinghamshire, UK). The human ΔN20 aggrus cDNA (61–486 bp) was cloned into a pGEX‐6P‐3 vector (ΔN20‐Aggrus). The substitution of amino acids and the deletion of the appropriate region in ΔN20 aggrus cDNA were accomplished using the QuikChange site‐directed mutagenesis kit (Stratagene, La Jolla, CA, USA). BL21 Escherichia coli (Invitrogen, Carlsbad, CA, USA) was transformed with these vectors, and GST‐tagged recombinant proteins were purified using Glutathione–Sepharose (GE Healthcare).
Hybridoma production, antibody purification, and Fab preparation. Six‐week‐old female BALB/c mice were immunized by neck subcutaneous injections of recombinant ΔN20‐Aggrus and TT679‐repeat with Freund’s complete adjuvant (Difco Laboratories, Detroit, MI, USA). Every other week, intraperitoneal immunization was performed. Spleen cells were fused with mouse myeloma P3U1 cells using polyethylene glycol 4000 (Merck, Whitehouse Station, NJ, USA). Antibodies were purified from ascites fluid by caprylic acid/ammonium sulfate precipitation method. The Fab fragment of mouse mAb P2‐0 was prepared using a Fab preparation kit (Takara, Shiga, Japan).
Flow cytometry. Cells were harvested and treated with antihuman Aggrus mAb or control mouse IgG. Flow cytometric analysis was performed using a Cytomics FC500 flow cytometry system (Beckman‐Coulter, Miami, FL, USA).
Western blot analysis. Cell lysates and recombinant Aggrus proteins were electrophoresed and blotted onto a nitrocellulose membrane, as previously described.( 11 ) We used antibodies to GST tag (Abcam, Cambridge, UK), human Aggrus, or β‐actin.
Surface plasmon resonance analysis. Biosensor analyses were performed using the Biacore X100 system (GE Healthcare). The recombinant Aggrus protein (R&D Systems, Minneapolis, MN, USA) was covalently attached to a CM5 sensor chip by the amine coupling method, according to the manufacturer’s instructions. Final levels of immobilization were approximately 2000 response units. All experiments were performed at 25°C at a constant flow rate of 30 μL/min driving buffer. The antibodies diluted in driving buffer were passed over the immobilized Aggrus for 1 min, and the driving buffer alone was passed for 2 min. Five concentrations of antibodies were passed over the chip in a single cycle, without regenerating the surface between injections. Sensorgrams were fit by global analysis using the Biacore X100 evaluation software. The equilibrium dissociation constant (K D) was determined from the bivalent analyte model.
ELISA. Synthetic peptides were immobilized on amino‐type ELISA plates (SUMILON, Tokyo, Japan). After blocking, the plates were incubated with control mouse IgG or P2‐0. The plates were then incubated with peroxidase‐conjugated anti‐mouse IgG antibody (MP Biomedicals, Aurora, OH, USA), following with the addition of the 1‐Step Ultra TMB‐ELISA reagent (Pierce, Rockford, IL, USA). In some experiments, the His‐tagged recombinant human CLEC‐2 protein (R&D Systems) was immobilized on Ni‐NTA HisSorb plates (Qiagen, Chatsworth, CA, USA). After blocking, the plates were further incubated with human IgG Fc‐tagged recombinant human Aggrus protein (R&D Systems) in the presence of control IgG or anti‐human Aggrus mAb.
Platelet aggregation assay. Cells were incubated with mouse or chimeric anti‐human Aggrus mAb or control mouse or human IgG (HT1080: 1 × 107 cells/mL; CHO: 2 × 107 cells/mL). Mouse platelet‐rich plasma was prepared from BALB/c mice, and a platelet aggregation assay was performed, as previously described,( 11 ) with a platelet aggregometer (MCM HEMA TRACER 313M; SSR Engineering, Kanagawa, Japan).
Experimental lung metastasis. Cells were harvested and incubated with anti‐human Aggrus antibodies or control mouse or human IgG (HT1080: 5 × 106 cells/mL; CHO: 1.25 × 106 cells/mL). Next, 200 μL of the cell suspension was intravenously inoculated into the lateral tail vein (HT1080: 6‐week‐old female CB‐17/Icr‐scid mice; CHO: 6‐week‐old female BALB/c‐nu/nu mice).
Development of chimeric antibody. Total RNA was extracted from P2‐0 mAb‐producing hybridoma cells. The cDNA of the heavy and light chains of P2‐0 were amplified by PCR, cloned into the pT7 Blue vector (Novagen, Madison, WI, USA), and sequenced. The cDNA were analyzed by online V‐Quest software provided by the International ImMunoGeneTics database (http://www.imgt.org/IMGT_vquest/share/textes/) for identification of the complementarity‐determining region (CDR). The NotI–NheI fragment containing heavy‐chain CDR, or the NotI–BsiWI fragment containing light‐chain CDR, was then subcloned into a heavy or κ light‐chain constant region of human IgG1 on a pRetro‐Q retroviral vector (Clontech, Mountain View, CA, USA), respectively. GP2‐293 packaging cells (Clontech) were cotransfected with each CDR construct and a vesicular stomatitis virus glycoprotein (VSV‐G) expression plasmid using Lipofectamine 2000 reagent (Invitrogen). The CHO cells were doubly infected with culture supernatants containing chimeric heavy and κ light chain‐expressing retroviruses. The infected cells were selected by medium containing 10 μg/mL of puromycin and 500 μg/mL of hygromycin B. The chimeric antibody was purified from the supernatant of the infected CHO cells using Protein A–Sepharose (Zymed, South San Francisco, CA, USA).
Statistical analysis. A Mann–Whitney U‐test was performed to determine the statistical significance in the metastasis assays. P‐values <0.05 were considered statistically significant. All statistical tests were two‐sided.
Results
Establishment of two novel mouse mAb against human Aggrus. We, and other groups, have previously established rat antibodies against human Aggrus using the TT679 antigen (EGGVAMPGAEDDVV; 38–51 aa).( 20 , 21 , 22 ) In the present study, we attempted to establish mouse mAb by immunizing mice with the TT679 antigen. However, we did not succeed in establishing mouse mAb. We supposed that the immunogenicity of the TT679 antigen was weak in mice. We then generated an eight times tandemly‐repeated TT679 (TT679‐repeat) or the recombinant ΔN20‐Aggrus protein, and immunized the mice with them. As a result, we succeeded in establishing P2‐0 (IgG1) from mice immunized with the TT679‐repeat. In addition, we cloned the HAG‐3 antibody (IgG1) from mice immunized with ΔN20‐Aggrus. As shown in Figure 1(a), both mAb specifically recognized the Aggrus protein, because both mAb recognized CHO cells stably transfected with the vector encoding Aggrus (CHO/Aggrus), but not CHO/mock. With regard to affinity, the binding capability of HAG‐3 was lower than that of P2‐0, because HAG‐3 could hardly detect endogenous Aggrus expressed in cancer cells (Fig. 1b). Human primary lung squamous cancers, human lung squamous H226 cells, and human fibrosarcoma HT1080 cells have been known to endogenously express Aggrus (Fig. S1).( 1 , 23 ) The surface plasmon resonance analysis indicated that P2‐0 showed higher affinity to the Aggrus protein (K D = 9.30 nM) than HAG‐3 did (K D = 430 nM) (Fig. 1c).
Figure 1.
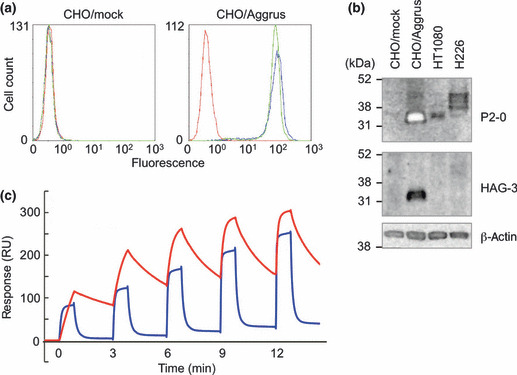
Characterization of two novel anti‐human Aggrus mAb, P2‐0 and HAG‐3. (a) Stably‐transfected CHO/mock and CHO/Aggrus were treated with control mouse IgG1 (control IgG) ( ), P2‐0 (
), P2‐0 ( ), and HAG‐3 (
), and HAG‐3 ( ). After incubation with the second antibody, Aggrus expression was analyzed by flow cytometry. (b) Cells were lysed and immunoblotted with the indicated antibodies. (c) Interaction between the human Aggrus protein and anti‐human Aggrus mAb was estimated by surface plasmon resonance analysis. Five different concentrations of antibodies (P2‐0, 6.25–100 nM; HAG‐3, 62.5–1000 nM) were passed over a sensor chip with the immobilized Aggrus protein for 1 min before the flow was switched to the buffer alone for another 2 min in a single cycle. Equilibrium dissociation constants (K
D) are shown. P2‐0 (K
D = 9.30 x 10−9 M) (
). After incubation with the second antibody, Aggrus expression was analyzed by flow cytometry. (b) Cells were lysed and immunoblotted with the indicated antibodies. (c) Interaction between the human Aggrus protein and anti‐human Aggrus mAb was estimated by surface plasmon resonance analysis. Five different concentrations of antibodies (P2‐0, 6.25–100 nM; HAG‐3, 62.5–1000 nM) were passed over a sensor chip with the immobilized Aggrus protein for 1 min before the flow was switched to the buffer alone for another 2 min in a single cycle. Equilibrium dissociation constants (K
D) are shown. P2‐0 (K
D = 9.30 x 10−9 M) ( ) and HAG‐3 (K
D = 4.30 x 10−7 M) (
) and HAG‐3 (K
D = 4.30 x 10−7 M) ( ).
).
Identification of the P2‐0 epitope on human Aggrus. We previously identified the conserved motif EDXXVTPG in the Aggrus protein as the platelet aggregation‐inducing (PLAG) domain critical for Aggrus’s platelet aggregation‐inducing ability.( 11 , 22 ) Human Aggrus has three PLAG domains (PLAG1–3) at the N‐terminal region. The examination of the reactivity of anti‐human Aggrus mAb to Aggrus‐deletion mutants (Fig. 2a) clarified that P2‐0 recognized the region around the PLAG2 and PLAG3 domains, and HAG‐3 recognized the region around the PLAG1 domain. Because NZ‐1, which possessed Aggrus‐neutralizing activity, was known to recognize the region around the PLAG2 domain, we further examined the P2‐0 recognition site in more detail. ELISA, using various synthetic peptides corresponding to PLAG2 and PLAG3 domains (Fig. 2b), and Western blot analysis, using point‐mutated Aggrus proteins (Fig. 2c), suggested that P2‐0 recognized the region containing 44–52 aa, particularly the perimeter structure around Gly45, Asp48, and Asp49.
Figure 2.
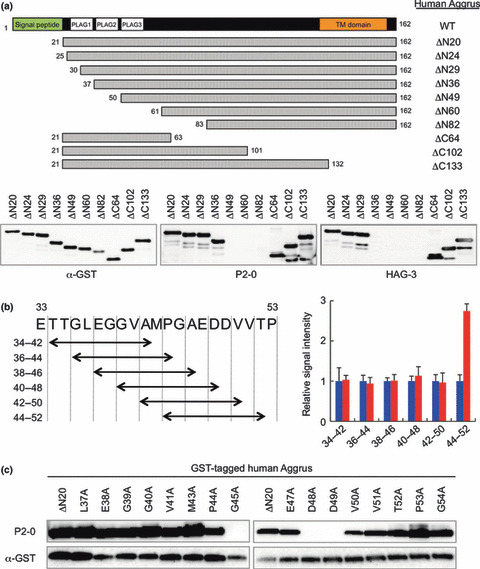
Analysis of the epitopes of P2‐0 and HAG‐3. (a) GST‐tagged recombinant Aggrus deletion mutants were immunoblotted with the indicated antibodies. Schematic representation of the generated Aggrus deletion mutants is shown on upper panel. (b) Synthetic peptides were immobilized on an ELISA plate and incubated with control mouse IgG and P2‐0. Schematic representation of the used synthetic nine amino‐acid peptides is shown in the left panel. Data are means ± standard deviations of triplicate determinations. Control IgG ( ) and P2‐0 (
) and P2‐0 ( ). (c) GST‐tagged recombinant human ΔN20‐Aggrus protein and its point mutants were electrophoresed and immunoblotted with anti‐GST and P2‐0 antibodies.
). (c) GST‐tagged recombinant human ΔN20‐Aggrus protein and its point mutants were electrophoresed and immunoblotted with anti‐GST and P2‐0 antibodies.
P2‐0 inhibits Aggrus–CLEC‐2 interaction and Aggrus‐induced platelet aggregation. Following the identification of the antibody recognition sites, we examined the effects of these antibodies on the Aggrus–CLEC‐2 interaction. The Aggrus–CLEC‐2 interaction could be detected by ELISA using recombinant human Aggrus and CLEC‐2 proteins purified from mammalian cells. We first examined whether the interaction was affected by the established anti‐Aggrus mAb. As shown in Figure 3(a), only P2‐0 inhibited the Aggrus–CLEC‐2 interaction in a concentration‐dependent manner. This result suggests that the P2‐0 epitope (44–52 aa) is critical for Aggrus binding to CLEC‐2. Because the Aggrus–CLEC‐2 interaction is critical for Aggrus‐induced platelet aggregation, we studied Aggrus‐induced mouse platelet aggregation in vitro in the presence of anti‐Aggrus mAb. As we reported previously, human Aggrus interacted with and induced the aggregation of mouse platelets.( 10 , 11 ) Parental CHO and CHO/mock cells are known to be incapable of inducing platelet aggregation.( 10 ) Thus, CHO/Aggrus‐induced platelet aggregation can be regarded as Aggrus‐induced platelet aggregation. In in vitro platelet aggregation analyses, we usually compare the time from the reaction starting point to the half‐maximum value point. Consistent with the result shown in Figure 3(a), Aggrus‐induced platelet aggregation was attenuated by P2‐0 addition in a concentration‐dependent manner (1 μg/mL, 5 min; 10 μg/mL, 9 min), but only slightly by HAG‐3 addition (1 μg/mL, 4 min; 10 μg/mL, 4 min) (Fig. 3b). Therefore, P2‐0 is an anti‐human Aggrus mAb that inhibits Aggrus–CLEC2 binding.
Figure 3.
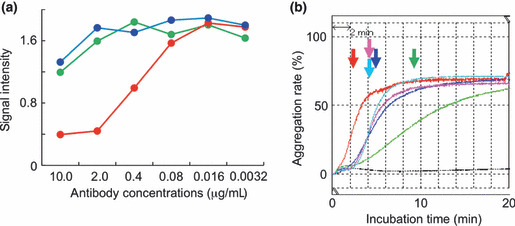
Effects of P2‐0 and HAG‐3 on the Aggrus–C‐type lectin‐like receptor‐2 (CLEC‐2) interaction and platelet aggregation. (a) Recombinant CLEC‐2 protein was immobilized on an ELISA plate and then incubated with the human IgG Fc‐tagged recombinant Aggrus protein in the presence of the indicated concentrations of control mouse IgG1 (control IgG) ( ), P2‐0 (
), P2‐0 ( ), and HAG‐3 (
), and HAG‐3 ( ). (b) CHO/Aggrus cells were incubated with control mouse IgG1 (control IgG; 10 μg/mL) (
). (b) CHO/Aggrus cells were incubated with control mouse IgG1 (control IgG; 10 μg/mL) ( ), P2‐0) (10 μg/mL [
), P2‐0) (10 μg/mL [ ] and 1 μg/mL [
] and 1 μg/mL [ ]) and HAG‐3 (10 μg/mL [
]) and HAG‐3 (10 μg/mL [ ] and 1 μg/mL [
] and 1 μg/mL [ ]), followed by incubation with mouse platelets. PBS was added as a negative control (black line). Arrows indicate the time points from the reaction starting point to the half‐maximum value point.
]), followed by incubation with mouse platelets. PBS was added as a negative control (black line). Arrows indicate the time points from the reaction starting point to the half‐maximum value point.
P2‐0 prevents Aggrus‐mediated experimental pulmonary metastasis. Next, we examined the effects of anti‐Aggrus mAb on experimental pulmonary metastasis of intravenously‐injected CHO/Aggrus cells. Although CHO cells have some metastatic ability, they become more metastatic upon the forced expression of Aggrus.( 10 , 11 ) This Aggrus‐induced experimental metastasis was almost completely inhibited by the concurrent administration of P2‐0 (Fig. 4a,b). The HAG‐3 administration showed a small inhibitory effect, but the number of metastatic foci was statistically insignificant compared to the number injected with control mouse IgG. This small inhibition of HAG‐3 might be due to its weak neutralizing ability (Fig. 3b) and its low affinity to the Aggrus protein (Fig. 1c). The Fab fragment of P2‐0 diminished the number of metastatic foci of CHO/Aggrus cells (Fig. 4c,d), and P2‐0 showed no influence on the metastasis of Aggrus‐negative A375M cells (Fig. S2), indicating that P2‐0 suppressed pulmonary metastasis by directly targeting Aggrus.
Figure 4.
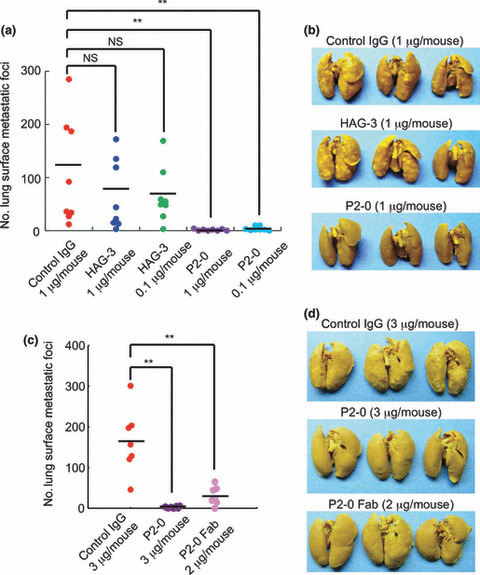
Prevention of experimental pulmonary metastasis by P2‐0. CHO/Aggrus cells were incubated with the indicated concentrations of control mouse IgG1 (control IgG), P2‐0, P2‐0 Fab fragment, and HAG‐3 (a–d). Cell suspension was intravenously inoculated into BALB/c‐nu/nu mice: n = 8 (a) and n = 7 (c). After 20 days, lung surface metastatic foci were counted. Numbers of metastatic foci (left panels) and representative pictures of the lungs (right panels) are shown. Bars, average. NS, not significant. **P < 0.005 by the Mann–Whitney U‐test.
Generation of the murine/human chimeric hP2‐0 antibody and its characterization. Because the subclass of P2‐0 is a mouse IgG1, P2‐0 might have some ADCC and CDC by interacting with the mouse immune system. Thus, the effectiveness of P2‐0 in Aggrus‐targeting therapy can be precisely examined in the mouse cancer model. However, we cannot extrapolate actual clinical safety simply from the results of a mouse model. Toxicological safety tests using more human‐like animals, such as monkeys, are necessary for clinical application, and the antibody also needs to become more like the human analog. Thus, we attempted to generate a murine/human chimeric P2‐0 antibody. We cloned the complementarity‐determining region of P2‐0 and ligated it into the constant region of human IgG1. Flow cytometric and surface plasmon resonance analyses showed that the generated hP2‐0 specifically bound to human Aggrus, with the affinity similar to P2‐0 (Fig. 5a,b; K D = 21.2 nM). As with the addition of P2‐0, the addition of hP2‐0 delayed Aggrus‐induced platelet aggregation and prevented the experimental metastasis of CHO/Aggrus in a concentration‐dependent manner (Fig. 5c,d). Moreover, we attempted to examine the pulmonary metastasis of actual cancer cells, that is, HT1080 cells. HT1080 cells endogenously expressed the Aggrus protein, as shown in 1, 5, and are known to induce platelet aggregation and possess metastatic ability in an Aggrus‐dependent fashion.( 23 ) As shown in Figure 5(d), a higher dose administration of hP2‐0 (30 μg/mouse) suppressed the pulmonary metastasis of HT1080 cells (Fig. 5d). Because the expression level of Aggrus in HT1080 cells was much lower than that in CHO/Aggrus (1, 5), a higher dose of hP2‐0 might be required for metastasis inhibition. We confirmed that hP2‐0 and P2‐0 recognized simian Aggrus (data not shown). These results suggest that hP2‐0 maintains the characteristic features of P2‐0 and can be utilized in preclinical safety tests involving monkeys.
Figure 5.
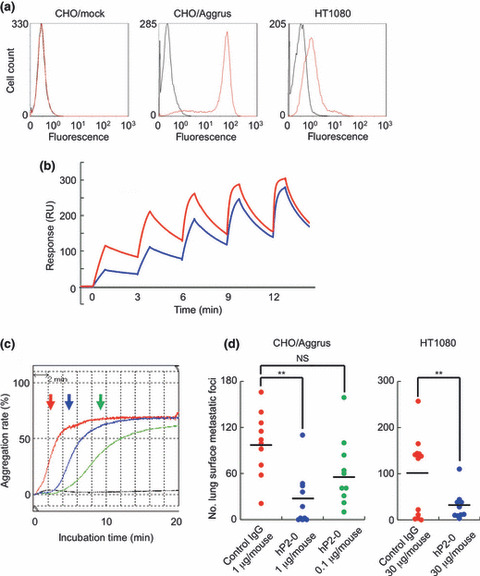
In vitro and in vivo evaluation of murine/human chimeric P2‐0 (hP2‐0). (a) Cells were treated with control human IgG1 ( ) and hP2‐0 (
) and hP2‐0 ( ). After incubation with the Alexa Fluor 488‐conjugated second antibody, Aggrus expression was analyzed by flow cytometry. (b) Interaction between the human Aggrus protein and hP2‐0 was estimated by SPR analysis. Five different concentrations of antibodies (6.25–100 nM) were passed over a sensor chip with the immobilized Aggrus protein for 1 min, before the flow was switched to the buffer alone for another 2 min in a single cycle. Equilibrium dissociation constants (K
D) are shown. P2‐0 (K
D = 9.30 x 10−9 M) [
). After incubation with the Alexa Fluor 488‐conjugated second antibody, Aggrus expression was analyzed by flow cytometry. (b) Interaction between the human Aggrus protein and hP2‐0 was estimated by SPR analysis. Five different concentrations of antibodies (6.25–100 nM) were passed over a sensor chip with the immobilized Aggrus protein for 1 min, before the flow was switched to the buffer alone for another 2 min in a single cycle. Equilibrium dissociation constants (K
D) are shown. P2‐0 (K
D = 9.30 x 10−9 M) [ ]) and hP2‐0 (K
D = 2.12 x 10−8 M [
]) and hP2‐0 (K
D = 2.12 x 10−8 M [ ]). (c) CHO/Aggrus cells were incubated with control IgG or hP2‐0, followed by incubation with mouse platelets. PBS was added as a negative control (black line). Arrows indicate the time points from the reaction starting point to the half‐maximum value point. Control IgG (10 μg/mL,
]). (c) CHO/Aggrus cells were incubated with control IgG or hP2‐0, followed by incubation with mouse platelets. PBS was added as a negative control (black line). Arrows indicate the time points from the reaction starting point to the half‐maximum value point. Control IgG (10 μg/mL,  ), hP2‐0 (10 μg/mL,
), hP2‐0 (10 μg/mL,  and 1 μg/mL,
and 1 μg/mL,  ). (d) CHO/Aggrus and HT1080 cells were incubated with control IgG and hP2‐0. Cell suspension was intravenously inoculated into BALB/c‐nu/nu (CHO, n = 10) or CB‐17/Icr‐scid (HT1080, n = 12 or 10) mice. After 20 (CHO) or 30 (HT1080) days, lung surface metastatic foci were counted. Bars, average. NS, not significant. **P < 0.005 by the Mann–Whitney U‐test.
). (d) CHO/Aggrus and HT1080 cells were incubated with control IgG and hP2‐0. Cell suspension was intravenously inoculated into BALB/c‐nu/nu (CHO, n = 10) or CB‐17/Icr‐scid (HT1080, n = 12 or 10) mice. After 20 (CHO) or 30 (HT1080) days, lung surface metastatic foci were counted. Bars, average. NS, not significant. **P < 0.005 by the Mann–Whitney U‐test.
Discussion
The relationship between platelets and cancer metastasis has long been investigated by many basic researchers, who have argued that cancer cell‐induced platelet aggregation promotes cancer metastasis.( 12 , 14 , 24 ) The relationship has also been reported by many clinical researchers. For example, many types of cancers are associated with a high incidence of venous thrombosis, and patients with distant metastasis have an increased risk.( 25 ) Moreover, the administration of anticoagulant drugs was reported to prolong survival in patients with advanced cancer.( 26 ) Detailed mechanisms of the relationship remain controversial, but a few suggestions exist. Platelets are believed to achieve enhanced cancer cell embolization in the microvasculature and protection from immunological assault in the circulation by forming a large cancer cell–platelet aggregate.( 12 , 13 , 14 ) Another mechanism is related to a wide variety of cytokines released from activated platelets. Some of these cytokines were reported to affect cancer cell growth directly or modify the tissue microenvironment.( 27 , 28 , 29 ) However, it is hard to believe that the administration of antiplatelet or anticoagulant drugs to cancer patients is a realistic therapy, as many patients suffer from thrombocytopenia due to platelet toxicity because of chemotherapy, and receive platelet transfusions. Thus, platelet‐targeting therapies must be specific for the pathological cancer cell–platelet interaction.
Aggrus has been shown to be engaged in cancer cell‐induced platelet aggregation, but its physiological role still remains poorly understood. Suzuki‐Inoue et al. ( 15 ) recently identified CLEC‐2 as one of counter‐receptors of Aggrus on platelets. C‐type lectin‐like receptor‐2 was reported to promote the formation of blood clots by hemophilic interaction on platelets.( 30 , 31 ) However, the Aggrus–CLEC‐2 interaction has been shown to be important in lymphatic development during fetal life,( 30 , 32 ) but its physiological role after birth is completely unknown. Presently, we believe that Aggrus and CLEC‐2 are not in a position where they can contact each other after birth. It is believed that cancer cells take over this interaction and utilize it pathologically. From this point of view, the Aggrus–CLEC‐2 interaction could be an ideal target for platelet‐targeting therapy.
At present, small molecule inhibitors of protein–protein interactions are extremely difficult to develop. Antibodies can inhibit protein–protein interactions more easily and specifically than small molecule inhibitors. In addition, antibodies have the collateral pharmacological effect of eliminating target cells by mobilizing the immune system, ADCC, and CDC, unlike small molecule inhibitors. We need to consider these three modes of action in antibody‐based cancer therapy. Although the recognition sites of two Aggrus antibodies established here were no more than 10 aa apart (Fig. 2a), their effects on the Aggrus–CLEC‐2 interaction varied significantly (Fig. 3a). Only P2‐0 suppressed the Aggrus–CLEC‐2 interaction, platelet aggregation, and experimental metastasis. This finding was attributed to the fact that P2‐0 bound the proximal region to the bioactive O‐glycosylation site at Thr52. The O‐glycosylation site at Thr52 in the PLAG3 domain was reported to be sialylated, which was required for recognition by CLEC‐2.( 19 ) We showed that substitution of the Thr52 residue by Ala diminished Aggrus‐induced platelet aggregation and pulmonary metastasis.( 10 ) However, it is still unknown whether P2‐0 binding affected CLEC‐2 access to Aggrus directly or the perimeter structure around the recognition site. The neutralization of platelet aggregation, ADCC, and CDC probably contribute to the metastasis inhibition by P2‐0. For the sufficient induction of ADCC and CDC, the species of antibody and the administered animal must be matched. Thus, in the mouse experimental metastasis model using P2‐0 (Fig. 4), ADCC and CDC might have some effects, but not in the mouse model using hP2‐0 (Fig. 5d). The metastasis inhibition of P2‐0 was stronger than that of hP2‐0 (4, 5). The medicinal benefit and safety of antibody‐based cancer therapy must be verified in the appropriate evaluation system considering the three modes of action. Accordingly, the use of P2‐0 in the mouse model and that of hP2‐0 in more human‐like animals might serve as powerful tools for assessing the efficacy and safety of Aggrus‐targeted cancer therapy.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Quantitative analysis of aggrus expression in tumorigenic and corresponding normal tissues from lung squamous cancer patients.
Fig. S2. Effects of P2‐0 antibody on the metastasis of Aggrus‐negative A375M cells.
Supporting info item
Acknowledgment
This study was supported in part by a grant from the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation, Japan (to NF).
References
- 1. Kato Y, Kaneko M, Sata M, Fujita N, Tsuruo T, Osawa M. Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet‐aggregation‐inducing factor in lung squamous cell carcinoma. Tumour Biol 2005; 26: 195–200. [DOI] [PubMed] [Google Scholar]
- 2. Martín‐Villar E, Scholl FG, Gamallo C et al. Characterization of human PA2.26 antigen (T1alpha‐2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int J Cancer 2005; 113: 899–910. [DOI] [PubMed] [Google Scholar]
- 3. Yuan P, Temam S, El‐Naggar A et al. Overexpression of podoplanin in oral cancer and its association with poor clinical outcome. Cancer 2006; 107: 563–9. [DOI] [PubMed] [Google Scholar]
- 4. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial‐mesenchymal transition: podoplanin‐mediated remodeling of the actin cytoskeleton. Cancer Cell 2006; 9: 261–72. [DOI] [PubMed] [Google Scholar]
- 5. Kimura N, Kimura I. Podoplanin as a marker for mesothelioma. Pathol Int 2005; 55: 83–6. [DOI] [PubMed] [Google Scholar]
- 6. Fukunaga M. Expression of D2‐40 in lymphatic endothelium of normal tissues and in vascular tumours. Histopathology 2005; 46: 396–402. [DOI] [PubMed] [Google Scholar]
- 7. Kato Y, Sasagawa I, Kaneko M, Osawa M, Fujita N, Tsuruo T. Aggrus: a diagnostic marker that distinguishes seminoma from embryonal carcinoma in testicular germ cell tumors. Oncogene 2004; 23: 8552–6. [DOI] [PubMed] [Google Scholar]
- 8. Mishima K, Kato Y, Kaneko MK et al. Podoplanin expression in primary central nervous system germ cell tumors: a useful histological marker for the diagnosis of germinoma. Acta Neuropathol 2006; 111: 563–8. [DOI] [PubMed] [Google Scholar]
- 9. Mishima K, Kato Y, Kaneko MK, Nishikawa R, Hirose T, Matsutani M. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol 2006; 111: 483–8. [DOI] [PubMed] [Google Scholar]
- 10. Kunita A, Kashima TG, Morishita Y et al. The platelet aggregation‐inducing factor aggrus/podoplanin promotes pulmonary metastasis. Am J Pathol 2007; 170: 1337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato Y, Fujita N, Kunita A et al. Molecular identification of Aggrus/T1alpha as a platelet aggregation‐inducing factor expressed in colorectal tumors. J Biol Chem 2003; 278: 51599–605. [DOI] [PubMed] [Google Scholar]
- 12. Malik AB. Pulmonary microembolism. Physiol Rev 1983; 63: 1114–207. [DOI] [PubMed] [Google Scholar]
- 13. Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor embolilabeled with 125 I‐5‐iodo‐2′‐deoxyuridine. J Natl Cancer Inst 1970; 45: 773–82. [PubMed] [Google Scholar]
- 14. Philippe C, Philippe B, Fouqueray B, Perez J, Lebret M, Baud L. Protection from tumor necrosis factor‐mediated cytolysis by platelets. Am J Pathol 1993; 143: 1713–23. [PMC free article] [PubMed] [Google Scholar]
- 15. Suzuki‐Inoue K, Kato Y, Inoue O et al. Involvement of the snake toxin receptor CLEC‐2, in podoplanin‐mediated platelet activation, by cancer cells. J Biol Chem 2007; 282: 25993–6001. [DOI] [PubMed] [Google Scholar]
- 16. Christou CM, Pearce AC, Watson AA et al. Renal cells activate the platelet receptor CLEC‐2 through podoplanin. Biochem J 2008; 411: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki‐Inoue K, Fuller GL, García A et al. A novel Syk‐dependent mechanism of platelet activation by the C‐type lectin receptor CLEC‐2. Blood 2006; 107: 542–9. [DOI] [PubMed] [Google Scholar]
- 18. Niwa R, Shoji‐Hosaka E, Sakurada M et al. Defucosylated chimeric anti‐CC chemokine receptor 4 IgG1 with enhanced antibody‐dependent cellular cytotoxicity shows potent therapeutic activity to T‐cell leukemia and lymphoma. Cancer Res 2004; 64: 2127–33. [DOI] [PubMed] [Google Scholar]
- 19. Kato Y, Kaneko MK, Kunita A et al. Molecular analysis of the pathophysiological binding of the platelet aggregation‐inducing factor podoplanin to the C‐type lectin‐like receptor CLEC‐2. Cancer Sci 2008; 99: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogasawara S, Kaneko MK, Price JE, Kato Y. Characterization of anti‐podoplanin monoclonal antibodies: critical epitopes for neutralizing the interaction between podoplanin and CLEC‐2. Hybridoma 2008; 27: 259–67. [DOI] [PubMed] [Google Scholar]
- 21. Kato Y, Kaneko MK, Kuno A et al. Inhibition of tumor cell‐induced platelet aggregation using a novel anti‐podoplanin antibody reacting with its platelet‐aggregation‐stimulating domain. Biochem Biophys Res Commun 2006; 349: 1301–7. [DOI] [PubMed] [Google Scholar]
- 22. Kaneko M, Kato Y, Kunita A, Fujita N, Tsuruo T, Osawa M. Functional sialylated O‐glycan to platelet aggregation on Aggrus (T1alpha/Podoplanin) molecules expressed in Chinese hamster ovary cells. J Biol Chem 2004; 279: 38838–43. [DOI] [PubMed] [Google Scholar]
- 23. Nakazawa Y, Sato S, Naito M et al. Tetraspanin family member CD9 inhibits Aggrus/podoplanin‐induced platelet aggregation and suppresses pulmonary metastasis. Blood 2008; 112: 1730–9. [DOI] [PubMed] [Google Scholar]
- 24. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease‐activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004; 104: 397–401. [DOI] [PubMed] [Google Scholar]
- 25. Blom JW, Vanderschoot JP, Oostindiër MJ, Osanto S, van der Meer FJ, Rosendaal FR. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost 2006; 4: 529–35. [DOI] [PubMed] [Google Scholar]
- 26. Klerk CP, Smorenburg SM, Otten HM et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23: 2130–5. [DOI] [PubMed] [Google Scholar]
- 27. Boucharaba A, Serre CM, Grès S et al. Platelet‐derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest 2004; 114: 1714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Massberg S, Konrad I, Schürzinger K et al. Platelets secrete stromal cell‐derived factor 1alpha and recruit bone marrow‐derived progenitor cells to arterial thrombi in vivo . J Exp Med 2006; 203: 1221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin DK, Shido K, Kopp HG et al. Cytokine‐mediated deployment of SDF‐1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 2006; 12: 557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki‐Inoue K, Inoue O, Ding G et al. Essential in vivo roles of the C‐type lectin receptor CLEC‐2: embryonic/neonatal lethality of CLEC‐2‐deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC‐2‐deficient platelets. J Biol Chem 2010; 285: 24494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. May F, Hagedorn I, Pleines I et al. CLEC‐2 is an essential platelet‐activating receptor in hemostasis and thrombosis. Blood 2009; 114: 3464–72. [DOI] [PubMed] [Google Scholar]
- 32. Uhrin P, Zaujec J, Breuss JM et al. Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood 2010; 115: 3997–4005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Quantitative analysis of aggrus expression in tumorigenic and corresponding normal tissues from lung squamous cancer patients.
Fig. S2. Effects of P2‐0 antibody on the metastasis of Aggrus‐negative A375M cells.
Supporting info item